Chronic Exposure to Particulate Nickel Induces Neoplastic Transformation in Human Lung Epithelial Cells

Abstract
:1. Introduction
2. Experimental Section
2.1. Chemicals and Reagents
2.2. Cells and Cell Culture
2.3. Nickel Subsulfide Preparation
2.4. Cytotoxicity Assay
2.5. Clastogenicity Assay
2.6. Transformation Assay
2.7. Statistical Analysis
3. Results and Discussion
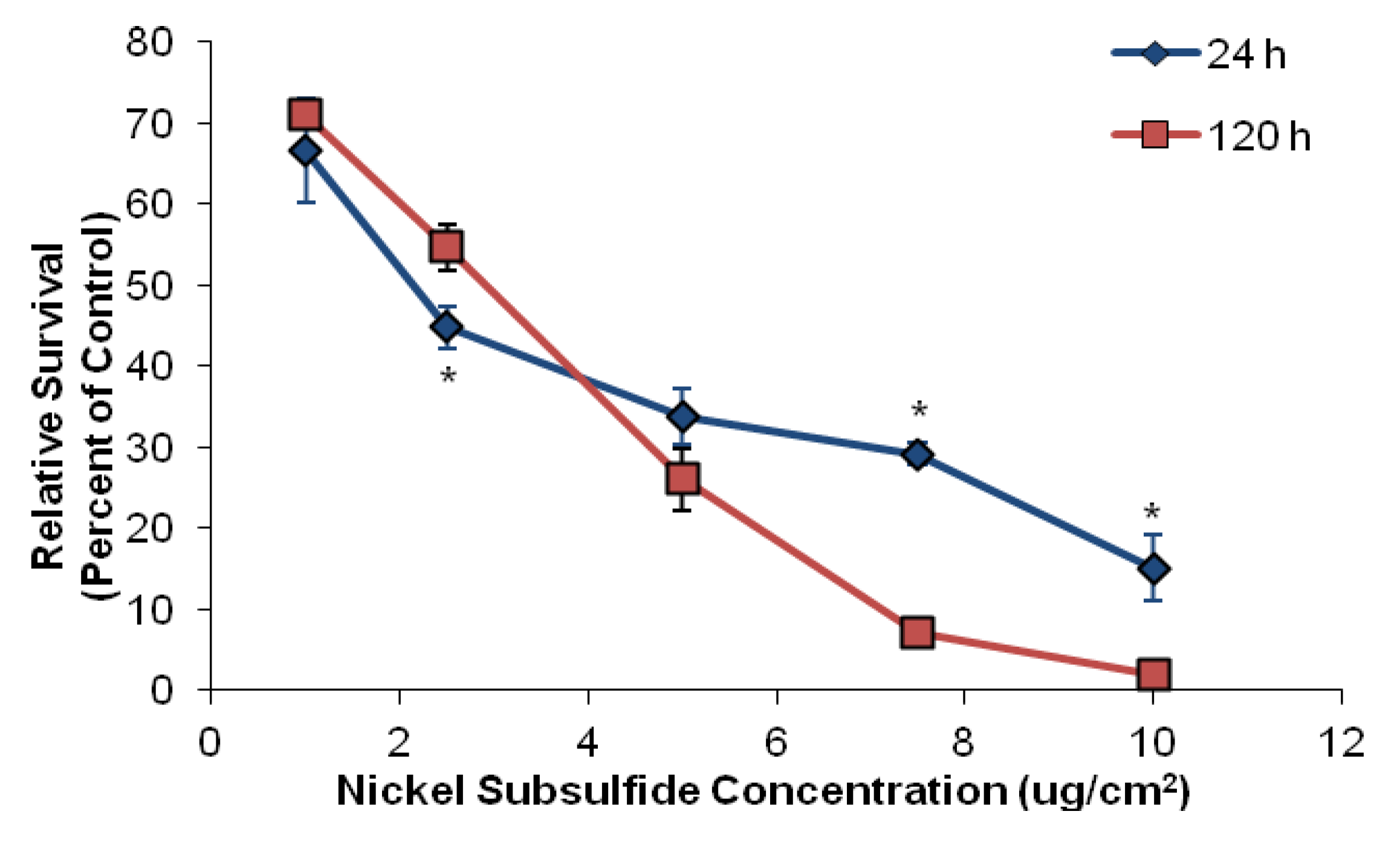
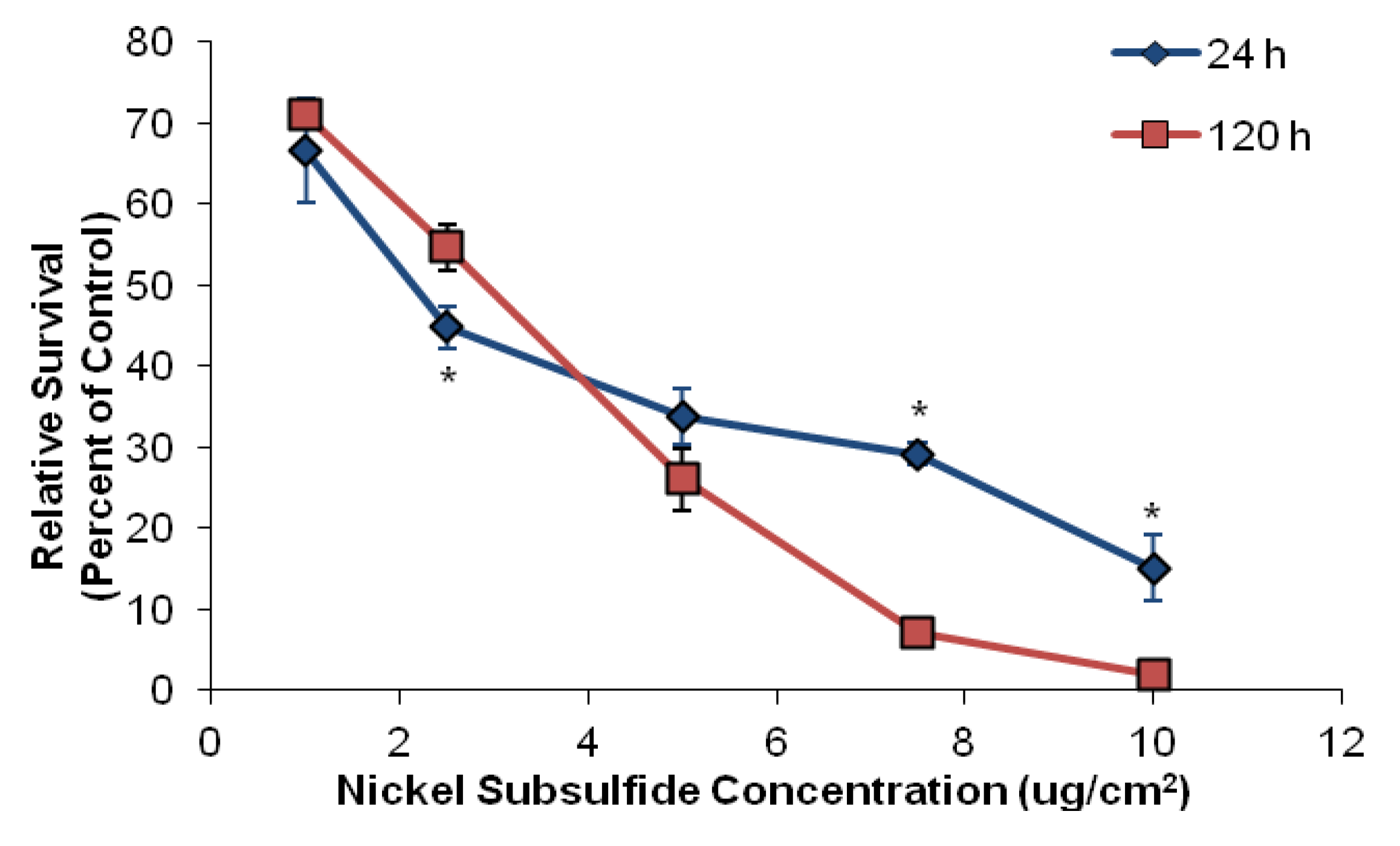
3.1. Nickel Subsulfide Is Cytotoxic to Human Lung Epithelial Cells
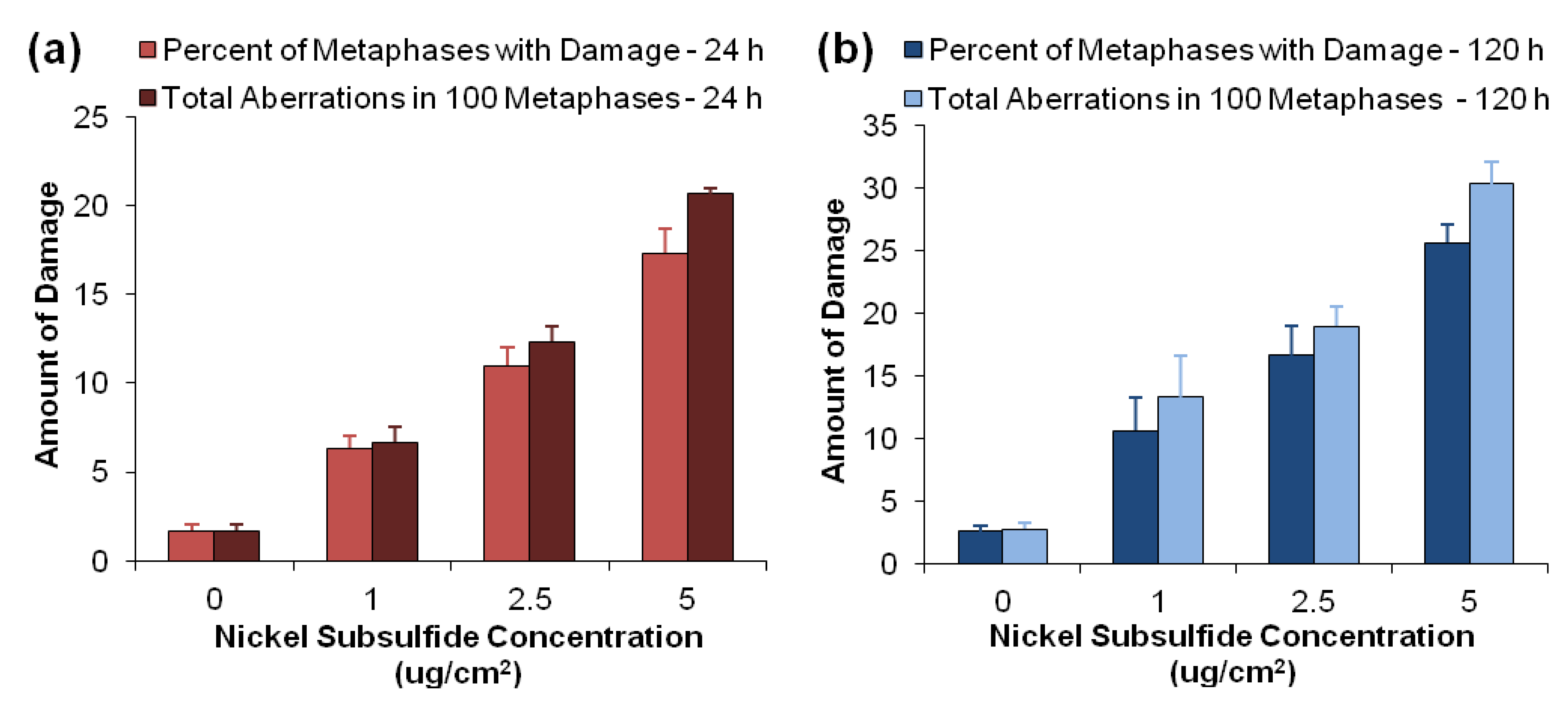
3.2. Nickel Subsulfide Is Clastogenic to Human Lung Epithelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ni3S2 concentration (μg/cm2) | Chromatid lesion | Isochromatid lesion | Chromatid exchange | Dicentric | Double minute | Centromere spreading | Total damage |
|---|---|---|---|---|---|---|---|
| 24 h Exposure | |||||||
| 0 | 1.7 ± 0.3 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 1.7 ± 0.3 |
| 1 | 5.3 ± 0.9 * | 0.3 ± 0.3 | 0 ± 0 | 0.7 ± 0.3 | 0.3 ± 0.3 | 0 ± 0 | 6.7 ± 0.9 * |
| 2.5 | 11.3 ± 1.3 * | 0.7 ± 0.7 | 0 ± 0 | 0 ± 0 | 0.3 ± 0.3 | 0 ± 0 | 12.3 ± 0.9 * |
| 5 | 18.7 ± 0.3 * | 1.3 ± 0.3 * | 0 ± 0 | 0 ± 0 | 0 ± 0.3 | 0.3 ± 0.3 | 21.7 ± 0.3 * |
| 120 h Exposure | |||||||
| 0 | 2.3 ± 0.5 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 2.8 ± 0.5 |
| 1 | 10.7 ± 3.7 * | 0.3 ± 0.3 | 0 ± 0 | 0.7 ± 0.3 | 1.7 ± 0.7 *,† | 0 ± 0 | 13.3 ± 3.3 * |
| 2.5 | 15.7 ± 4.1 * | 1.0 ± 0.5 | 0 ± 0 | 1.3 ± 0.3 *,† | 1.3 ± 0.7 | 0 ± 0 | 19.0 ± 1.5 *,† |
| 5 | 26.3 ± 1.5 *,† | 0.3 ± 0.3 | 0 ± 0 | 1.3 ± 1.3 | 1.0 ± 0 * | 1 ± 0.6 | 30.3 ± 1.8 *,† |
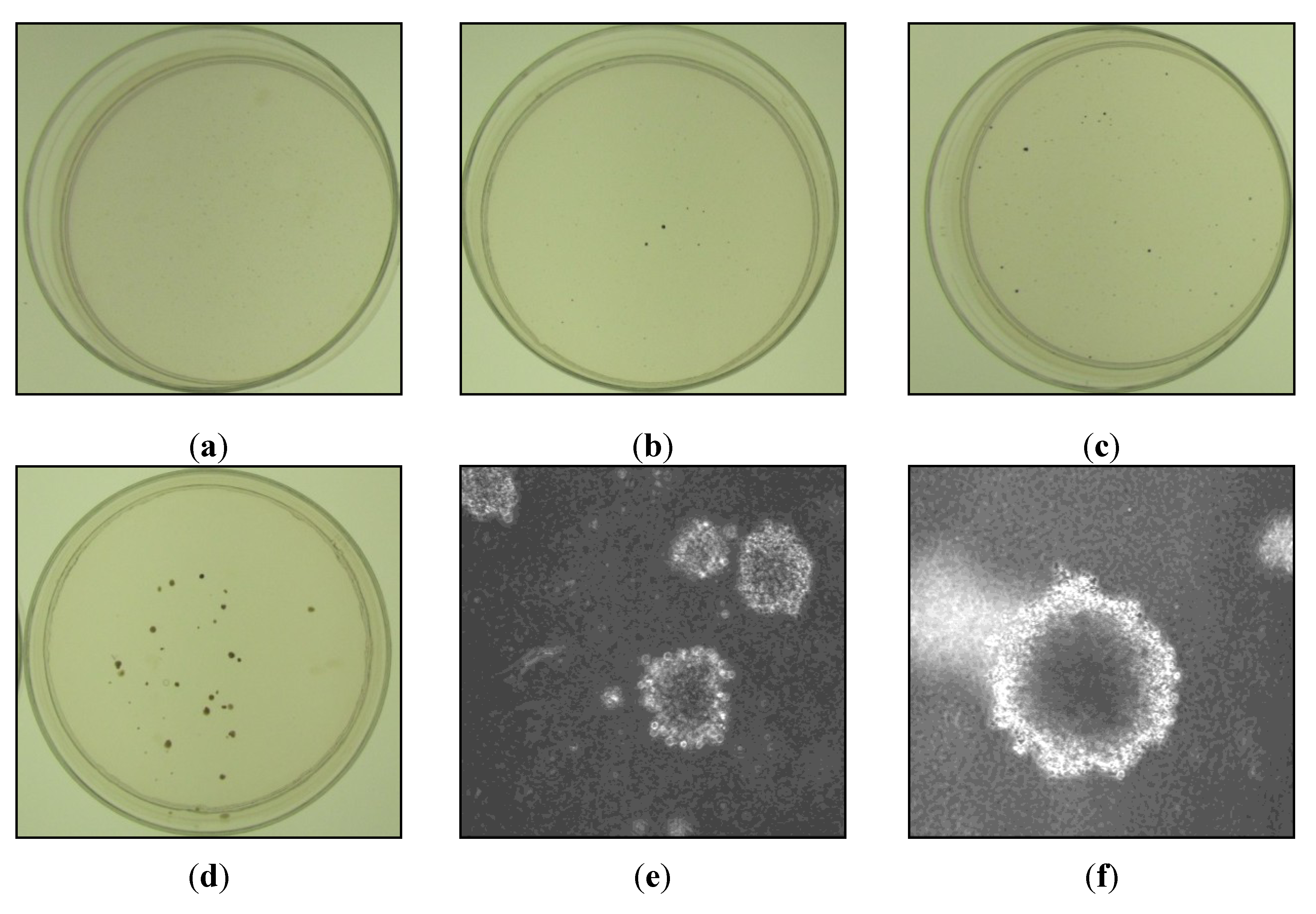
3.3. Nickel Subsulfide Induces Transformation in Human Lung Epithelial Cells

| Nickel subsulfide concentration (μg/cm2) | Exposure time | Foci frequency foci/dish | Number of foci isolated | Percentage of foci with growth in soft agar |
|---|---|---|---|---|
| 0 | 120 h | 0 (0/20) | 27/27 † | 0% |
| 1 | 120 h | 2.55 (51/20) * | 51/51 | 61% (26/43) |
| 2.5 | 120 h | 2.9 (29/10) * | 27/29 | 100% (27/27) |
| 5 | 120 h | 2.35 (47/20) * | 44/47 | 70% (31/44) |

| Nickel subsulfide concentration (μg/cm2) | Foci tested in soft agar | Percent of foci with growth in soft agar * | ||||
|---|---|---|---|---|---|---|
| 0 colonies/dish | 1–10 colonies/dish | 11–20 colonies/dish | 21–30 colonies/dish | >30 colonies/dish | ||
| 1 | 43 | 39% (17) | 26% (11) | 35% (15) | 0% (0) | 0% (0) |
| 2.5 | 27 | 0% (0) | 7% (2) | 67% (18) | 22% (6) | 4% (1) |
| 5 | 44 | 30% (13) | 2% (1) | 7% (3) | 25% (11) | 36% (16) |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Agency for Toxic Substances and Disease Registry. Toxicological Profile for Nickel; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2005. [Google Scholar]
- International Association for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. In Chromium, Nickel and Welding; International Agency for Research on Cancer, World Health Organization: Lyon, France, 1990; Volume 49, pp. 257–445. [Google Scholar]
- International Committee on Nickel Carcinogenesis in Man. Report of the International Committee on Nickel Carcinogenesis in Man. Scandinavian J. Work Environ. Healt 1990, 16, 1–82. [Google Scholar] [CrossRef]
- National Toxicology Program. (NTP) National Toxicology Program technical report on the toxicology and carcinogenesis studies of nickel oxide (CAS No. 1313–99–1) in F344/N rats and B6C3F1 mice (inhalation studies). Natl. Toxicol. Program Tech. Rep. Ser. 1996, 451, 1–381. [Google Scholar]
- National Toxicology Program. (NTP) National Toxicology Program technical report on the toxicology and carcinogenesis studies of nickel subsulfide (CAS No. 12035-72-2) in F344/N rats and B6C3F1 mice (inhalation studies). Natl. Toxicol. Program Tech. Rep. Ser. 1996, 453, 1–365. [Google Scholar]
- Abbracchio, M.P.; Simmons-Hansen, J.; Costa, M. Cytoplasmic dissolution of phagocytized crystalline nickel sulfide particles: A prerequisite for nuclear uptake of nickel. J. Toxicol. Environ. Health 1982, 9, 663–676. [Google Scholar] [CrossRef]
- Cangul, H.; Broday, L.; Salnikow, K.; Sutherland, J.; Peng, W.; Zhang, Q.; Poltaratsky, V.; Yee, H.; Zoroddu, M.A.; Costa, M. Molecular mechanisms of nickel carcinogenesis. Toxicol. Lett. 2002, 127, 69–75. [Google Scholar] [CrossRef]
- Sunderman, F.W., Jr.; Morgan, L.G.; Andersen, A.; Ashley, D.; Forouhar, F.A. Histopathology of sinonasal and lung cancers in nickel refinery workers. Ann. Clinical. Lab. Sci. 1989, 19, 44–50. [Google Scholar]
- Akslen, L.A.; Myking, A.O.; Mørkve, O.; Gulsvik, A.; Raithel, H.J.; Schaller, K.H. Increased content of chromium and nickel in lung tissues from patients with bronchial carcinoma. Pathol. Res. Pract. 1990, 186, 717–722. [Google Scholar] [CrossRef]
- Ji, W.D.; Chen, J.K.; Lu, J.C.; Wu, Z.L.; Yi, F.; Feng, S.M. Alterations of FHIT gene and P16 gene in nickel transformed human bronchial epithelial cells. Biomed. Environ. Sci. 2006, 19, 277–284. [Google Scholar]
- Willey, J.C.; Broussoud, A.; Sleemi, A.; Bennet, W.P.; Cerutti, P.; Harris, C.C. Immortilization of normal human bronchial epithelial cells by human papillomaviruses 16 or 18. Cancer Res. 1991, 51, 5370–5377. [Google Scholar]
- Costa, M.; Abbracchio, M.P.; Simmons-Hansen, J. Factors influencing the phagocytosis, neoplastic transformation, and cytotoxicity of particulate nickel compounds in tissue culture systems. Toxicol. Appl. Pharmacol. 1981, 60, 313–323. [Google Scholar] [CrossRef]
- Wise, J.P., Sr.; Wise, S.S.; Little, J.E. The cytotoxicity and genotoxicity of particulate and soluble hexavalent chromium in human lung cells. Mutat. Res. 2002, 517, 221–229. [Google Scholar] [CrossRef]
- Xie, H.; Holmes, A.L.; Wise, S.; Huang, S.; Peng, C.; Wise, J.P., Sr. Neoplastic transformation of human bronchial cells by lead chromate particles. Am. J. Respir. Cell Mol. Biol. 2007, 37, 544–552. [Google Scholar] [CrossRef]
- Rothman, K.J. Statistics in nonrandomized studies. Epidemiology 1990, 1, 417–418. [Google Scholar] [CrossRef]
- Hildebrand, H.F.; D’Hooghe, M.C.; Shirali, P.; Bailly, C.; Kerckaert, J.P. Uptake and biological transformation of beta NiS and alpha Ni3S2 by human embryonic pulmonary epithelial cells (L132) in culture. Carcinogenesis 1990, 11, 1943–1950. [Google Scholar]
- Sen, P.; Costa, M. Induction of chromosomal damage in Chinese hamster ovary cells by soluble and particulate nickel compounds: Preferential fragmentation of the heterochromatic long arm of the X-chromosome by carcinogenic crystalline NiS particles. Cancer Res. 1985, 45, 2320–2325. [Google Scholar]
- Sen, P.; Costa, M. Pathway of nickel uptake influences its interaction with heterochromatic DNA. Toxicol. Appl. Pharmacol. 1986, 84, 278–285. [Google Scholar] [CrossRef]
- Sen, P.; Conway, K.; Costa, M. Comparison of the localization of chromosome damage induced by calcium chromate and nickel compounds. Cancer Res. 1987, 47, 2142–2147. [Google Scholar]
- Lin, X.H.; Sugiyama, M.; Costa, M. Differences in the effect of vitamin E on nickel sulfide or nickel chloride-induced chromosomal aberrations in mammalian cells. Mutat. Res. 1991, 260, 159–164. [Google Scholar] [CrossRef]
- Landolph, J.R. Molecular mechanisms of transformation of C3H/10T1/2 C1 8 mouse embryo cells and diploid human fibroblasts by carcinogenic metal compounds. Environ. Health Perspect. 1994, 102, 119–125. [Google Scholar]
- Huang, X.; Zhuang, Z.; Frenkel, K.; Klein, C.B.; Costa, M. The role of nickel and nickel-mediated reactive oxygen species in the mechanism of nickel carcinogenesis. Environ. Health Perspect. 1994, 102, 281–284. [Google Scholar]
- Britten, R.J. Rate of DNA sequence evolution between different taxonomic groups. Science 1986, 231, 1393–1398. [Google Scholar]
- Dávila-Rodríguez, M.I.; Cortés Gutiérrez, E.I.; Cerda Flores, R.M.; Pita, M.; Fernández, J.L.; López-Fernández, C.; Gosálvez, J. Constitutive heterochromatin polymorphisms in human chromosomes identified by whole comparative genomic hybridization. Eur. J. Histochem. 2011, 55, e28. [Google Scholar]
- Murnane, J.P. Telomere dysfunction and chromosome instability. Mutat. Res. 2012, 730, 28–36. [Google Scholar] [CrossRef]
- Biedermann, K.A.; Landolph, J.R. Induction of anchorage independence in human diploid foreskin fibroblasts by carcinogenic metal salts. Cancer Res. 1987, 47, 3815–3823. [Google Scholar]
- Miura, T.; Patierno, S.R.; Sakuramoto, T.; Landolph, J.R. Morphological and neoplastic transformation of C3H/10T1/2 Cl 8 mouse embryo cells by insoluble carcinogenic nickel compounds. Environ. Mol. Mutagen. 1989, 14, 65–78. [Google Scholar] [CrossRef]
- Lu, H.; Shi, X.; Costa, M.; Huang, C. Carcinogenic effect of nickel compounds. Mol. Cell. Biochem. 2005, 279, 45–67. [Google Scholar] [CrossRef]
- Zhang, Q.; Salnikow, K.; Kluz, T.; Chen, L.C.; Su, W.C.; Costa, M. Inhibition and reversal of nickel-induced transformation by the histone deacetylase inhibitor trichostatin A. Toxicol. Appl. Pharmacol. 2003, 192, 201–211. [Google Scholar] [CrossRef]
- Reznikoff, C.A.; Bertram, J.S.; Brankow, D.W.; Heidelberger, C. Quantitative and qualitative studies of chemical transformation of cloned C3H mouse embryo cells sensitive to postconfluence inhibition of cell division. Cancer Res. 1973, 33, 3239–3249. [Google Scholar]
- Xie, X.; LaCerte, C.; Thompson, W.D.; Wise, J.P., Sr. Depleted uranium induces neoplastic transformation in human lung epithelial cells. Chem. Res. Toxicol. 2010, 33, 373–378. [Google Scholar]
- Patierno, S.R.; Banh, D.; Landolph, J.R. Transformation of C3H/10T1/2 mouse embryo cells to focus formation and anchorage independence by insoluble lead chromate but not soluble calcium chromate: relationship to mutagenesis and internalization of lead chromate particles. Cancer Res. 1988, 48, 5280–5288. [Google Scholar]
- Carney, D.N.; Matthews, M.J.; Ihde, D.C.; Bunn, P.A., Jr.; Cohen, M.H.; Makuch, R.W.; Gazdar, A.F.; Minna, J.D. Influence of histologic subtype of small cell carcinoma of the lung on clinical presentation, response to therapy, and survival. J. Natl. Cancer Inst. 1980, 65, 1225–1230. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Holmes, A.L.; The, T.; Thompson, K.; Mason, M.; Kandpal, S.; Zheng, T.; Wise, J.P., Sr. Chronic Exposure to Particulate Nickel Induces Neoplastic Transformation in Human Lung Epithelial Cells. Toxics 2013, 1, 46-59. https://doi.org/10.3390/toxics1010046
Holmes AL, The T, Thompson K, Mason M, Kandpal S, Zheng T, Wise JP Sr. Chronic Exposure to Particulate Nickel Induces Neoplastic Transformation in Human Lung Epithelial Cells. Toxics. 2013; 1(1):46-59. https://doi.org/10.3390/toxics1010046
Chicago/Turabian StyleHolmes, Amie L., Therry The, Kelsey Thompson, Michael Mason, Sanjeev Kandpal, Tongzhang Zheng, and John Pierce Wise, Sr. 2013. "Chronic Exposure to Particulate Nickel Induces Neoplastic Transformation in Human Lung Epithelial Cells" Toxics 1, no. 1: 46-59. https://doi.org/10.3390/toxics1010046