
Determination of Trichloroethylene in Water by Liquid–Liquid Microextraction Assisted Solid Phase Microextraction

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Instruments
2.3. Sample Preparation
3. Results and Discussion
3.1. LLME–SPME Method Optimization
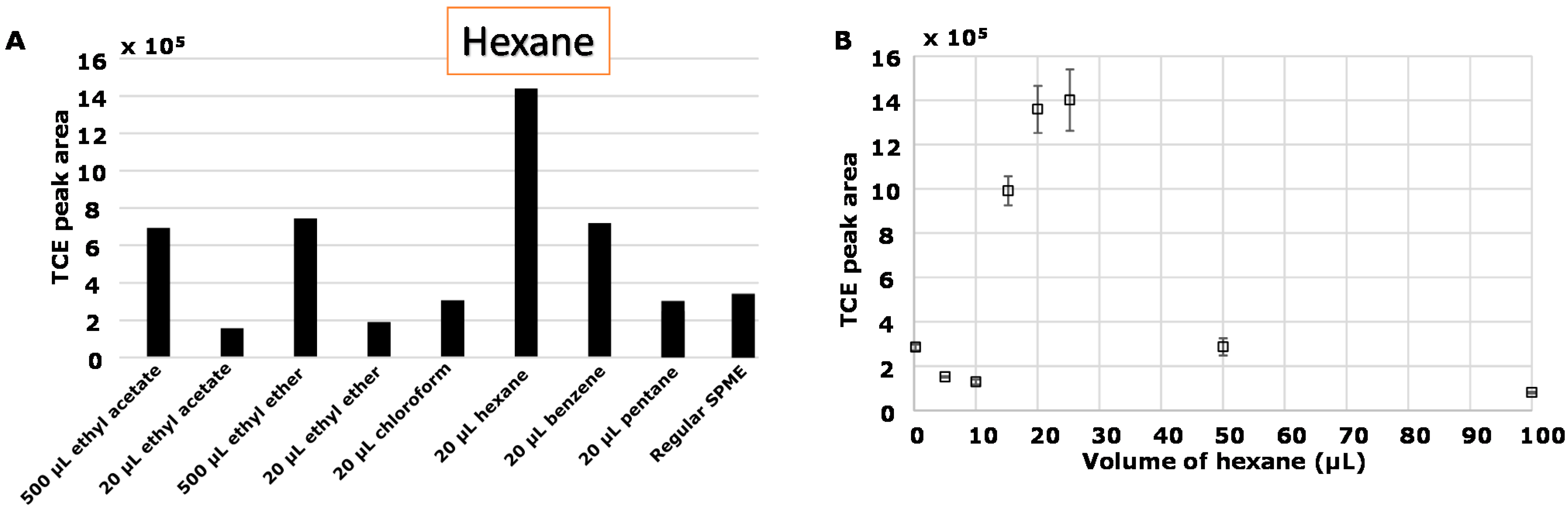
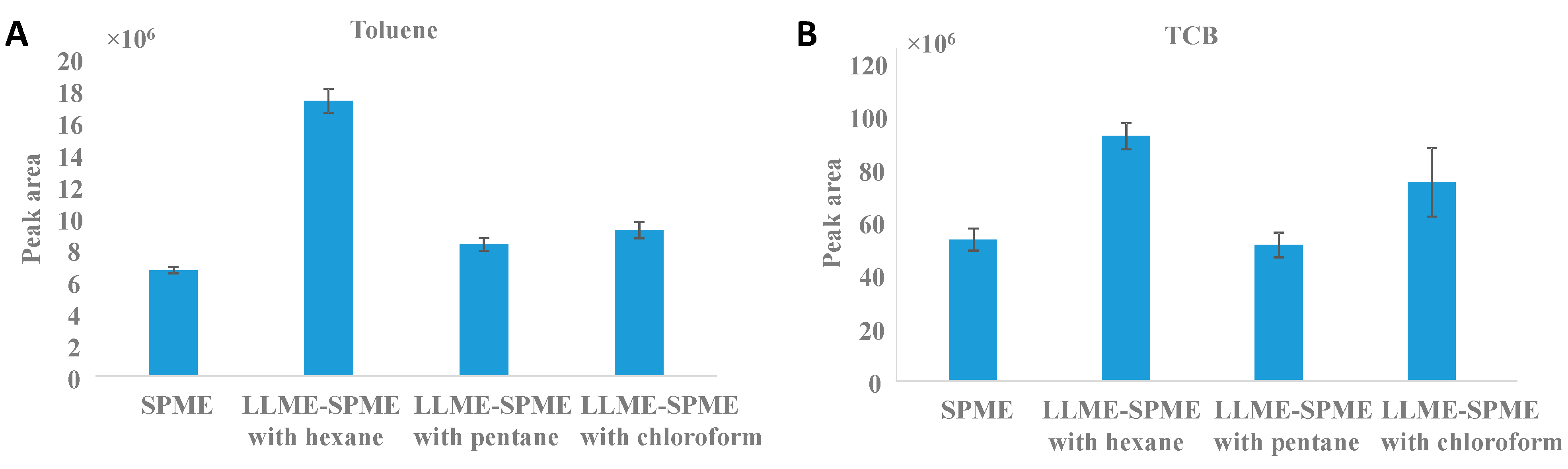
3.1.1. Selection of Organic Solvent


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | RI * | Density (g cm−3) |
|---|---|---|
| TCE | 694 | 1.46 |
| Ethyl ether | 495 | 0.71 |
| Ethyl acetate | 600 | 0.9 |
| Hexane | 600 | 0.65 |
| Chloroform | 628 | 1.49 |
| Benzene | 650 | 0.88 |
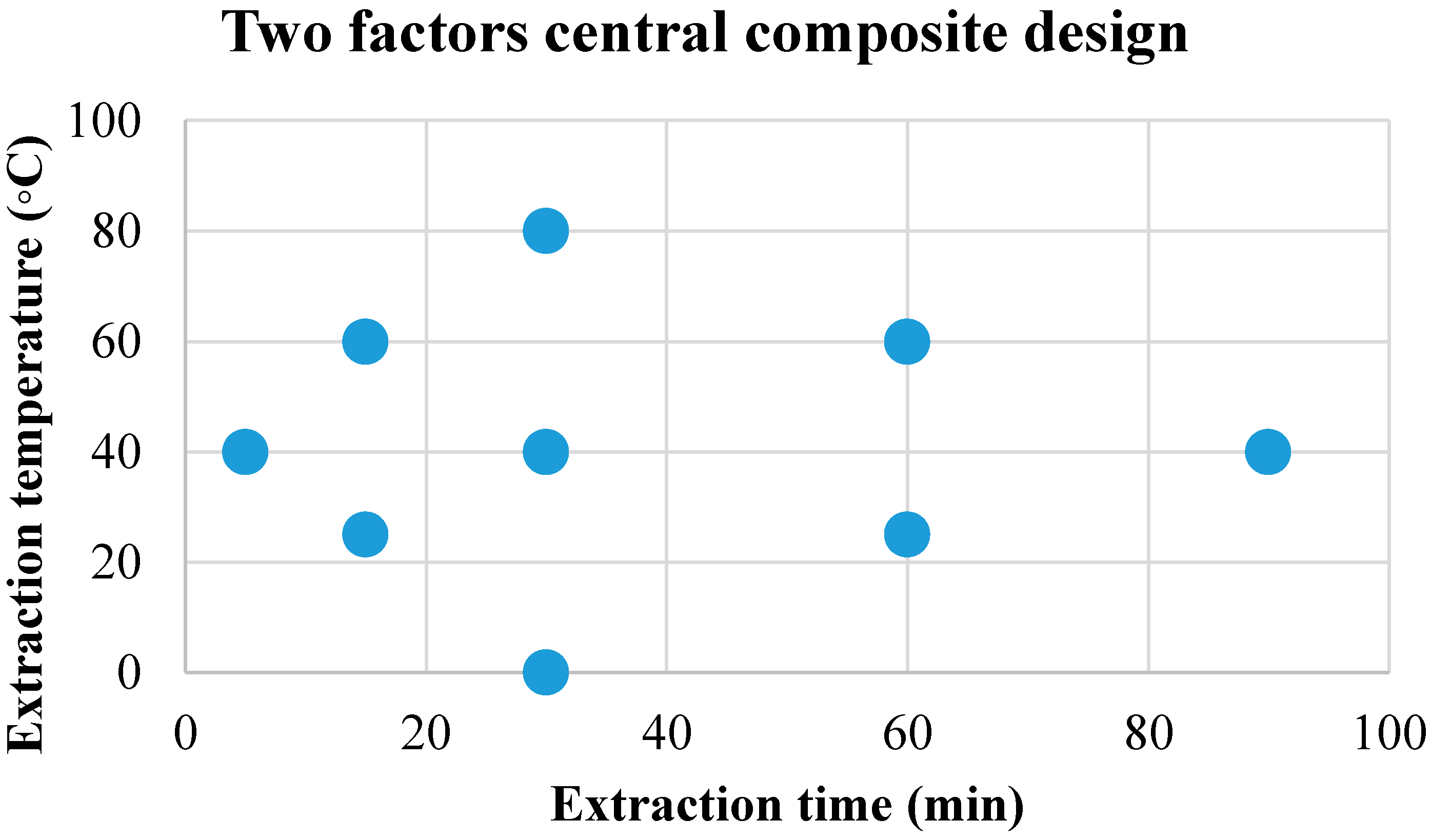
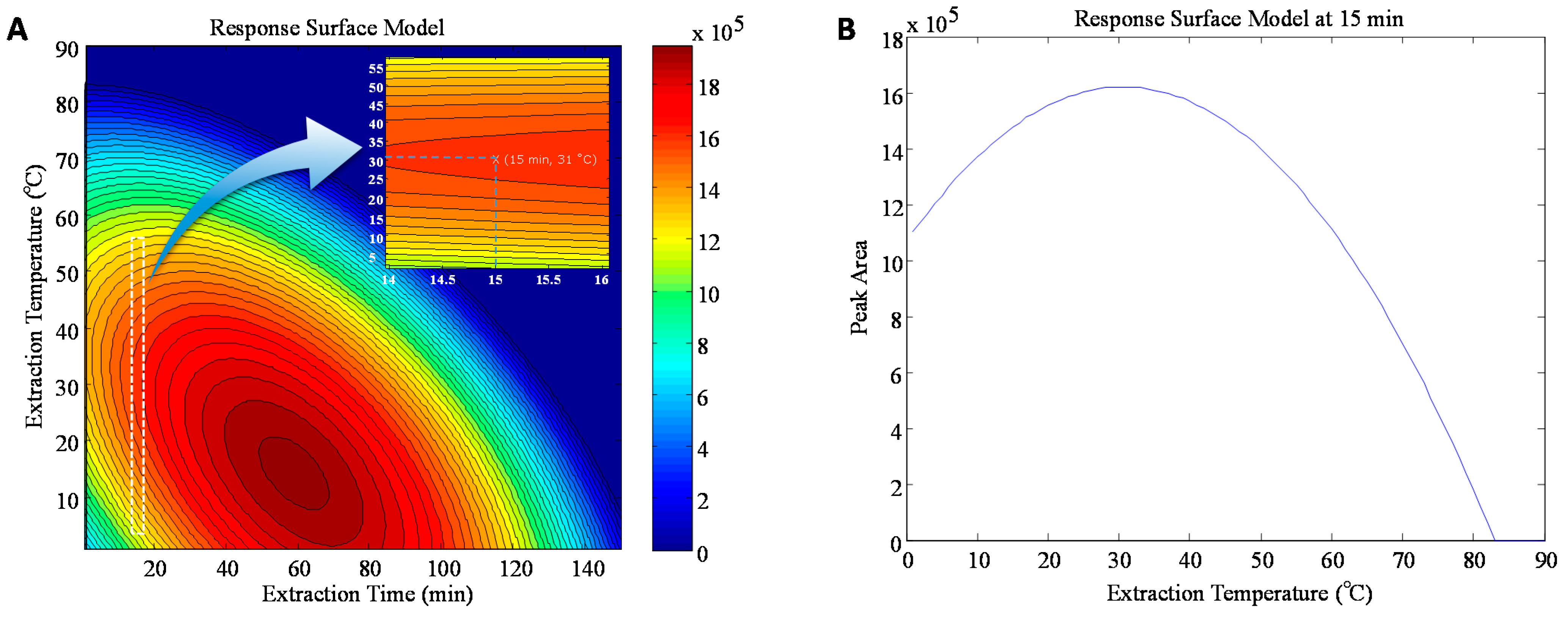
3.1.2. Effect of Extraction Temperature and Time on LLME–SPME


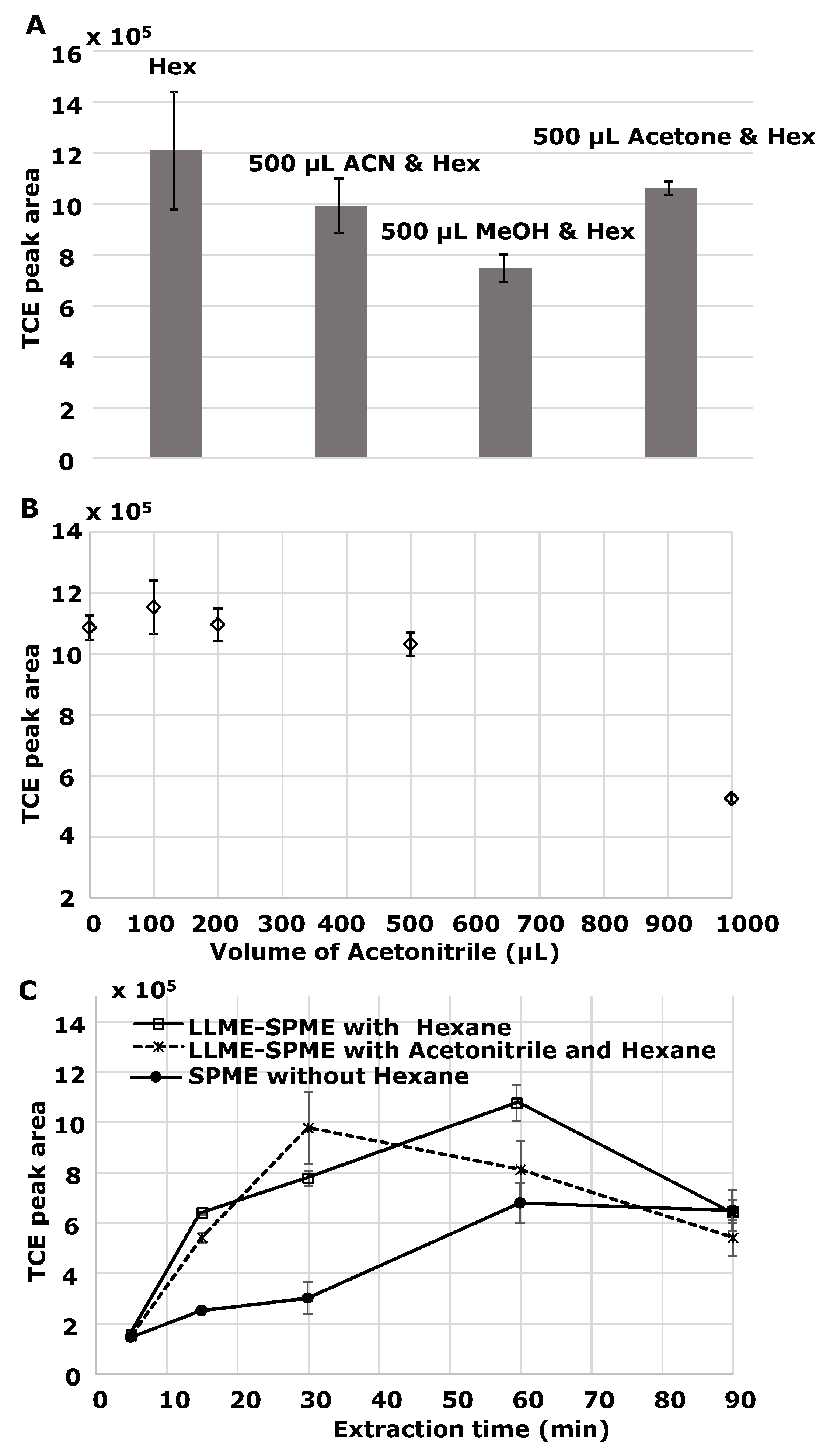
3.1.3. Effect of Dispersive Solvents

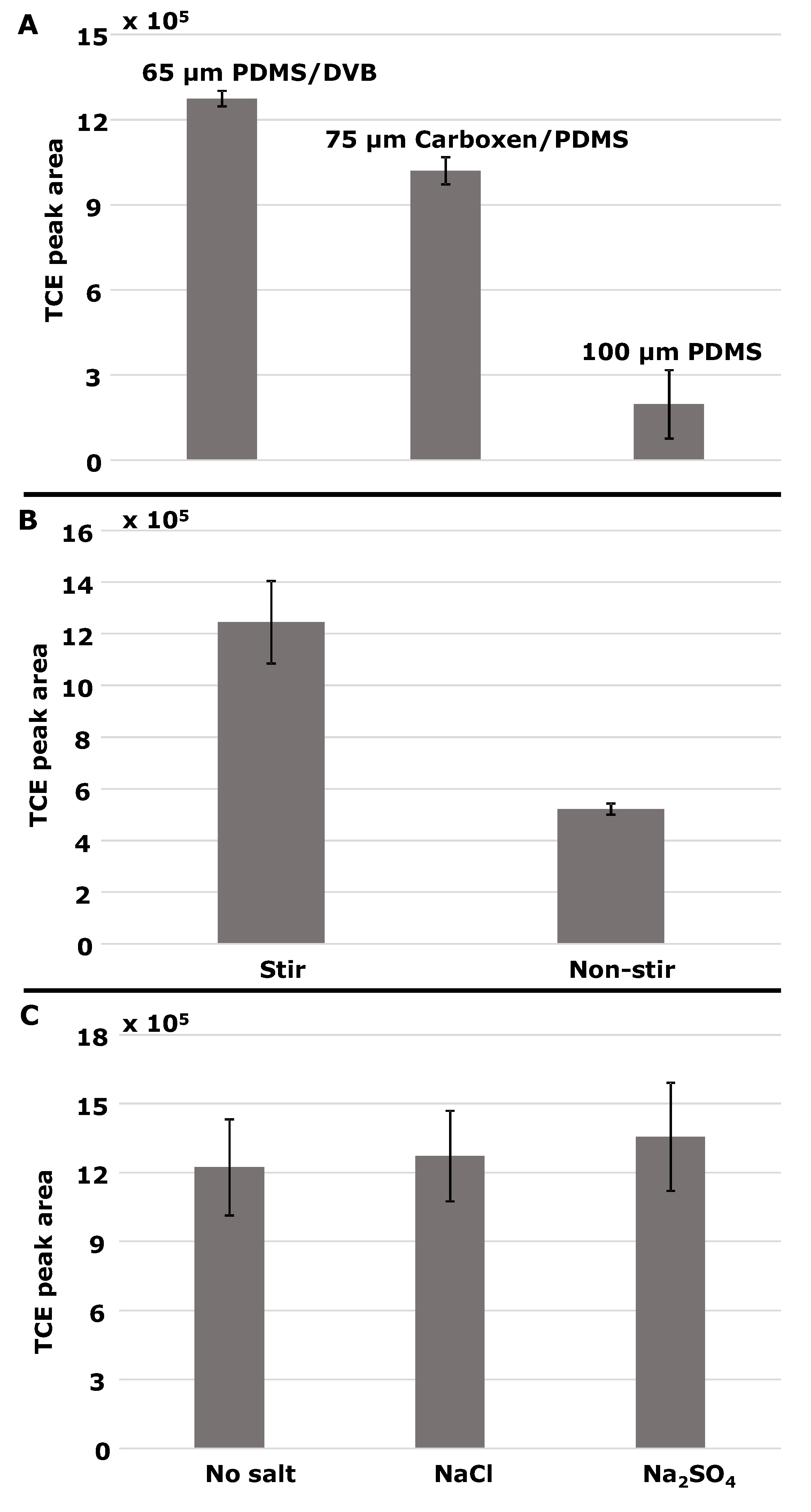
3.1.4. Other Factors: SPME Fiber, Stirring, and Salting Out

3.2. Recoveries and Enrichment Factors
| TCE concentration (ng mL−1) | TCE in the vial (ng) | SPME | LLME-SPME | Enrichment factor | ||
|---|---|---|---|---|---|---|
| TCE on column (ng) | Recovery (%) | TCE on column (ng) | Recovery (%) | |||
| 10 | 100 | 1.1 ± 0.1 | 11 | 2.9 ± 0.4 | 29 | 2.6 ± 0.2 |
| 100 | 1000 | 17 ± 1 | 17 | 41 ± 2 | 41 | 2.4 ± 0.4 |
| 300 | 3000 | 48 ± 3 | 16 | 107 ± 14 | 36 | 2.2 ± 0.3 |
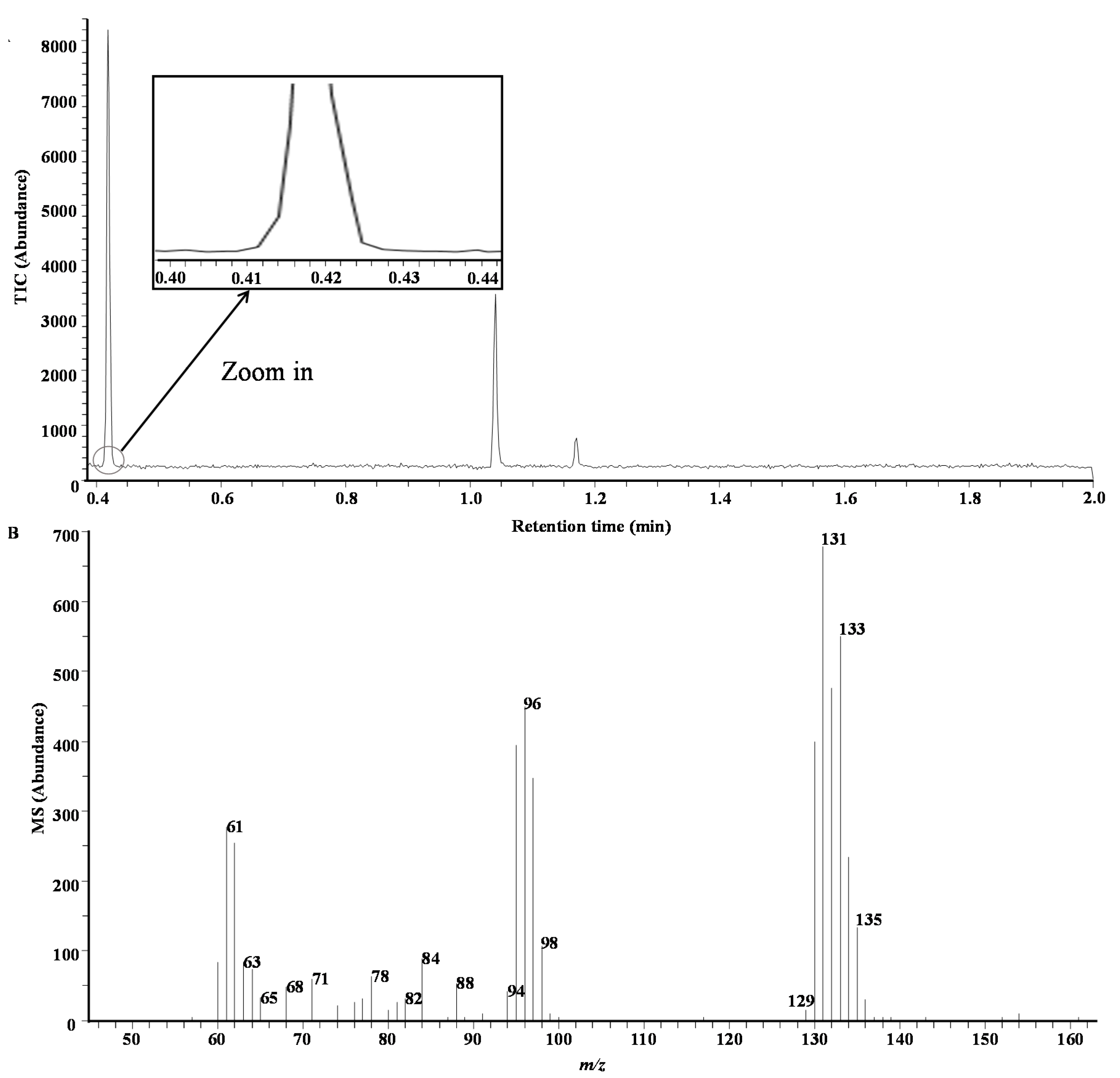
3.3. Application of Extraction Method Using Portable GC/MS Instrument and Validation
| Added concentration (ng mL−1) | Measured concentration (ng mL−1) | RSD (%) | RE (%) |
|---|---|---|---|
| 10 (LLOQ) | 10 ± 1 | 10.8 | 2 |
| 30 | 28 ± 3 | 11.9 | −7 |
| 100 | 88 ± 5 | 5.9 | −12 |
| 300 | 330 ± 15 | 4.4 | 10 |

3.4. Effectiveness of LLME–SPME on Other Volatile Organic Contaminants in Water

4. Conclusion
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Bakke, B.; Stewart, P.A.; Waters, M.A. Uses of and exposure to trichloroethylene in US industry: A systematic literature review. J. Occup. Environ. Hyg. 2007, 4, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Beamer, P.I.; Luik, C.E.; Abrell, L.; Campos, S.; Martinez, M.E.; Saez, A.E. Concentration of trichloroethylene in breast milk and household water from Nogales, Arizona. Environ. Sci. Technol. 2012, 46, 9055–9061. [Google Scholar] [CrossRef] [PubMed]
- Zogorski, J.S.; Carter, J.M.; Ivahnenko, T.; Lapham, W.W.; Moran, M.J.; Rowe, B.L.; Squillace, P.J.; Toccalino, P.L. Volatile Organic Compounds in the Nation’s Ground Water and Drinking-Water Supply Wells; U.S. Geological Survey: Reston, VA, USA, 2006; p. 16. [Google Scholar]
- Forkert, P.G.; Lash, L.; Tardif, R.; Tanphaichitr, N.; Vandevoort, C.; Moussa, M. Identification of trichloroethylene and its metabolites in human seminal fluid of workers exposed to trichloroethylene. Drug Metab. Dispos. 2003, 31, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Munch, D.J.; Hautman, D.P. USEPA Method 551.1: Determination of Chlorination Disinfection Byproducts, Chlorinated Solvents, and Halogenated Pesticides/Herbicides in Drinking Water by Liquid-Liquid Extraction and Gas Chromatography with Electroncapture Detection; US Environmental Protection Agency: Cincinatti, OH, USA, 1995. [Google Scholar]
- Dehon, B.; Humbert, L.; Devisme, L.; Stievenart, M.; Mathieu, D.; Houdret, N.; Lhermitte, M. Tetrachloroethylene and trichloroethylene fatality: Case report and simple headspace SPME-capillary gas chromatographic determination in tissues. J. Anal. Toxicol. 2000, 24, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Cervera, M.I.; Beltran, J.; Lopez, F.J.; Hernandez, F. Determination of volatile organic compounds in water by headspace solid-phase microextraction gas chromatography coupled to tandem mass spectrometry with triple quadrupole analyzer. Anal. Chim. Acta 2011, 704, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Jakubowska, N.; Henkelmann, B.; Schramm, K.W.; Namiesnik, J. Optimization of a novel procedure for determination of VOCs in water and human urine samples based on SBSE coupled with TD-GC-HRMS. J. Chromatogr. Sci. 2009, 47, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Muralidhara, S.; Bruckner, J.V.; Bartlett, M.G. Trace level determination of trichloroethylene in biological samples by headspace solid-phase microextraction gas chromatography/negative chemical ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Poli, D.; Manini, P.; Andreoli, R.; Franchini, I.; Mutti, A. Determination of dichloromethane, trichloroethylene and perchloroethylene in urine samples by headspace solid phase microextraction gas chromatography–mass spectrometry. J. Chromatogr. B 2005, 820, 95–102. [Google Scholar] [CrossRef]
- Xu, N.; Vandegrift, S.; Sewell, G.W. Determination of chloroethenes in environmental biological samples using gas chromatography coupled with solid phase micro extraction. Chromatographia 1996, 42, 313–317. [Google Scholar] [CrossRef]
- Arthur, C.L.; Pawliszyn, J. Solid phase microextraction with thermal desorption using fused silica optical fibers. Anal. Chem. 1990, 62, 2145–2148. [Google Scholar] [CrossRef]
- Risticevic, S.; Niri, V.H.; Vuckovic, D.; Pawliszyn, J. Recent developments in solid-phase microextraction. Anal. Bioanal. Chem. 2009, 393, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Vas, G.; Vekey, K. Solid-phase microextraction: A powerful sample preparation tool prior to mass spectrometric analysis. J. Mass Spectrom. 2004, 39, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Ai, J. Solid phase microextraction for quantitative analysis in nonequilibrium situations. Anal. Chem. 1997, 69, 1230–1236. [Google Scholar] [CrossRef]
- Risticevic, S.; Lord, H.; Gorecki, T.; Arthur, C.L.; Pawliszyn, J. Protocol for solid-phase microextraction method development. Nat. Protoc. 2010, 5, 122–139. [Google Scholar] [CrossRef] [PubMed]
- NIST. Nist standard reference database number 69. Avalable online: http://webbook.nist.gov/chemistry/ (acessed on 14 October 2014).
- Garrido Frenich, A.; Romero-Gonzalez, R.; Martinez Vidal, J.L.; Martinez Ocana, R.; Baquero Feria, P. Comparison of solid phase microextraction and hollow fiber liquid phase microextraction for the determination of pesticides in aqueous samples by gas chromatography triple quadrupole tandem mass spectrometry. Anal. Bioanal. Chem. 2011, 399, 2043–2059. [Google Scholar] [CrossRef] [PubMed]
- Massart, D.L.; Vandeginste, B.G.M.; Deming, S.N.; Michotte, Y.; Kaufman, L. Chemometrics: A Textbook; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Rezaee, M.; Assadi, Y.; Hosseinia, M.R.M.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid-liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.T.; Lin, D.L.; Liu, R.H. Isotopic analogs as internal standards for quantitative analyses by GC/MS—Evaluation of cross-contribution to ions designated for the analyte and the isotopic internal standard. Forensic Sci. Int. 2001, 121, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Harrington, P.B. Application of classical least squares to resolve overlapping mass spectral peak clusters between trichloroethylene and its deuterated internal standard. Rapid Commun. Mass Spectrom. 2015, in press. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Harrington, P.D.B. Determination of Trichloroethylene in Water by Liquid–Liquid Microextraction Assisted Solid Phase Microextraction. Chromatography 2015, 2, 66-78. https://doi.org/10.3390/chromatography2010066
Zhang M, Harrington PDB. Determination of Trichloroethylene in Water by Liquid–Liquid Microextraction Assisted Solid Phase Microextraction. Chromatography. 2015; 2(1):66-78. https://doi.org/10.3390/chromatography2010066
Chicago/Turabian StyleZhang, Mengliang, and Peter De B. Harrington. 2015. "Determination of Trichloroethylene in Water by Liquid–Liquid Microextraction Assisted Solid Phase Microextraction" Chromatography 2, no. 1: 66-78. https://doi.org/10.3390/chromatography2010066