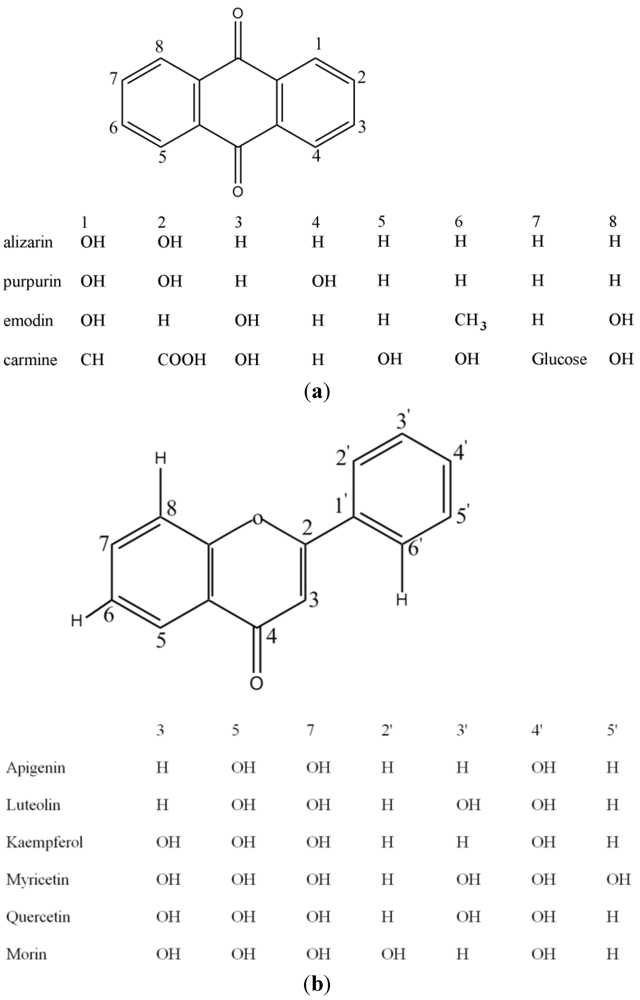
Micellar Electrokinetic Chromatography with Laser-Induced Fluorescence Detection for Separation of Red and Yellow Historical Dyes

Abstract
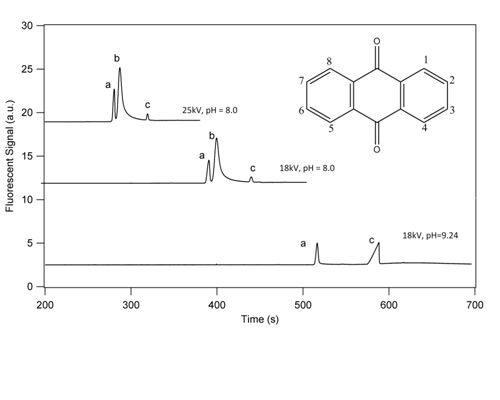
:
1. Introduction
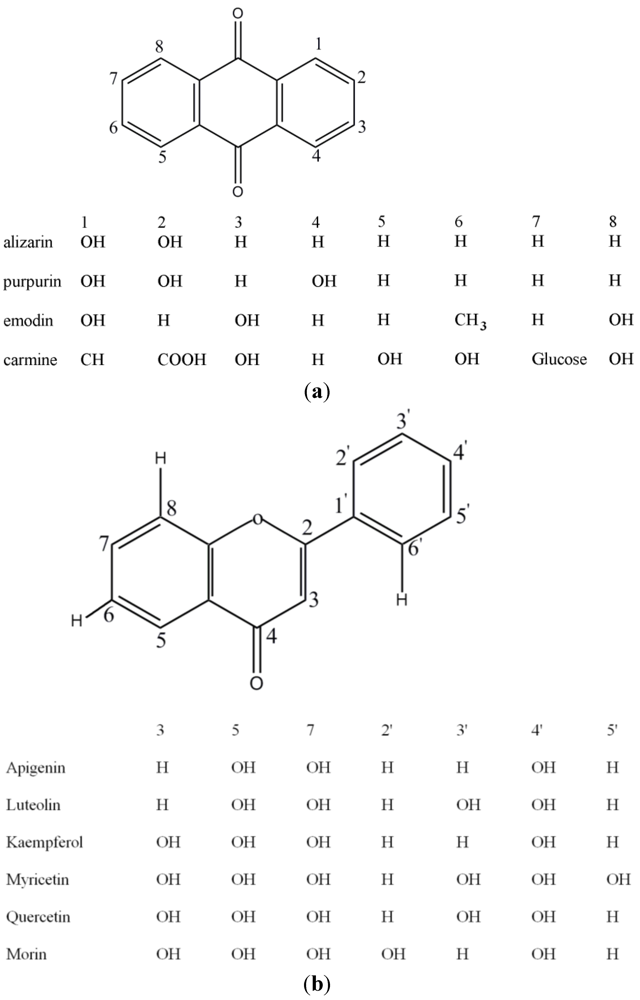
2. Experimental Section
Reagents
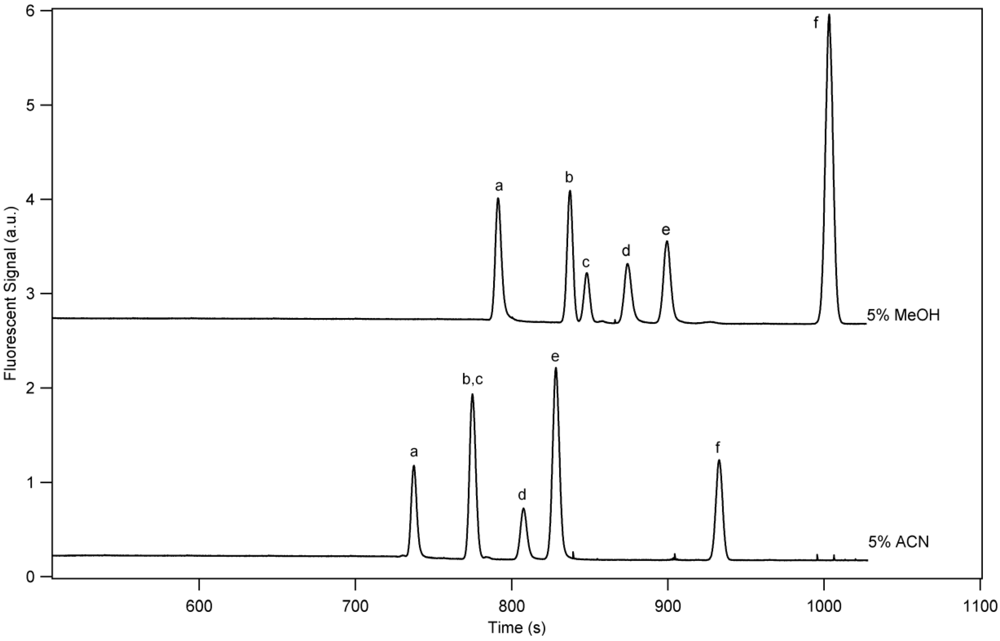
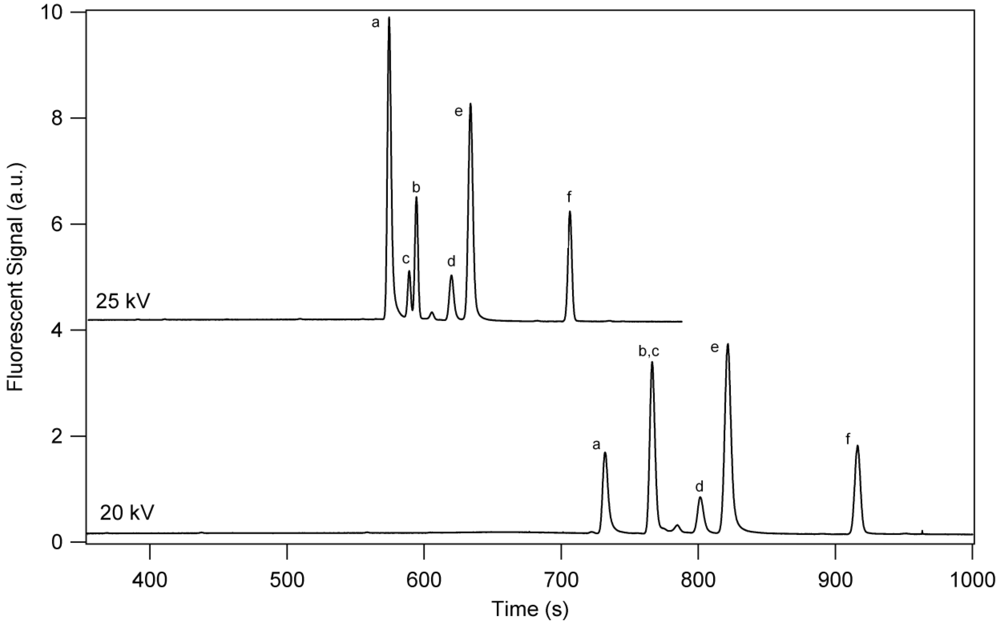
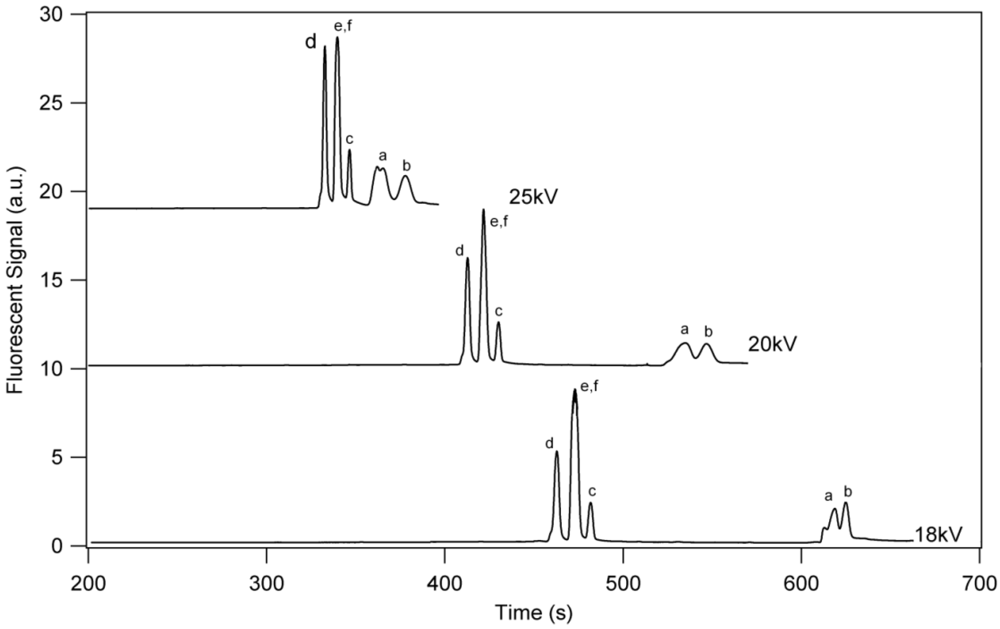
3. Results and Discussion
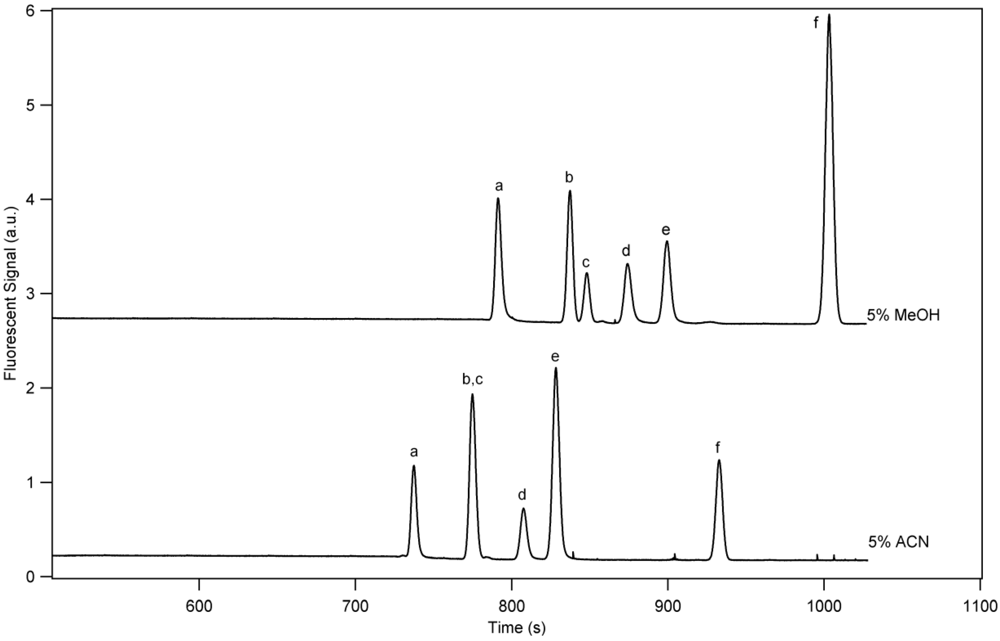
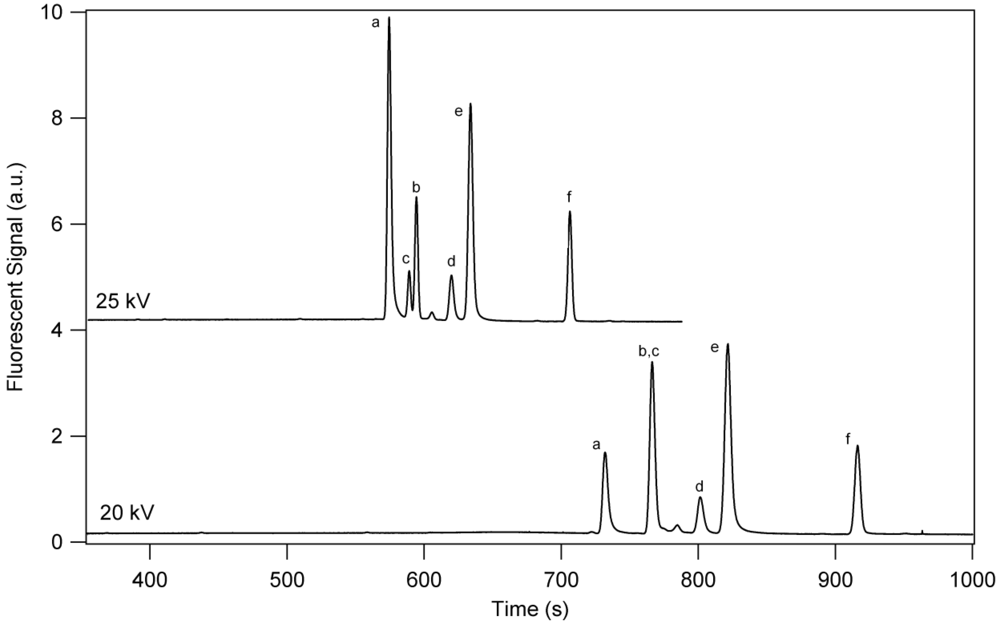
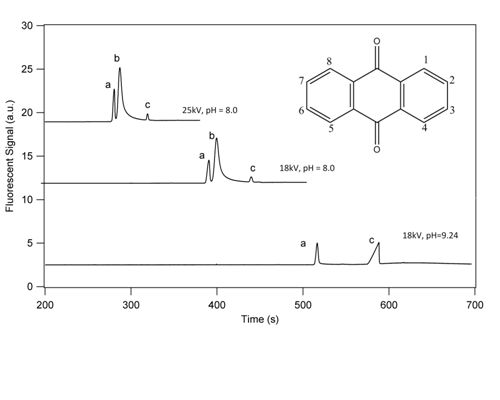
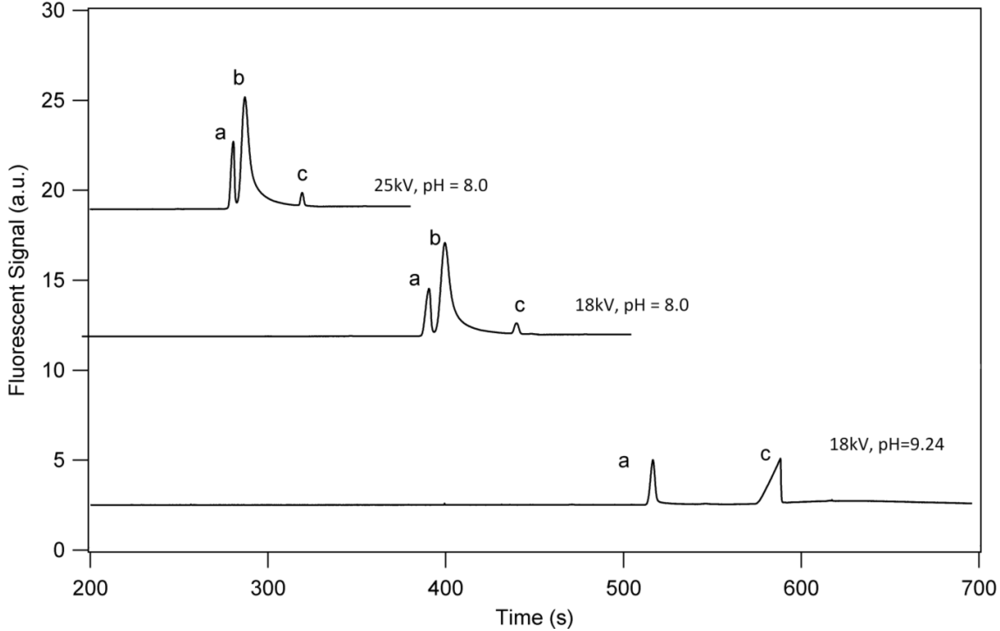
3.1. Separation Properties
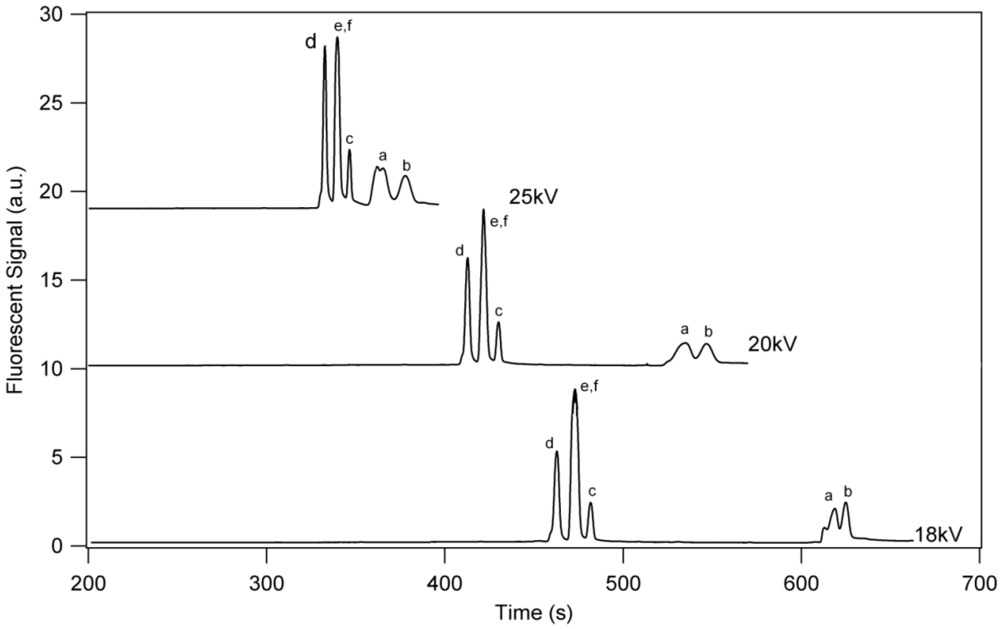
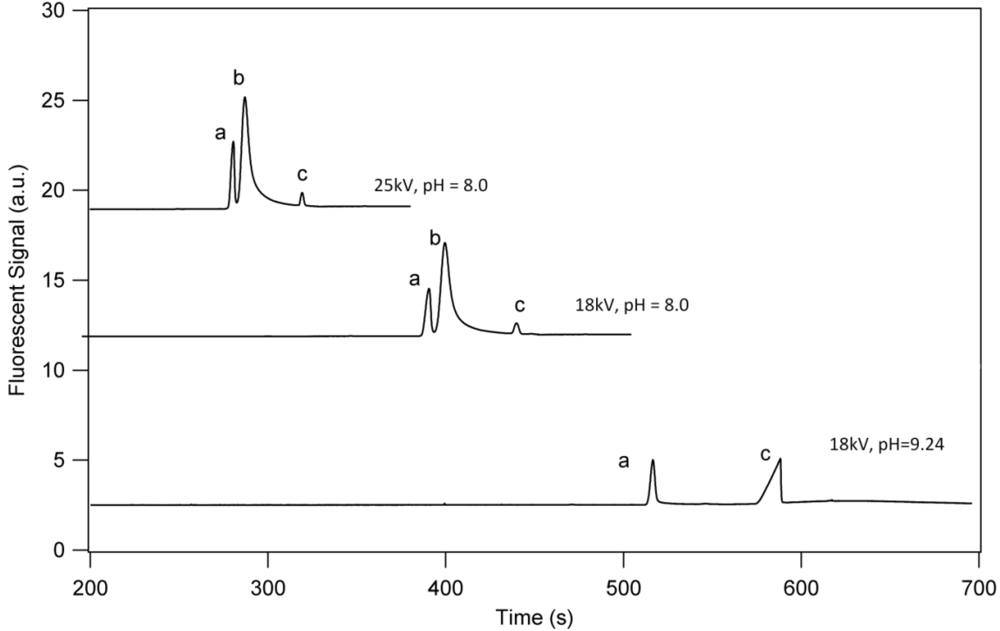
3.2. Detection Limits
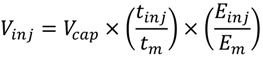

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH = 9.240% ACN | pH = 9.255% ACN | pH = 8.00% ACN | ||||
|---|---|---|---|---|---|---|
| ng·mL−1 | pg | ng·mL−1 | pg | ng·mL−1 | pg | |
| Alizarin | 14 | 0.2 | 39 | 0.9 | 70 | 2.4 |
| Purpurin | ND | ND | ND | ND | 13 | 0.5 |
| Emodin | 40 | 1.0 | 12 | 0.2 | 38 | 1.1 |
| Kaempferol | 10 | 0.2 | 0.7 | 0.01 | 6.3 | 0.2 |
| Apigenin | 23 | 0.4 | 9.0 | 0.2 | 20 | 0.5 |
| Luteolin | 17 | 0.3 | 7.2 | 0.1 | 19 | 0.5 |
| Carmine | ND | ND | 230 | 3.8 | 400 | 12 |
| Myricetin | 51 | 0.9 | 15 | 0.2 | 5.7 | 0.2 |
| Quercetin | 12 | 0.2 | 1.8 | 0.03 | 2.9 | 0.1 |
| Morin | 9.0 | 0.1 | 0.7 | 0.01 | 2.4 | 0.1 |
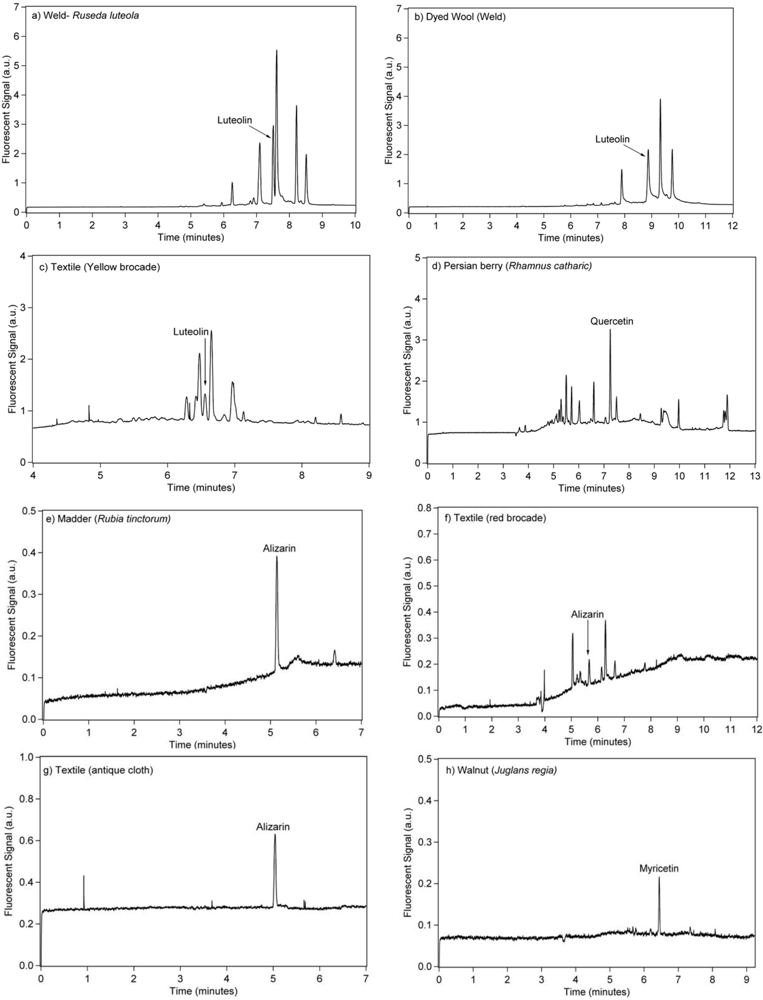
3.3. Extraction of Natural Dyes in Field Samples

4. Conclusions
Acknowledgments
Conflict of Interest
References
- Grierson, S. Dyeing and Dyestuffs; Shire Publications Ltd.: Bucks, UK, 1989. [Google Scholar]
- Sanz Rodríguez, E.; Arteaga Rodríguez, A.; García Rodríguez, M.A.; del Egido, M.; Cámara, C.; Bailão, A.; Garcia, M. Identification of natural dyes in historical coptic textiles from the national archaeological museum of spain. E-Conserv. Mag. 2010, 15, 32–45. [Google Scholar]
- Cardon, D. Natural Dyes. Sources, Tradition, Technology and Science; Archetype Publications: London, UK, 2007. [Google Scholar]
- Bechtold, T.; Mussak, R. Handbook of Natural Colorants; Wiley: Chichester, UK, 2009. [Google Scholar]
- Peggie, D. The Development and Application of Analytical methods for the Identification of Dyes on Historical Textiles; University of Edinburgh: Edinburgh, UK, 2006. [Google Scholar]
- Degano, I.; Ribechini, E.; Modugno, F.; Colombini, M.P. Analytical methods for the characterization of organic dyes in artworks and in historical textiles. Appl. Spectrosc. Rev. 2009, 44, 363–410. [Google Scholar] [CrossRef]
- Rie, E.R. Fluorescence of paint and varnish layers (Part I). Stud. Conserv. 1982, 27, 1–7. [Google Scholar] [CrossRef]
- Wallert, A. Unusual pigments on a greek marble basin. Stud. Conserv. 1995, 40, 177–188. [Google Scholar] [CrossRef]
- Miliani, C.; Romani, A.; Favaro, G. A spectrophotometric and fluorimetric study of some anthraquinoid and indigoid colorants used in artistic paintings. Spectrochim. Acta Part A 1998, 54, 581–588. [Google Scholar] [CrossRef]
- Clementi, C.; Miliani, C.; Romani, A.; Santamaria, U.; Morresi, F.; Mlynarska, K.; Favaro, G. In-situ fluorimetry: A powerful non-invasive diagnostic technique for natural dyes used in artefacts Part II. Identification of orcein and indigo in Renaissance tapestries. Spectrochim. Acta Part A 2009, 71, 2057–2062. [Google Scholar] [CrossRef]
- Low, M.J.D.; Baer, N.S. Application of infrared fourier transform spectroscopy to problems in conservation. Stud. Conserv. 1977, 22, 116–128. [Google Scholar] [CrossRef]
- Shearer, J.C.; Peters, D.C.; Hoepfner, G.; Newton, T. FT-IR in the service of art conservation. Anal. Chem. 1983, 55, 874A–880A. [Google Scholar]
- Gillard, R.D.; Hardman, S.M.; Thomas, R.G.; Watkinson, D.E. The detection of dyes by FTIR microscopy. Stud. Conserv. 1994, 39, 187–192. [Google Scholar] [CrossRef]
- Edwards, H.G.M. Illumination of a mediaeval mystery: The FT-Raman spectroscopic analysis of red pigment from a mediaeval corbel in the church st clement of rome, fiskerton. J. Mol. Struct. 2003, 661, 271–277. [Google Scholar] [CrossRef]
- Edwards, H.G.M.; Villar, S.E.J.; Eremin, K.A. Raman spectroscopic analysis of pigments from dynastic egyptian funerary artefacts. J. Raman Spectrosc. 2004, 35, 786–795. [Google Scholar] [CrossRef]
- Kharbade, B.V.; Agrawal, O.P. Analysis of natural dyes in indian historic textiles. Stud. Conserv. 1988, 33, 1–8. [Google Scholar] [CrossRef]
- Scott, D.; Khandekar, N.; Schilling, M.; Turner, N.; Taniguchi, Y.; Khanjian, H. Technical examination of a fifteenth-century German illuminated manuscript on paper: A case study in the identification of materials. Stud. Conserv. 2001, 46, 93–108. [Google Scholar] [CrossRef]
- Wouters, J.; Verhecken, A. The coccid insect dyes: HPLC and computerized diode-array analysis of dyed yarns. Stud. Conserv. 1989, 34, 189–200. [Google Scholar] [CrossRef]
- Colombini, M.P.; Carmignani, A.; Modugno, F.; Frezzato, F.; Olchini, A.; Brecoulaki, H.; Vassilopoulou, V.; Karkanas, P. Integrated analytical techniques for the study of ancient greek polychromy. Talanta 2004, 63, 839–848. [Google Scholar] [CrossRef]
- Domenech-Carbo, M.; Casas-Catalan, M.; Domenech-Carbo, A.; Mateo-Castro, R.; Gimeno-Adelantado, J.; Bosch-Reig, F. Analytical study of canvas painting collection from the basilica de la virgen de los desamparados using SEM/EDX, FT-IR, GC and electrochemical techniques. Fresenius J. Anal. Chem. 2001, 369, 571–575. [Google Scholar] [CrossRef]
- Puchalska, M.; Orlinska, M.; Ackacha, M.; Polec-Pawlak, K.; Jarosz, M. Identification of anthraquinone coloring matters in natural red dyes by electrospray mass spectrometry coupled to capillary electrophoresis. J. Mass Spectrom. 2003, 38, 1252–1258. [Google Scholar]
- Surowiec, I.; Pawelec, K.; Rezeli, M.; Kilar, F.; Trojanowicz, M. Capillary electrophoretic determination of main components of natural dyes with ms detection. J. Sep. Sci. 2008, 31, 2457–2462. [Google Scholar] [CrossRef]
- Trojanowicz, M.; Wojcik, L.; Urbaniak-Walczak, K. Identification of natural dyes in historical coptic textiles by capillary electrophoresis with diode array detection. Chem. Anal. 2003, 48, 607–620. [Google Scholar]
- Maguregui, M.I.; Alonso, R.M.; Barandiaran, M.; Jimenez, R.M.; Garcia, N. Micellar electrokinetic chromatography method for the determination of several natural red dyestuff and lake pigments used in art work. J. Chromatogr. A 2007, 1154, 429–436. [Google Scholar] [CrossRef]
- Lopez-Montes, A.; Blanc Garcia, R.; Espejo, T.; Huertas-Perez, J.F.; Navalon, A.; Luis Vilchez, J. Simultaneous identification of natural dyes in the collection of drawings and maps from the royal chancellery archives in Granada (Spain) by CE. Electrophoresis 2007, 28, 1243–1251. [Google Scholar]
- Goltz, D.M.; Ahmadi, S.; Absalan, G.; Craig, D.B. Separation of historical dyes using capillary electrophoresis with laser-induced fluorescence detection. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2054–2065. [Google Scholar]
- Colombini, M.P.; Andreotti, A.; Baraldi, C.; Degano, I.; Łucejko, J.J. Colour fading in textiles: A model study on the decomposition of natural dyes. Microchem. J. 2007, 85, 174–182. [Google Scholar] [CrossRef]
- Craig, D.; Arriaga, E.; Banks, P.; Zhang, Y.; Renborg, A.; Palcic, M.; Dovichi, N. Fluorescence-based enzymatic assay by capillary electrophoresis laser-induced fluorescence detection for the determination of a few beta-galactosidase molecules. Anal. Biochem. 1995, 226, 147–153. [Google Scholar]
- Rice, C.L.; Whitehead, R. Electrokinetic flow in a narrow cylindrical capillary. J. Phys. Chem. 1965, 69, 4017–4024. [Google Scholar]
- Jorgenson, J.W.; Lukacs, K.D. Capillary zone electrophoresis. Science 1983, 222, 266–272. [Google Scholar]
- McGhie, T.K. Analysis of sugarcane flavonoids by capillary zone electrophoresis. J. Chromatogr. 1993, 634, 107–112. [Google Scholar] [CrossRef]
- Ng, C.L.; Ong, C.P.; Lee, H.; Li, S.F.Y. Systematic optimization of micellar electrokinetic chromatographic-separation of flavonoids. Chromatographia 1992, 34, 166–172. [Google Scholar]
- McGhie, T.K.; Markham, K.R. Separation of flavonols by capillary electrophoresis—The effect of structure on electrophoretic mobility. Phytochem. Anal. 1994, 5, 121–126. [Google Scholar] [CrossRef]
- Delgado, C.; Tomasbarberan, F.A.; Talou, T.; Gaset, A. Capillary electrophoresis as an alternative to HPLC for determination of honey flavonoids. Chromatographia 1994, 38, 71–78. [Google Scholar]
- Khaled, M.Y.; Anderson, M.R.; McNair, H.M. Micellar electrokinetic capillary chromatography of pungent compounds using simultaneous online ultraviolet and electrochemical detection. J. Chromatogr. Sci. 1993, 31, 259–264. [Google Scholar]
- Jimidar, M.; Hamoir, T.P.; Foriers, A.; Massart, D.L. Comparison of capillary zone electrophoresis with high-performance liquid chromatography for the determination of additives in foodstuffs. J. Chromatogr. A 1993, 636, 179–186. [Google Scholar] [CrossRef]
- Terabe, S. Selectivity manipulation in micellar electrokinetic chromatography. J. Pharm. Biomed. Anal. 1992, 10, 705–715. [Google Scholar] [CrossRef]
- Kum, S.; Jagasia, R.; Haldar, B.C. Acid dissociation constants of morin in ethanol-water mixture. J. Indian Chem. Soc. 1963, 40, 287–292. [Google Scholar]
- Tyukavkina, N.A.; Pogodaeva, N.N. Ultraviolet-absorption of flavonoids. VIII. Ionization-constants of kempferol and quercetin. Khim. Prirod. Soedin. 1975, 6, 708–711. [Google Scholar]
- Herrero-Martinez, J.; Sanmartin, M.; Roses, M.; Bosch, E.; Rafols, C. Determination of dissociation constants of flavonoids by capillary electrophoresis. Electrophoresis 2005, 26, 1886–1895. [Google Scholar] [CrossRef]
- Harris, D.C. Quantitative Chemical Analysis; W.H. Freeman and Company: New York, NY, USA, 2003. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ahmadi, S.; Craig, D.B.; Goltz, D.M. Micellar Electrokinetic Chromatography with Laser-Induced Fluorescence Detection for Separation of Red and Yellow Historical Dyes. Chromatography 2014, 1, 9-23. https://doi.org/10.3390/chromatography1010009
Ahmadi S, Craig DB, Goltz DM. Micellar Electrokinetic Chromatography with Laser-Induced Fluorescence Detection for Separation of Red and Yellow Historical Dyes. Chromatography. 2014; 1(1):9-23. https://doi.org/10.3390/chromatography1010009
Chicago/Turabian StyleAhmadi, Shokoufeh, Douglas B. Craig, and Douglas M. Goltz. 2014. "Micellar Electrokinetic Chromatography with Laser-Induced Fluorescence Detection for Separation of Red and Yellow Historical Dyes" Chromatography 1, no. 1: 9-23. https://doi.org/10.3390/chromatography1010009
APA StyleAhmadi, S., Craig, D. B., & Goltz, D. M. (2014). Micellar Electrokinetic Chromatography with Laser-Induced Fluorescence Detection for Separation of Red and Yellow Historical Dyes. Chromatography, 1(1), 9-23. https://doi.org/10.3390/chromatography1010009