
Synthetic Proteins and Peptides for the Direct Interrogation of α-Synuclein Posttranslational Modifications

{kind=link}
{kind=link}
{kind=link}
Abstract
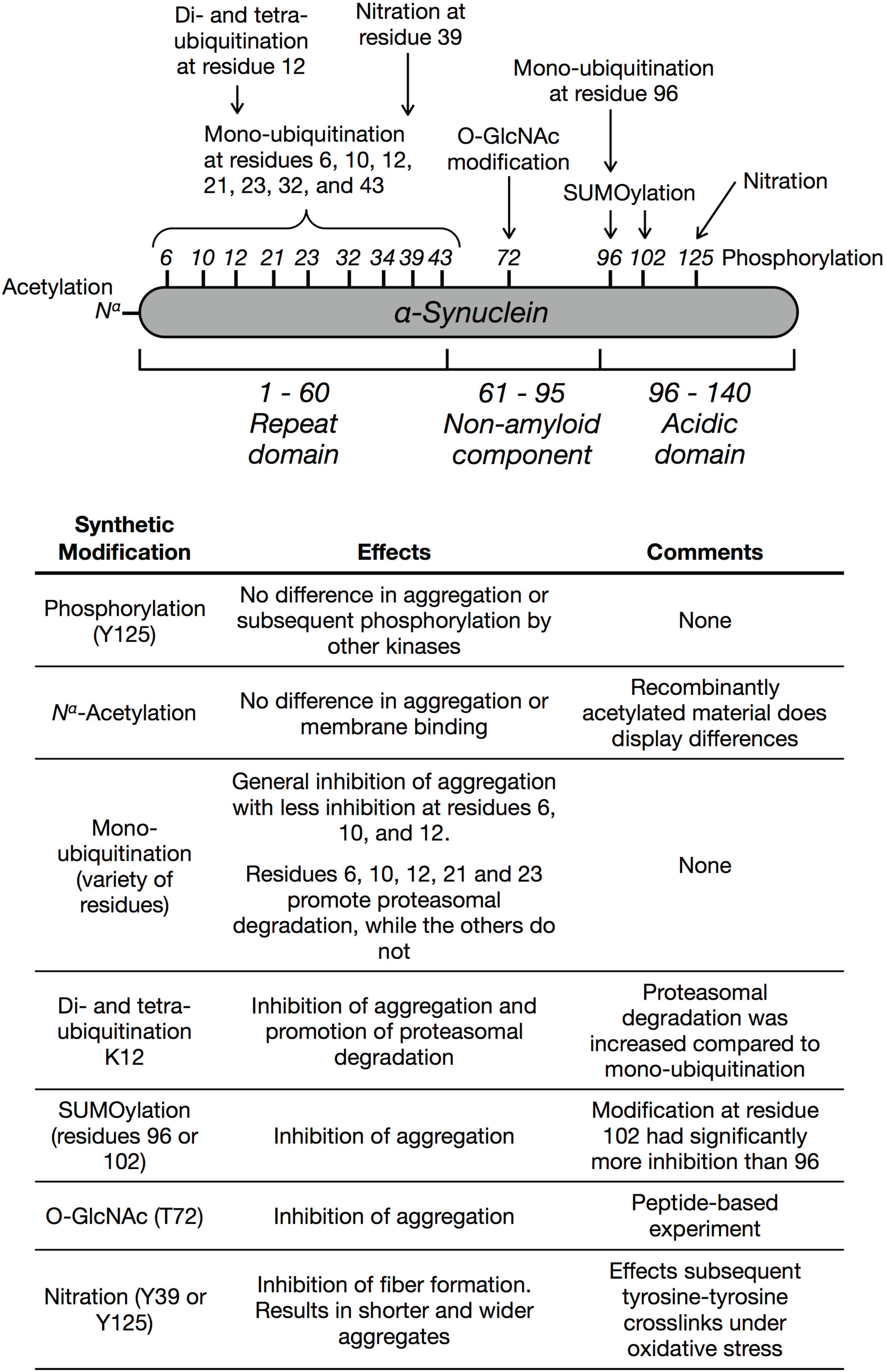
:1. Introduction
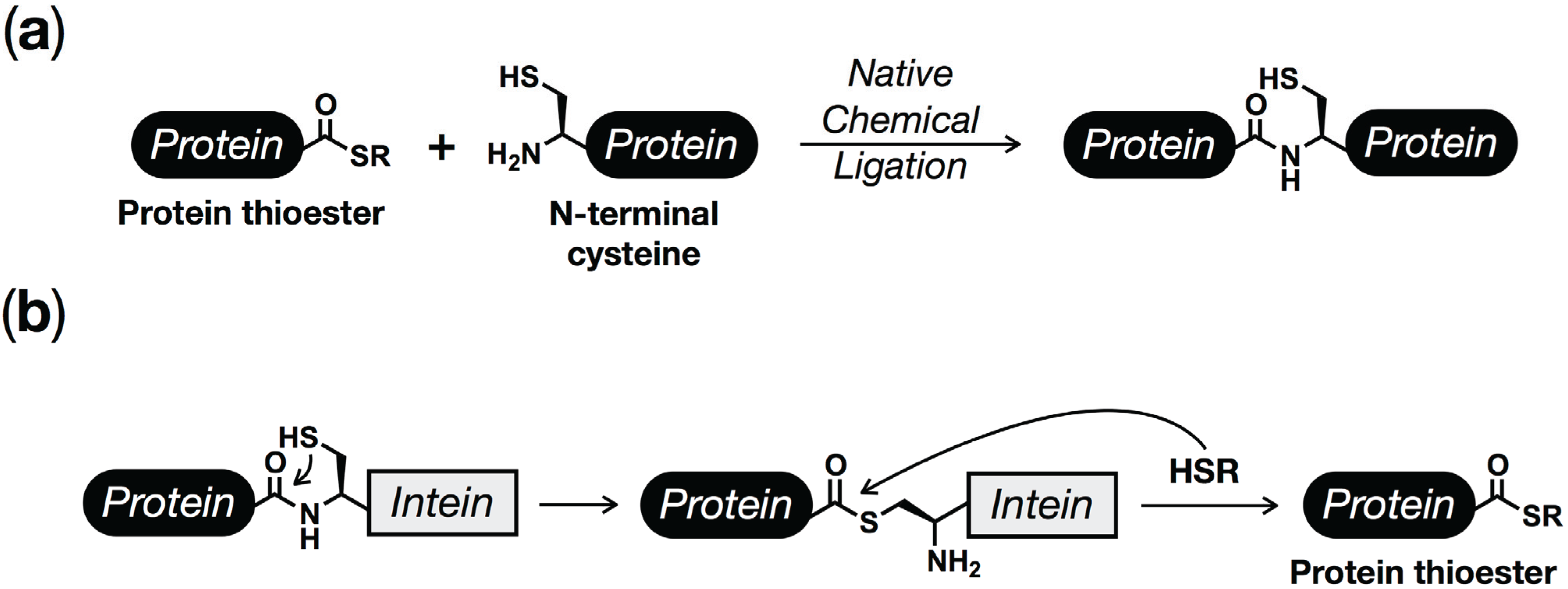
2. Protein (Semi)Synthesis by Native and Expressed Protein Ligation
3. Phosphorylation
4. Acetylation
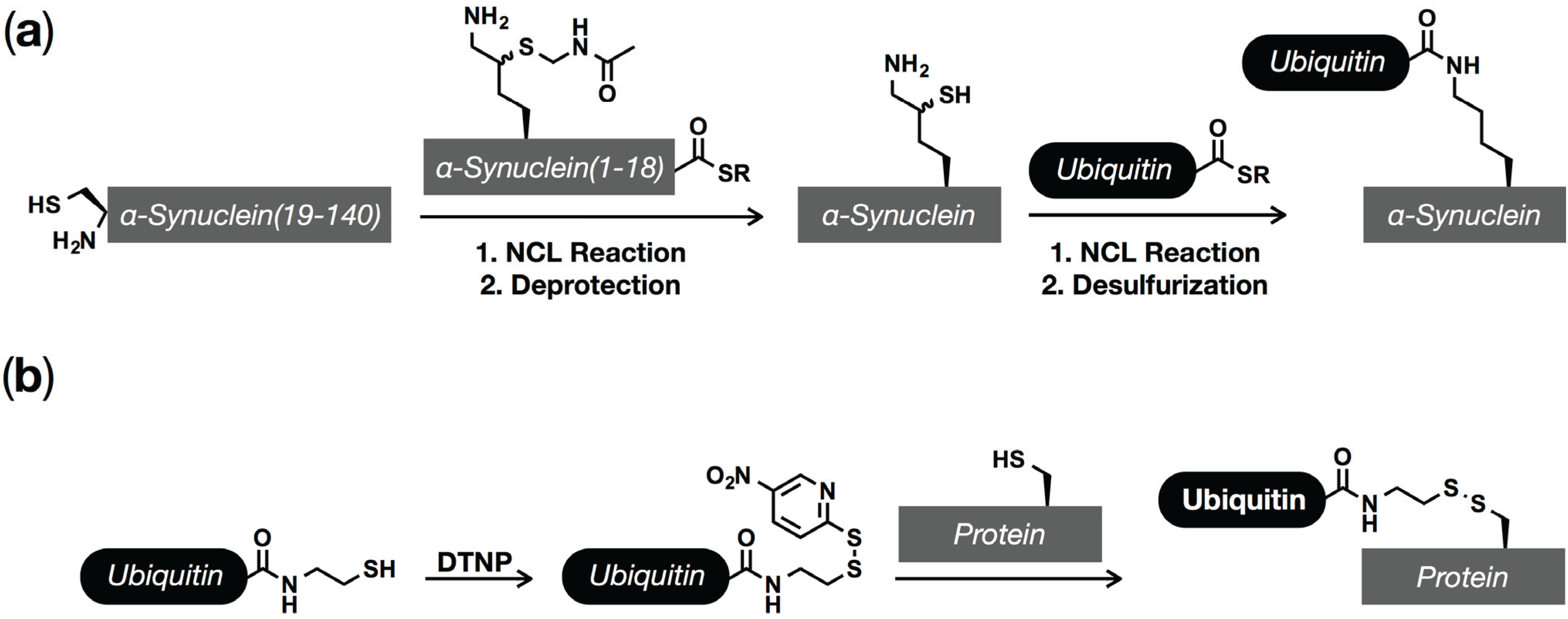
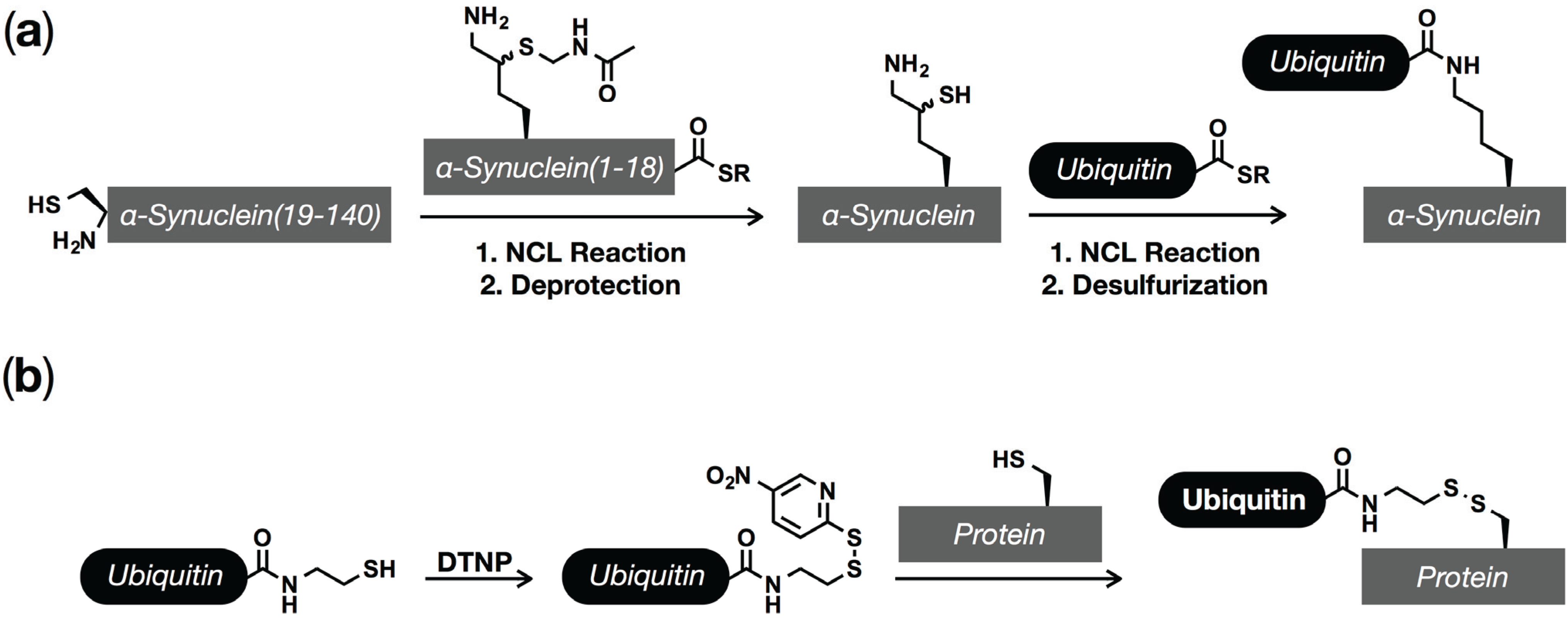
5. Ubiquitination
6. SUMOylation
7. O-GlcNAc Modification
8. Nitration
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Heron, M. National vital statistics reports. Natl. Vital Stat. Rep. 2012, 60, 1–94. [Google Scholar] [PubMed]
- Goetz, C.G.; Poewe, W.; Rascol, O.; Sampaio, C. Evidence-based medical review update: Pharmacological and surgical treatments of Parkinson’s disease: 2001 to 2004. Mov. Disord. 2005, 20, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.M.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Mbefo, M.K.; Fares, M.-B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M.-Y. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. Alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed Tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Paik, S.R.; Yang, C.-H. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry 2002, 41, 13782–13790. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Interactions between metals and alpha-synuclein—Function or artefact? FEBS J. 2007, 274, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109–140 on alpha-synuclein aggregation in vitro. 2004, 43, 16233–16242. [Google Scholar] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet Neurol. 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003. [Google Scholar] [CrossRef]
- Fink, A.L. The aggregation and fibrillation of α-synuclein. Acc. Chem. Res. 2006, 39, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A.-L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.A.; Angot, E.; Brundin, P. A deadly spread: Cellular mechanisms of alpha-synuclein transfer. Cell. Death Differ. 2011, 18, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jäkälä, P.; Hartmann, T.; Price, D.L.; et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [PubMed]
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fredenburg, R.A.; Lansbury, P.T.; Fernandez, C.O.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Invest. 2009, 119, 3257–3265. [Google Scholar] [PubMed]
- Hejjaoui, M.; Butterfield, S.; Fauvet, B.; Vercruysse, F.; Cui, J.; Dikiy, I.; Prudent, M.; Olschewski, D.; Zhang, Y.; Eliezer, D.; et al. Elucidating the role of C-terminal post-translational modifications using protein semisynthesis strategies: α-Synuclein phosphorylation at tyrosine 125. J. Am. Chem. Soc. 2012, 134, 5196–5210. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Tortosa, E.; Newell, K.; Irizarry, M.C.; Sanders, J.L.; Hyman, B.T. Alpha-synuclein immunoreactivity in dementia with Lewy bodies: Morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 2000, 99, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Udeshi, N.D.; O’Malley, M.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 2010, 9, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, J.F.; Gong, C.-X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.W.; Purvine, S.O.; Wang, Z.; Camp, D.G.; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Fournier, M.; Lashuel, H.A. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Prog. Brain Res. 2010, 183, 115–145. [Google Scholar] [PubMed]
- Schmid, A.W.; Fauvet, B.; Moniatte, M.; Lashuel, H.A. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol. Cell. Proteomics 2013, 12, 3543–3558. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.; Muir, T.; Clark-Lewis, I.; Kent, S. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Paulus, H. Protein splicing and related forms of protein autoprocessing. Annu. Rev. Biochem. 2000, 69, 447–496. [Google Scholar] [CrossRef] [PubMed]
- Hirata, R.; Ohsumk, Y.; Nakano, A.; Kawasaki, H.; Suzuki, K.; Anraku, Y. Molecular structure of a gene, VMA1, encoding the catalytic subunit of H+-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 6726–6733. [Google Scholar] [PubMed]
- Kane, P.M.; Yamashiro, C.T.; Wolczyk, D.F.; Neff, N.; Goebl, M.; Stevens, T.H. Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H+-adenosine triphosphatase. Science 1990, 250, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Aranko, A.S.; Oeemig, J.S.; Kajander, T.; Iwaï, H. Intermolecular domain swapping induces intein-mediated protein alternative splicing. Nat. Chem. Biol. 2013, 9, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Q.; Perler, F.B. The mechanism of protein splicing and its modulation by mutation. EMBO J. 1996, 15, 5146–5153. [Google Scholar] [PubMed]
- Muir, T.W.; Sondhi, D.; Cole, P.A. Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. USA 1998, 95, 6705–6710. [Google Scholar] [CrossRef] [PubMed]
- Severinov, K.; Muir, T. Expressed protein ligation, a novel method for studying protein-protein interactions in transcription. J. Biol. Chem. 1998, 273, 16205–16209. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.C.; Benner, J.; Xu, M.Q. Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 1998, 7, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Muir, T.W. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 2003, 72, 249–289. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kanuka, H.; Fujiwara, H.; Koyama, A.; Hasegawa, M.; Miura, M.; Iwatsubo, T. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 2003, 336, 155–158. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Sullivan, L.F.; Chen, W.; Kondrikova, G.; Manfredsson, F.P.; Mandel, R.J.; Muzyczka, N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2008, 105, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Da Silveira, S.A.; Schneider, B.L.; Cifuentes-Diaz, C.; Sage, D.; Abbas-Terki, T.; Iwatsubo, T.; Unser, M.; Aebischer, P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 872–887. [Google Scholar]
- Mbefo, M.K.; Paleologou, K.E.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Schneider, B.L.; Aebischer, P.; Lashuel, H.A. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E3945–E3954. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Roberts, W.K. Evidence that approximately eighty per cent of the soluble proteins from Ehrlich ascites cells are Nα-acetylated. J. Biol. Chem. 1976, 251, 1009–1014. [Google Scholar] [PubMed]
- Arnesen, T.; van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [PubMed]
- Starheim, K.K.; Gevaert, K.; Arnesen, T. Protein N-terminal acetyltransferases: When the start matters. Trends Biochem. Sci. 2012, 37, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Fares, M.-B.; Samuel, F.; Dikiy, I.; Tandon, A.; Eliezer, D.; Lashuel, H.A. Characterization of semisynthetic and naturally Nα-acetylated α-synuclein in vitro and in intact cells: Implications for aggregation and cellular properties of α-synuclein. J. Biol. Chem. 2012, 287, 28243–28262. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of N-terminal acetylation of α-synuclein on its random coil and lipid binding properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Trexler, A.J.; Rhoades, E. N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, G.M.; Janowska, M.K.; Kang, L.; Baum, J. Exploring the accessible conformations of N-terminal acetylated α-synuclein. FEBS Lett. 2013, 587, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Welchman, R.; Gordon, C.; Mayer, R. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Schulman, B.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A.P. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Kuzuhara, S.; Mori, H.; Izumiyama, N.; Yoshimura, M.; Ihara, Y. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 1988, 75, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.; Blanchard, A.; Morrell, K.; Lennox, G.; Reynolds, L.; Billett, M.; Landon, M.; Mayer, R.J. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J. Pathol. 1988, 155, 9–15. [Google Scholar] [PubMed]
- Manetto, V.; Perry, G.; Tabaton, M.; Mulvihill, P.; Fried, V.A.; Smith, H.T.; Gambetti, P.; Autilio-Gambetti, L. Ubiquitin is associated with abnormal cytoplasmic filaments characteristic of neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1988, 85, 4501–4505. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [PubMed]
- Sampathu, D.; Giasson, B.; Pawlyk, A.; Trojanowski, J.; Lee, V. Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am. J. Pathol. 2003, 163, 91–100. [Google Scholar] [CrossRef]
- Tofaris, G.K. Ubiquitination of alpha-synuclein in lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003, 278, 44405–44411. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Iwatsubo, T.; Hasegawa, M. Ubiquitination of alpha-synuclein. Biochemistry 2005, 44, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wheeler, T.; Li, L.; Chin, L. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2008, 17, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.K.M.; Wong, A.K.Y.; Lee, Y.W.; Wan, O.W.; Edwin Chan, H.Y.; Chung, K.K.K. The role of ubiquitin linkages on α-synuclein induced-toxicity in a Drosophila model of Parkinson’s disease. J. Neurochem. 2009, 110, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.-W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [Google Scholar] [CrossRef] [PubMed]
- Hejjaoui, M.; Haj-Yahya, M.; Kumar, K.S.A.; Brik, A.; Lashuel, H.A. Towards elucidation of the role of ubiquitination in the pathogenesis of Parkinson’s disease with semisynthetic ubiquitinated α-synuclein. Angew. Chem. Int. Ed. 2010, 50, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Ajish Kumar, K.S.; Haj-Yahya, M.; Olschewski, D.; Lashuel, H.A.; Brik, A. Highly efficient and chemoselective peptide ubiquitylation. Angew. Chem. Int. Ed. 2009, 48, 8090–8094. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Mcginty, R.K.; Fierz, B.; Muir, T.W. Disulfide-directed histone ubiquitylation reveals plasticity in hDot1L activation. Nat. Chem. Biol. 2010, 6, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ai, Y.; Wang, J.; Haracska, L.; Zhuang, Z. Chemically ubiquitylated PCNA as a probe for eukaryotic translesion DNA synthesis. Nat. Chem. Biol. 2010, 6, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471. [Google Scholar] [CrossRef] [PubMed]
- Shabek, N.; Herman-Bachinsky, Y.; Buchsbaum, S.; Lewinson, O.; Haj-Yahya, M.; Hejjaoui, M.; Lashuel, H.A.; Sommer, T.; Brik, A.; Ciechanover, A. The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol. Cell 2012, 48, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yahya, M.; Fauvet, B.; Herman-Bachinsky, Y.; Hejjaoui, M.; Bavikar, S.N.; Karthikeyan, S.V.; Ciechanover, A.; Lashuel, H.A.; Brik, A. Synthetic polyubiquitinated α-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. USA 2013, 110, 17726–17731. [Google Scholar] [CrossRef] [PubMed]
- Abeywardana, T.; Lin, Y.H.; Rott, R.; Engelender, S.; Pratt, M.R. Site-specific differences in proteasome-dependent degradation of monoubiquitinated α-synuclein. Chem. Biol. 2013, 20, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Weishaupt, J.H. Sumoylation in neurodegenerative diseases. Cell. Mol. Life Sci. 2013, 70, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.-H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-H.; Galanis, A.; Witty, J.; Sharrocks, A.D. An extended consensus motif enhances the specificity of substrate modification by SUMO. EMBO J. 2006, 25, 5083–5093. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jang, W.H.; Quezado, M.M.; Oh, Y.; Chung, K.C.; Junn, E.; Mouradian, M.M. Proteasome inhibition induces α-synuclein SUMOylation and aggregate formation. J. Neurol. Sci. 2011, 307, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Pountney, D.L.; Chegini, F.; Shen, X.; Blumbergs, P.C.; Gai, W.P. SUMO-1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neurosci. Lett. 2005, 381, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.B.; Goodwin, J.; Norazit, A.; Meedeniya, A.C.B.; Richter-Landsberg, C.; Gai, W.P.; Pountney, D.L. SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox. Res. 2013, 23, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Kim, Y.M.; Mouradian, M.M.; Chung, K.C. Human Polycomb protein 2 promotes α-synuclein aggregate formation through covalent SUMOylation. Brain Res. 2011, 1381, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Shahpasandzadeh, H.; Popova, B.; Kleinknecht, A.; Fraser, P.E.; Outeiro, T.F.; Braus, G.H. Interplay between sumoylation and phosphorylation for protection against α-synuclein inclusions. J. Biol. Chem. 2014, 289, 31224–31240. [Google Scholar] [CrossRef] [PubMed]
- Abeywardana, T.; Pratt, M.R. Extent of inhibition of α-synuclein aggregation in vitro by SUMOylation is conjugation site- and SUMO isoform-selective. Biochemistry 2015, 54, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Vocadlo, D.J. O-GlcNAc processing enzymes: Catalytic mechanisms, substrate specificity, and enzyme regulation. Curr. Opin. Chem. Biol. 2012, 16, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.L.; Gloster, T.M.; Yuzwa, S.A.; Vocadlo, D.J. Insights into O-linked N-acetylglucosamine (O-GlcNAc) processing and dynamics through kinetic analysis of O-GlcNAc transferase and O-GlcNAcase activity on protein substrates. J. Biol. Chem. 2012, 287, 15395–15408. [Google Scholar] [CrossRef] [PubMed]
- Du, H.-N.; Li, H.-T.; Zhang, F.; Lin, X.-J.; Shi, J.-H.; Shi, Y.-H.; Ji, L.-N.; Hu, J.; Lin, D.-H.; Hu, H.-Y. Acceleration of alpha-synuclein aggregation by homologous peptides. FEBS Lett. 2006, 580, 3657–3664. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lim, D.; Kim, J.Y.; Kang, S.J.; Kim, Y.-H.; Im, H. Beta-sheet-breaking peptides inhibit the fibrillation of human alpha-synuclein. Biochem. Biophys. Res. Commun. 2009, 387, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Marotta, N.P.; Cherwien, C.A.; Abeywardana, T.; Pratt, M.R. O-GlcNAc modification prevents peptide-dependent acceleration of α-synuclein aggregation. ChemBioChem 2012, 13, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Crow, J.P. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem. Soc. Trans. 1993, 21, 330–334. [Google Scholar] [PubMed]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Duda, J.E.; Giasson, B.I.; Chen, Q.; Gur, T.L.; Hurtig, H.I.; Stern, M.B.; Gollomp, S.M.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am. J. Pathol. 2000, 157, 1439–1445. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratt, M.R.; Abeywardana, T.; Marotta, N.P. Synthetic Proteins and Peptides for the Direct Interrogation of α-Synuclein Posttranslational Modifications. Biomolecules 2015, 5, 1210-1227. https://doi.org/10.3390/biom5031210
Pratt MR, Abeywardana T, Marotta NP. Synthetic Proteins and Peptides for the Direct Interrogation of α-Synuclein Posttranslational Modifications. Biomolecules. 2015; 5(3):1210-1227. https://doi.org/10.3390/biom5031210
Chicago/Turabian StylePratt, Matthew R., Tharindumala Abeywardana, and Nicholas P. Marotta. 2015. "Synthetic Proteins and Peptides for the Direct Interrogation of α-Synuclein Posttranslational Modifications" Biomolecules 5, no. 3: 1210-1227. https://doi.org/10.3390/biom5031210