Sumoylation and the DNA Damage Response

Abstract
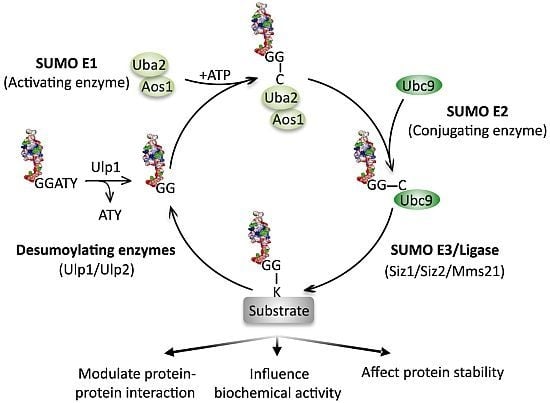
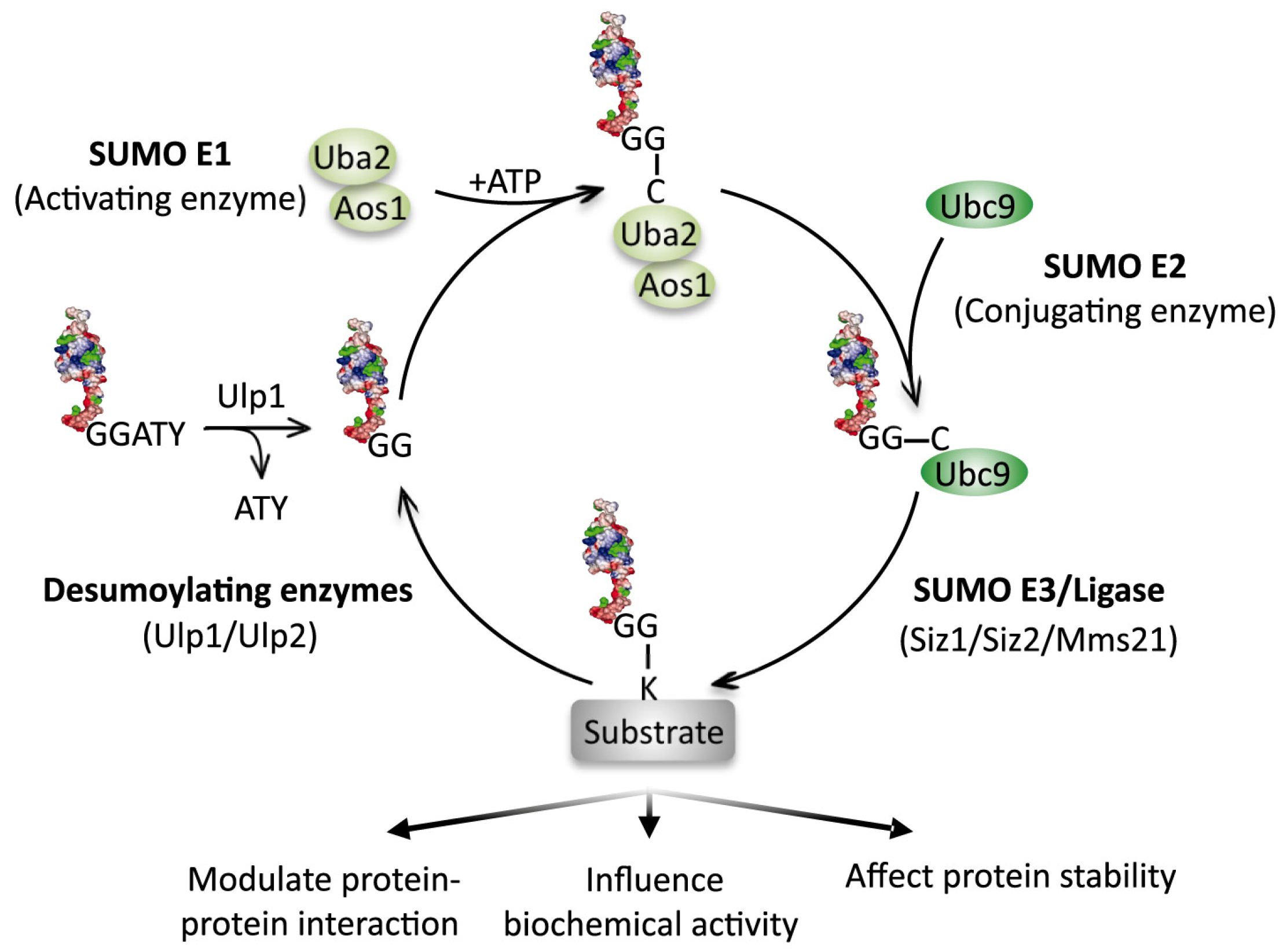
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
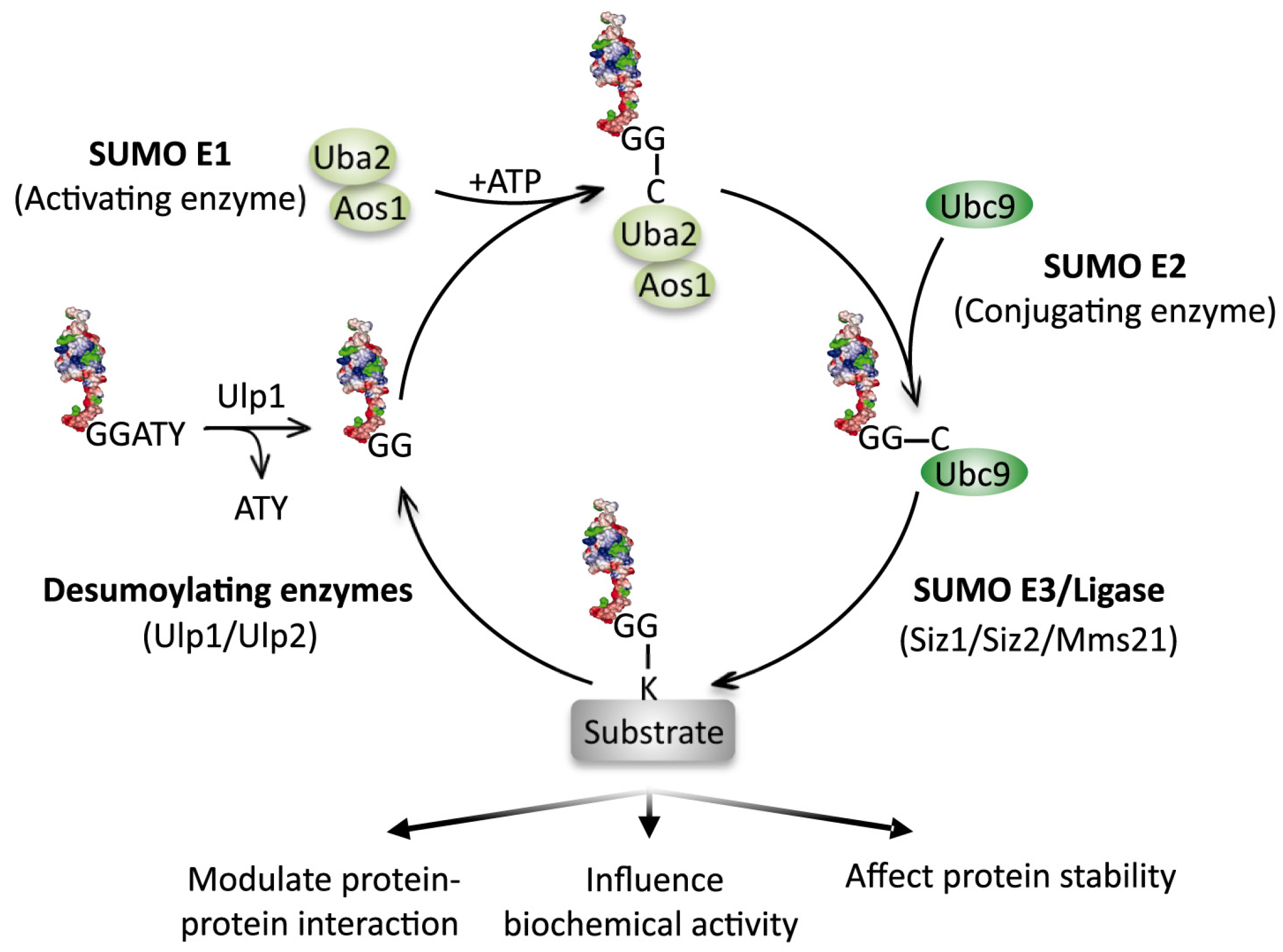
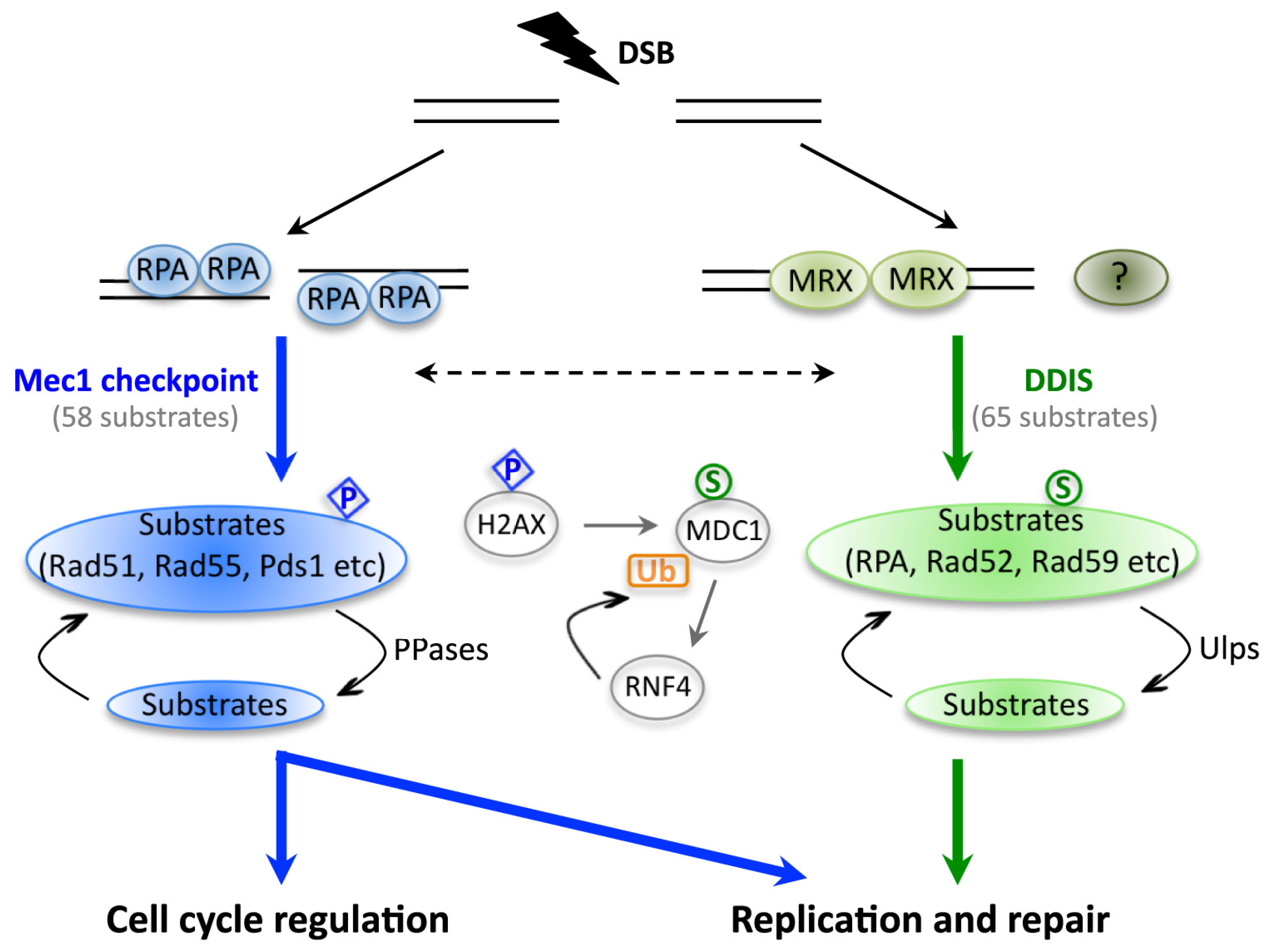
2. The Significance of DNA Damage-Induced Sumoylation (DDIS)
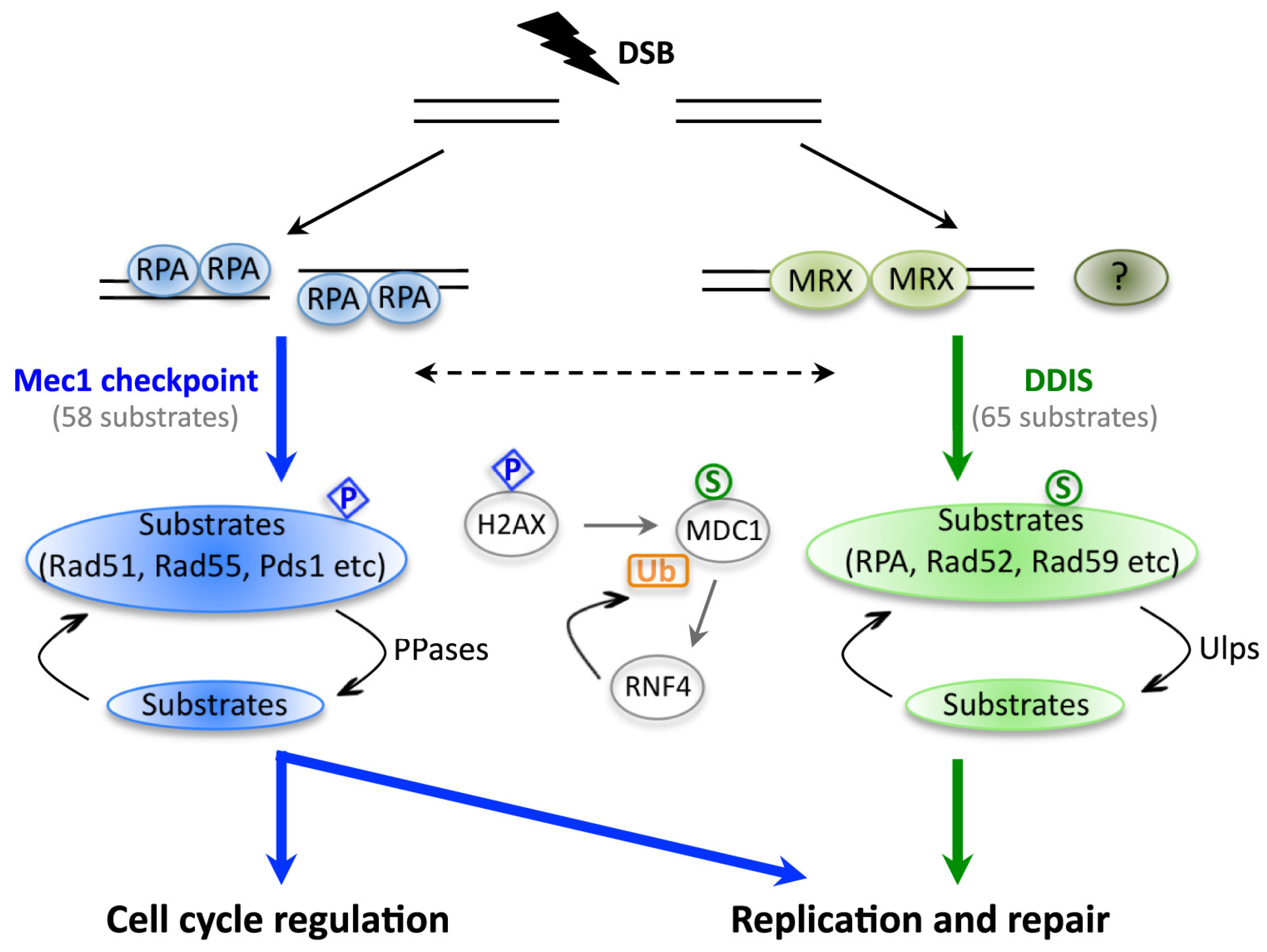
3. The Effects of Sumoylation on DNA Repair Proteins
4. Sumoylation Dynamics
5. Crosstalk with other Posttranslational Modifications
6. Concluding Remarks
Conflict of Interest Statement
Acknowledgments
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Wang, Y.; Dasso, M. SUMOylation and deSUMOylation at a glance. J. Cell Sci. 2009, 122, 4249–4252. [Google Scholar] [CrossRef]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Mailand, N. The ubiquitin- and SUMO-dependent signaling response to DNA double-strand breaks. FEBS Lett. 2011, 585, 2914–2919. [Google Scholar] [CrossRef]
- Dou, H.; Huang, C.; van Nguyen, T.; Lu, L.S.; Yeh, E.T. SUMOylation and de-SUMOylation in response to DNA damage. FEBS Lett. 2011, 585, 2891–2896. [Google Scholar]
- Ulrich, H.D. Ubiquitin and SUMO in DNA repair at a glance. J. Cell Sci. 2012, 125, 249–254. [Google Scholar] [CrossRef]
- Zlatanou, A.; Stewart, G.S. A PIAS-ed view of DNA double strand break repair focuses on SUMO. DNA Repair 2010, 9, 588–592. [Google Scholar] [CrossRef]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar]
- Morris, J.R. SUMO in the mammalian response to DNA damage. Biochem. Soc. Trans. 2010, 38, 92–97. [Google Scholar] [CrossRef]
- Praefcke, G.J.; Hofmann, K.; Dohmen, R.J. SUMO playing tag with ubiquitin. Trends Biochem. Sci. 2012, 37, 23–31. [Google Scholar] [CrossRef]
- Nagai, S.; Davoodi, N.; Gasser, S.M. Nuclear organization in genome stability: SUMO connections. Cell Res. 2011, 21, 474–485. [Google Scholar] [CrossRef]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar]
- Morris, J.R.; Boutell, C.; Keppler, M.; Densham, R.; Weekes, D.; Alamshah, A.; Butler, L.; Galanty, Y.; Pangon, L.; Kiuchi, T.; Ng, T.; Solomon, E. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 2009, 462, 886–890. [Google Scholar]
- Altmannová, V.; Eckert-Boulet, N.; Arneric, M.; Kolesár, P.; Chaloupkova, R.; Damborsky, J.; Sung, P.; Zhao, X.; Lisby, M.; Krejčí, L. Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 2010, 38, 4708–4721. [Google Scholar]
- Altmannová, V.; Kolesár, P.; Krejčí, L. SUMO wrestles with recombination. Biomolecules 2012, 2, 350–375. [Google Scholar]
- Bayer, P.; Arndt, A.; Metzger, S.; Mahajan, R.; Melchior, F.; Jaenicke, R.; Becker, J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998, 280, 275–286. [Google Scholar]
- Song, J.; Durrin, L.; Wilkinson, T.A.; Krontiris, T.G.; Chen, Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 14373–14378. [Google Scholar]
- Cremona, C.A.; Sarangi, P.; Yang, Y.; Hang, L.E.; Rahman, S.; Zhao, X. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the Mec1 checkpoint. Mol. Cell 2012, 45, 422–432. [Google Scholar]
- Silver, H.R.; Nissley, J.A.; Reed, S.H.; Hou, Y.M.; Johnson, E.S. A role for SUMO in nucleotide excision repair. DNA Repair 2011, 10, 1243–1251. [Google Scholar]
- Dou, H.; Huang, C.; Singh, M.; Carpenter, P.B.; Yeh, E.T. Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol. Cell 2010, 39, 333–345. [Google Scholar] [CrossRef]
- Luo, K.; Zhang, H.; Wang, L.; Yuan, J.; Lou, Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2010, 31. [Google Scholar]
- Sacher, M.; Pfander, B.; Hoege, C.; Jentsch, S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 2006, 8, 1284–1290. [Google Scholar]
- Branzei, D.; Sollier, J.; Liberi, G.; Zhao, X.; Maeda, D.; Seki, M.; Enomoto, T.; Ohta, K.; Foiani, M. Ubc9- and Mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 2006, 127, 509–522. [Google Scholar]
- Maeda, D.; Seki, M.; Onoda, F.; Branzei, D.; Kawabe, Y.; Enomoto, T. Ubc9 is required for damage-tolerance and damage-induced interchromosomal homologous recombination in S.cerevisiae. DNA Repair 2004, 3, 335–341. [Google Scholar]
- Burgess, R.C.; Rahman, S.; Lisby, M.; Rothstein, R.; Zhao, X. The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Mol. Cell. Biol. 2007, 27, 6153–6162. [Google Scholar]
- Hardeland, U.; Steinacher, R.; Jiricny, J.; Schar, P. Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 2002, 21, 1456–1464. [Google Scholar] [CrossRef]
- Potts, P.R.; Yu, H. Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol. Cell. Biol. 2005, 25, 7021–7032. [Google Scholar] [CrossRef]
- Andrews, E.A.; Palecek, J.; Sergeant, J.; Taylor, E.; Lehmann, A.R.; Watts, F.Z. Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol. Cell. Biol. 2005, 25, 185–196. [Google Scholar] [CrossRef]
- Zhao, X.; Blobel, G. A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. USA 2005, 102, 4777–4782. [Google Scholar] [CrossRef]
- Zhou, W.; Ryan, J.J.; Zhou, H. Global analyses of sumoylated proteins in Saccharomyces cerevisiae. Induction of protein sumoylation by cellular stresses. J. Biol. Chem. 2004, 279, 32262–32268. [Google Scholar]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2009, 2. ra24. [Google Scholar] [CrossRef]
- Lee, Y.J.; Mou, Y.; Maric, D.; Klimanis, D.; Auh, S.; Hallenbeck, J.M. Elevated global SUMOylation in Ubc9 transgenic mice protects their brains against focal cerebral ischemic damage. PLosOne 2011, 6, e25852. [Google Scholar]
- Li, S.J.; Hochstrasser, M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol. Cell. Biol. 2000, 20, 2367–2377. [Google Scholar] [CrossRef]
- Mullen, J.R.; Kaliraman, V.; Ibrahim, S.S.; Brill, S.J. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 2001, 157, 103–118. [Google Scholar]
- Danielsen, J.R.; Povlsen, L.K.; Villumsen, B.H.; Streicher, W.; Nilsson, J.; Wikstrom, M.; Bekker-Jensen, S.; Mailand, N. DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding Zinc finger. J. Cell Biol. 2012, 197, 179–187. [Google Scholar]
- Chen, S.H.; Albuquerque, C.P.; Liang, J.; Suhandynata, R.T.; Zhou, H. A proteome-wide analysis of kinase-substrate network in the DNA damage response. J. Biol. Chem. 2010, 285, 12803–12812. [Google Scholar]
- Jeram, S.M.; Srikumar, T.; Pedrioli, P.G.; Raught, B. Using mass spectrometry to identify ubiquitin and ubiquitin-like protein conjugation sites. Proteomics 2009, 9, 922–934. [Google Scholar]
- Hsiao, H.H.; Meulmeester, E.; Urlaub, H. Identification of endogenous SUMO1 accepter sites by mass spectrometry. Methods Mol. Biol. 2012, 893, 431–441. [Google Scholar] [CrossRef]
- Blomster, H.A.; Imanishi, S.Y.; Siimes, J.; Kastu, J.; Morrice, N.A.; Eriksson, J.E.; Sistonen, L. In vivo identification of sumoylation sites by a signature tag and cysteine-targeted affinity purification. J. Biol. Chem. 2010, 285, 19324–19329. [Google Scholar]
- Wohlschlegel, J.A.; Johnson, E.S.; Reed, S.I.; Yates, J.R., 3rd. Global analysis of protein sumoylation in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 45662–45668. [Google Scholar]
- Lisby, M.; Rothstein, R. Choreography of recombination proteins during the DNA damage response. DNA Repair 2009, 8, 1068–1076. [Google Scholar] [CrossRef]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar]
- Parnas, O.; Zipin-Roitman, A.; Pfander, B.; Liefshitz, B.; Mazor, Y.; Ben-Aroya, S.; Jentsch, S.; Kupiec, M. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010, 29, 2611–2622. [Google Scholar]
- Armstrong, A.A.; Mohideen, F.; Lima, C.D. Recognition of SUMO-modified PCNA requires tandem receptor motifs in Srs2. Nature 2012, 483, 59–63. [Google Scholar]
- Kolesar, P.; Sarangi, P.; Altmannova, V.; Zhao, X.; Krejci, L. Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res. 2012. [Google Scholar]
- Li, Y.J.; Stark, J.M.; Chen, D.J.; Ann, D.K.; Chen, Y. Role of SUMO:SIM-mediated protein-protein interaction in non-homologous end joining. Oncogene 2010, 29, 3509–3518. [Google Scholar] [CrossRef]
- Li, Y.J.; Perkins, A.L.; Su, Y.; Ma, Y.; Colson, L.; Horne, D.A.; Chen, Y. Gold nanoparticles as a platform for creating a multivalent poly-SUMO chain inhibitor that also augments ionizing radiation. Proc. Natl. Acad. Sci. USA 2012, 109, 4092–4097. [Google Scholar]
- Yang, K.; Moldovan, G.L.; Vinciguerra, P.; Murai, J.; Takeda, S.; D’Andrea, A.D. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011, 25, 1847–1858. [Google Scholar] [CrossRef]
- Heideker, J.; Prudden, J.; Perry, J.J.; Tainer, J.A.; Boddy, M.N. SUMO-targeted ubiquitinligase, Rad60, and Nse2 SUMO ligase suppress spontaneous Top1-mediated DNA damage and genome instability. PLoSGenet. 2011, 7, e1001320. [Google Scholar]
- Jacquiau, H.R.; van Waardenburg, R.C.; Reid, R.J.; Woo, M.H.; Guo, H.; Johnson, E.S.; Bjornsti, M.A. Defects in SUMO (small ubiquitin-related modifier) conjugation and deconjugation alter cell sensitivity to DNA topoisomerase I-induced DNA damage. J. Biol. Chem. 2005, 280, 23566–23575. [Google Scholar]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef]
- Shen, T.H.; Lin, H.K.; Scaglioni, P.P.; Yung, T.M.; Pandolfi, P.P. The mechanisms of PML-nuclear body formation. Mol. Cell 2006, 24, 331–339. [Google Scholar] [CrossRef]
- Hattersley, N.; Shen, L.; Jaffray, E.G.; Hay, R.T. The SUMO protease SENP6 is a direct regulator of PML nuclear bodies. Mol. Biol. Cell 2011, 22, 78–90. [Google Scholar] [CrossRef]
- Nagai, S.; Dubrana, K.; Tsai-Pflugfelder, M.; Davidson, M.B.; Roberts, T.M.; Brown, G.W.; Varela, E.; Hediger, F.; Gasser, S.M.; Krogan, N.J. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitinligase. Science 2008, 322, 597–602. [Google Scholar]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD mediates NF-κB activation in response to DNA damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef]
- Huang, T.T.; Wuerzberger-Davis, S.M.; Wu, Z.H.; Miyamoto, S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell 2003, 115, 565–576. [Google Scholar] [CrossRef]
- Lee, M.H.; Mabb, A.M.; Gill, G.B.; Yeh, E.T.; Miyamoto, S. NF-κB induction of the SUMO protease SENP2: A negative feedback loop to attenuate cell survival response to genotoxic stress. Mol. Cell 2011, 43, 180–191. [Google Scholar]
- Schwartz, D.C.; Felberbaum, R.; Hochstrasser, M. The Ulp2 SUMO protease is required for cell division following termination of the DNA damage checkpoint. Mol. Cell. Biol. 2007, 27, 6948–6961. [Google Scholar] [CrossRef]
- Lee, M.T.; Bakir, A.A.; Nguyen, K.N.; Bachant, J. The SUMO isopeptidase Ulp2p is required to prevent recombination-induced chromosome segregation lethality following DNA replication stress. PLoS Genet. 2011, 7, e1001355. [Google Scholar] [CrossRef]
- Chen, X.; Ding, B.; LeJeune, D.; Ruggiero, C.; Li, S. Rpb1 sumoylation in response to UV radiation or transcriptional impairment in yeast. PLoSOne 2009, 4, e5267. [Google Scholar]
- Ulrich, H.D. Mutual interactions between the SUMO and ubiquitin systems: A plea of no contest. Trends Cell Biol. 2005, 15, 525–532. [Google Scholar] [CrossRef]
- Moldovan, G.L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar]
- Heideker, J.; Perry, J.J.; Boddy, M.N. Genome stability roles of SUMO-targeted ubiquitin ligases. DNA Repair 2009, 8, 517–524. [Google Scholar] [CrossRef]
- Yin, Y.; Seifert, A.; Chua, J.S.; Maure, J.F.; Golebiowski, F.; Hay, R.T. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 2012, 26, 1196–1208. [Google Scholar] [CrossRef]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Jackson, S.P. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012, 26, 1179–1195. [Google Scholar]
- Ismail, I.H.; Gagne, J.P.; Caron, M.C.; McDonald, D.; Xu, Z.; Masson, J.Y.; Poirier, G.G.; Hendzel, M.J. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Research 2012, 40, 5497–5510. [Google Scholar] [CrossRef]
- De la Vega, L.; Grishina, I.; Moreno, R.; Kruger, M.; Braun, T.; Schmitz, M.L. A Redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol. Cell 2012, 46, 472–483. [Google Scholar]
- Ullmann, R.; Chien, C.D.; Avantaggiati, M.L.; Muller, S. An acetylation switch regulates SUMO-dependent protein interaction networks. Mol. Cell 2012, 46, 759–770. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cremona, C.A.; Sarangi, P.; Zhao, X. Sumoylation and the DNA Damage Response. Biomolecules 2012, 2, 376-388. https://doi.org/10.3390/biom2030376
Cremona CA, Sarangi P, Zhao X. Sumoylation and the DNA Damage Response. Biomolecules. 2012; 2(3):376-388. https://doi.org/10.3390/biom2030376
Chicago/Turabian StyleCremona, Catherine A., Prabha Sarangi, and Xiaolan Zhao. 2012. "Sumoylation and the DNA Damage Response" Biomolecules 2, no. 3: 376-388. https://doi.org/10.3390/biom2030376
APA StyleCremona, C. A., Sarangi, P., & Zhao, X. (2012). Sumoylation and the DNA Damage Response. Biomolecules, 2(3), 376-388. https://doi.org/10.3390/biom2030376