Break-Induced Replication and Genome Stability

Abstract
:1. Introduction
2. Double-Strand Break Repair Mechanisms
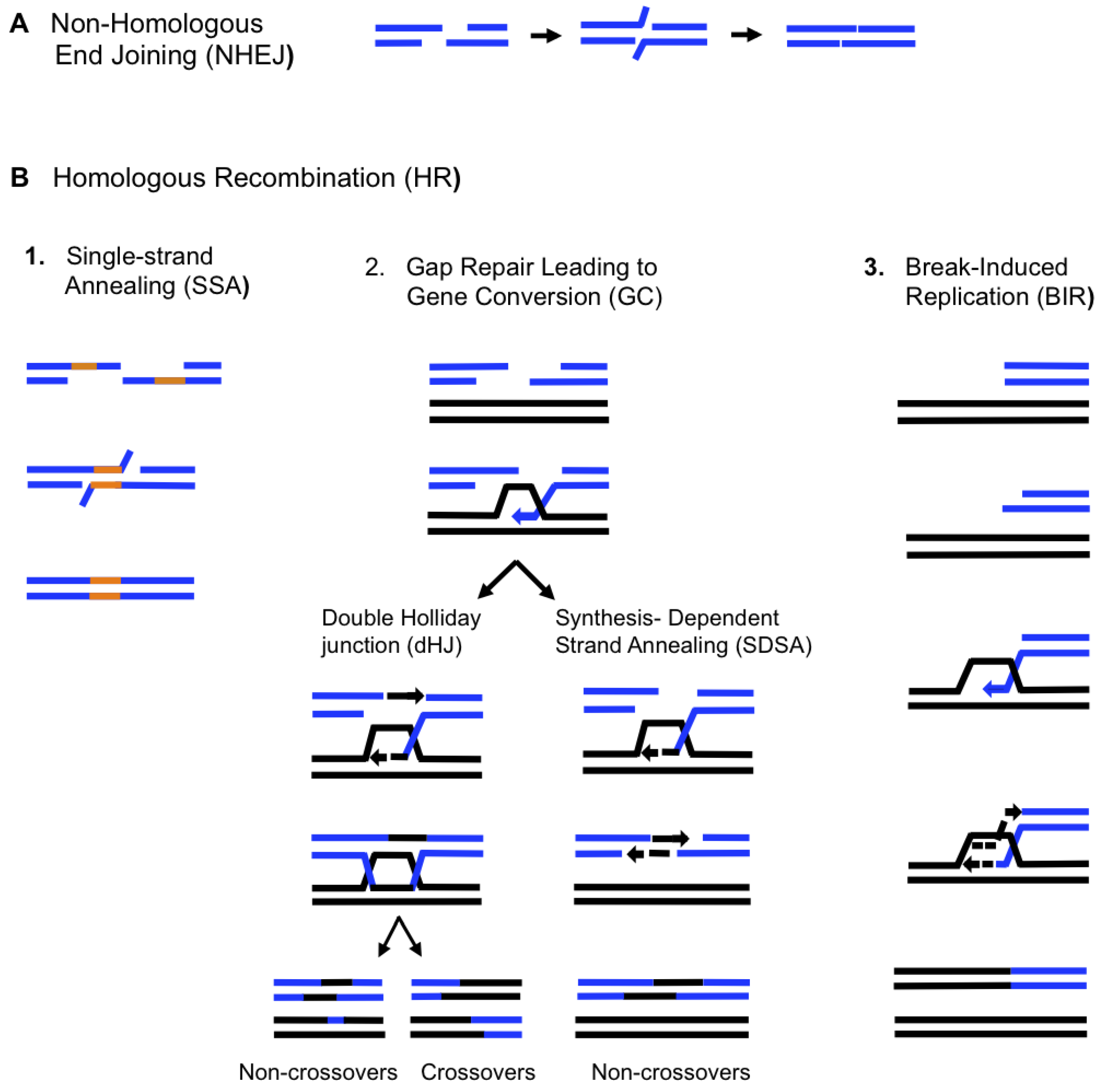
3. Recombination-Dependent Replication (RDR) in Bacteriophages and Prokaryotes
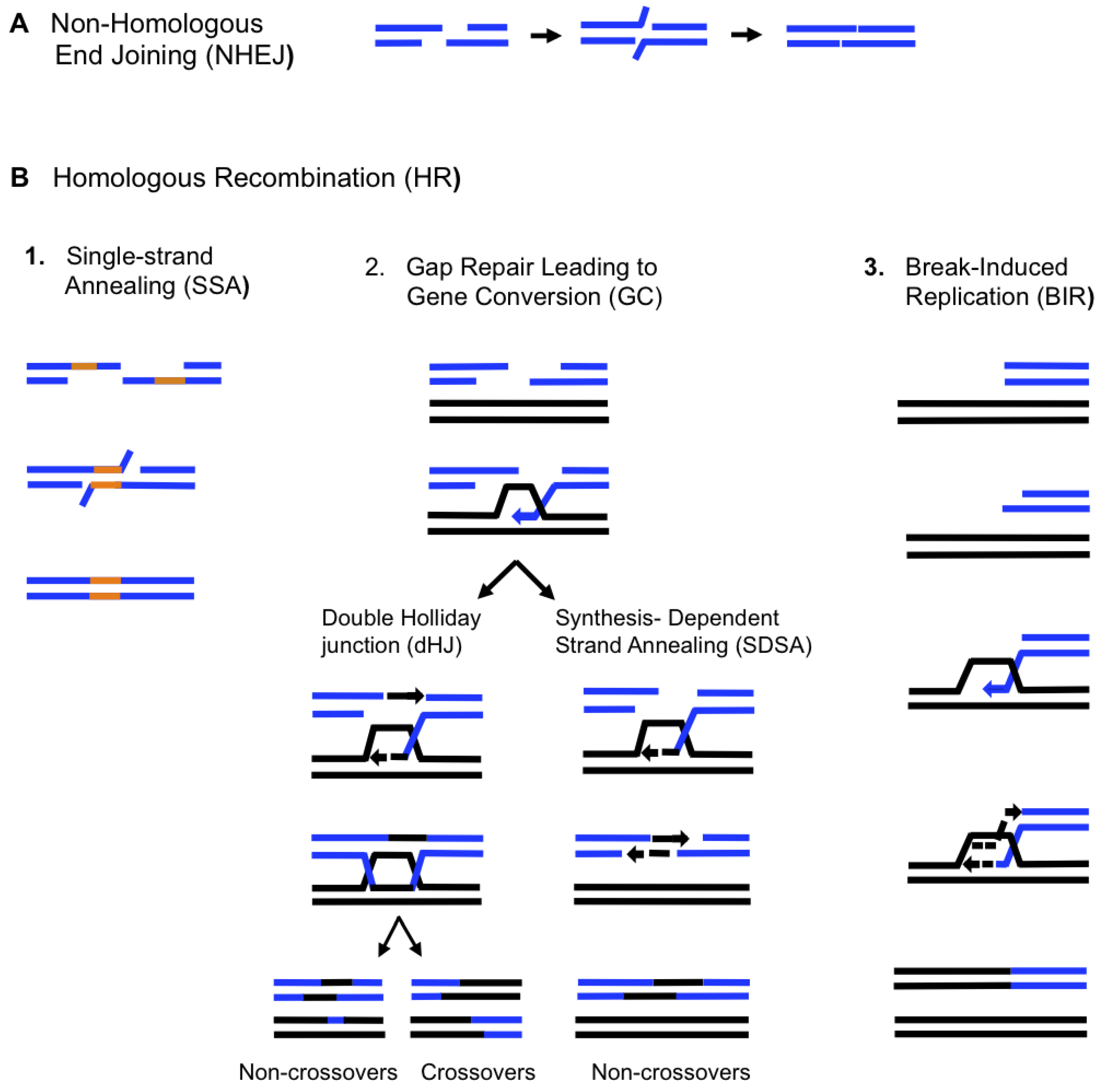
4. BIR in Eukaryotes
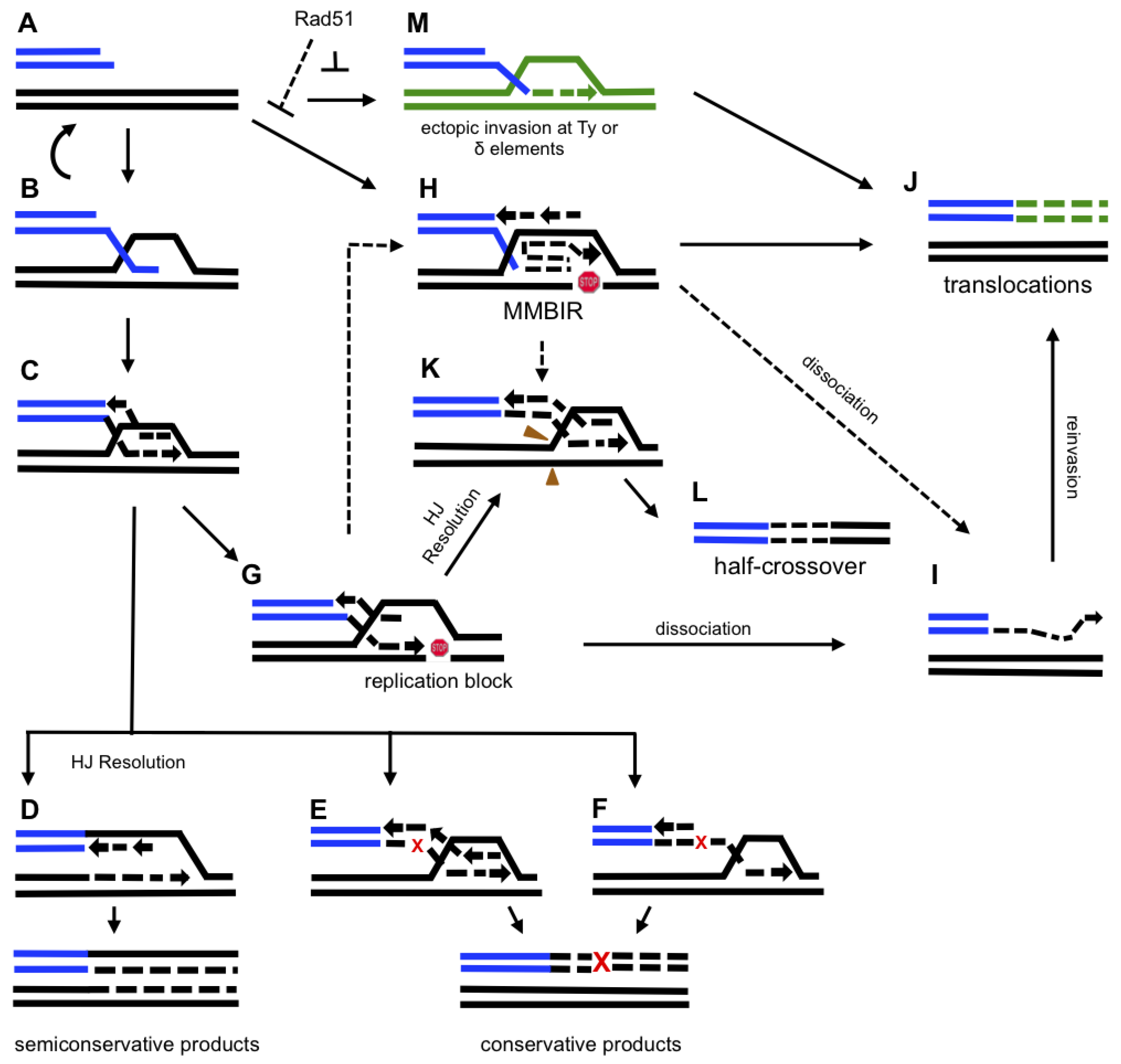
5. Molecular Mechanism of BIR
5.1. Initiation of BIR

{kind=link}
{kind=link}
{kind=link}
| Proteins | Requirement of the protein in the repair pathway | |
|---|---|---|
| Gap repair (Gene Conversion) | BIR* | |
| Rad52 | Required [15,86,87] | Required [33,77] |
| Rad51 | Required [15,86,87] | Required [33,39] |
| Rad55, Rad57 | Required [15,86,87] | Required [78] |
| Rad54 | Required [15,86,87] | Required [78,79] |
| Pol32 | Not required [34] / required **[31] | Required [27,29,34] |
| Mcm 2-7 | Not required [88] | Required [89] |
| PCNA | Required [88,90] | Required [89] |
| Polα/Primase | Not required [88] | Required [34] |
| Polδ | One is required (Polδ and Polε can substitute each other) [90] | Required [27,29,34] |
| Polε | Required***[34] | |
| Cdc45 | Not required [88] | Required [89] |
| GINS, Cdt1, Cdc7 | ? | Required [89] |
| Cdc6 | ? | Not required [89] |
| ORC | Not required [90] | Not required [89] |
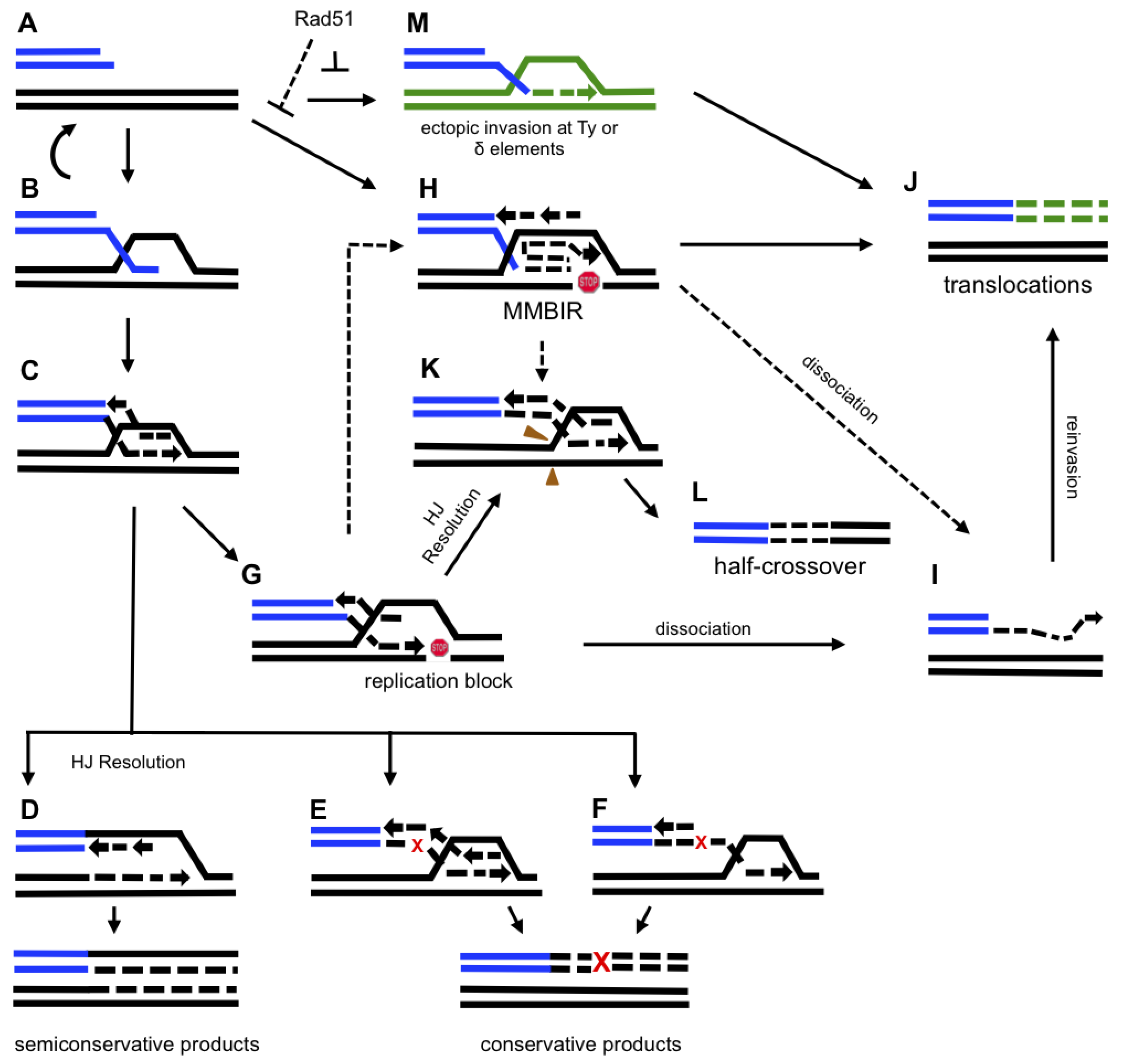
5.2. DNA Synthesis Associated with BIR
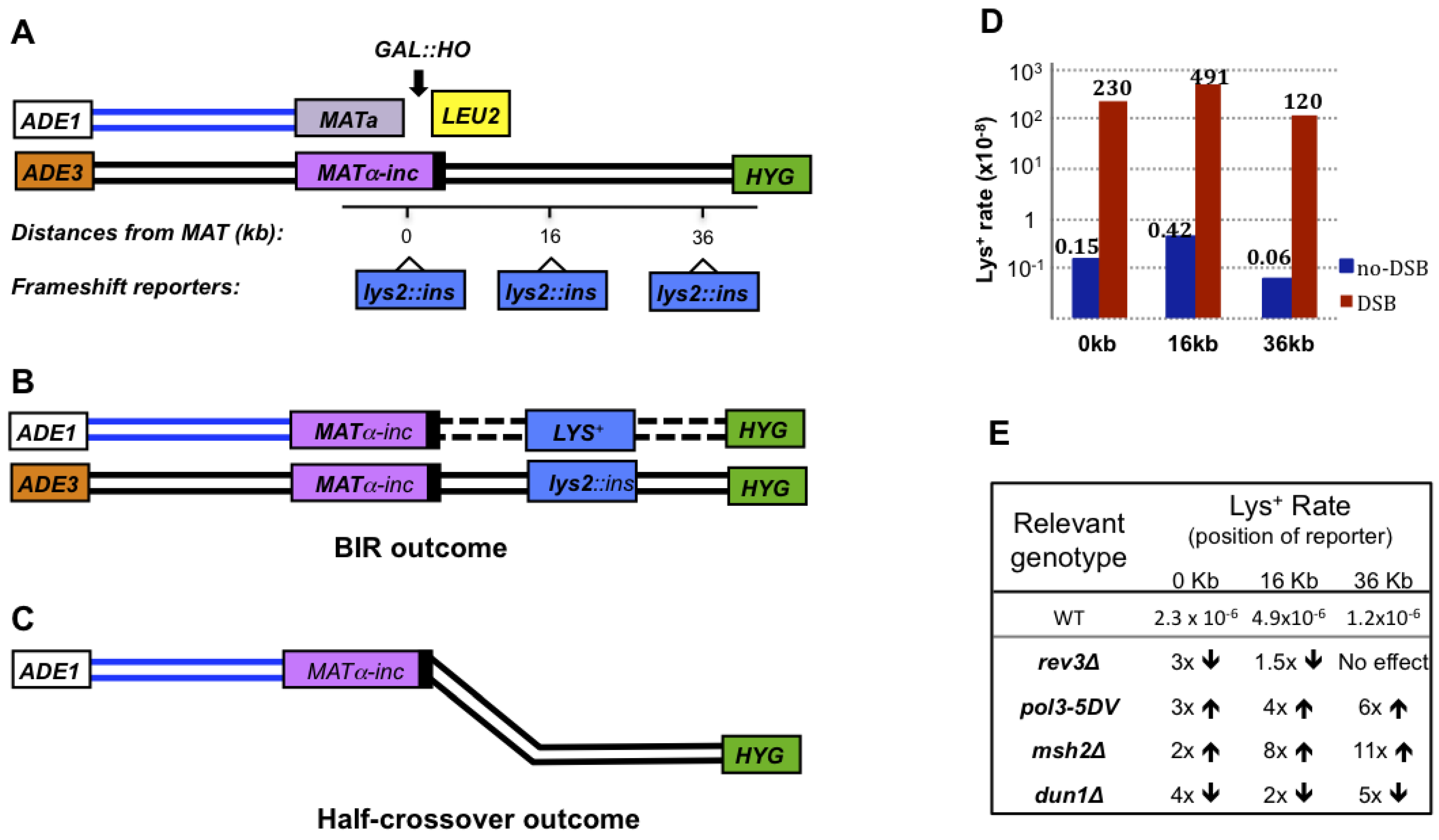
6. Mutagenesis Associated with BIR.
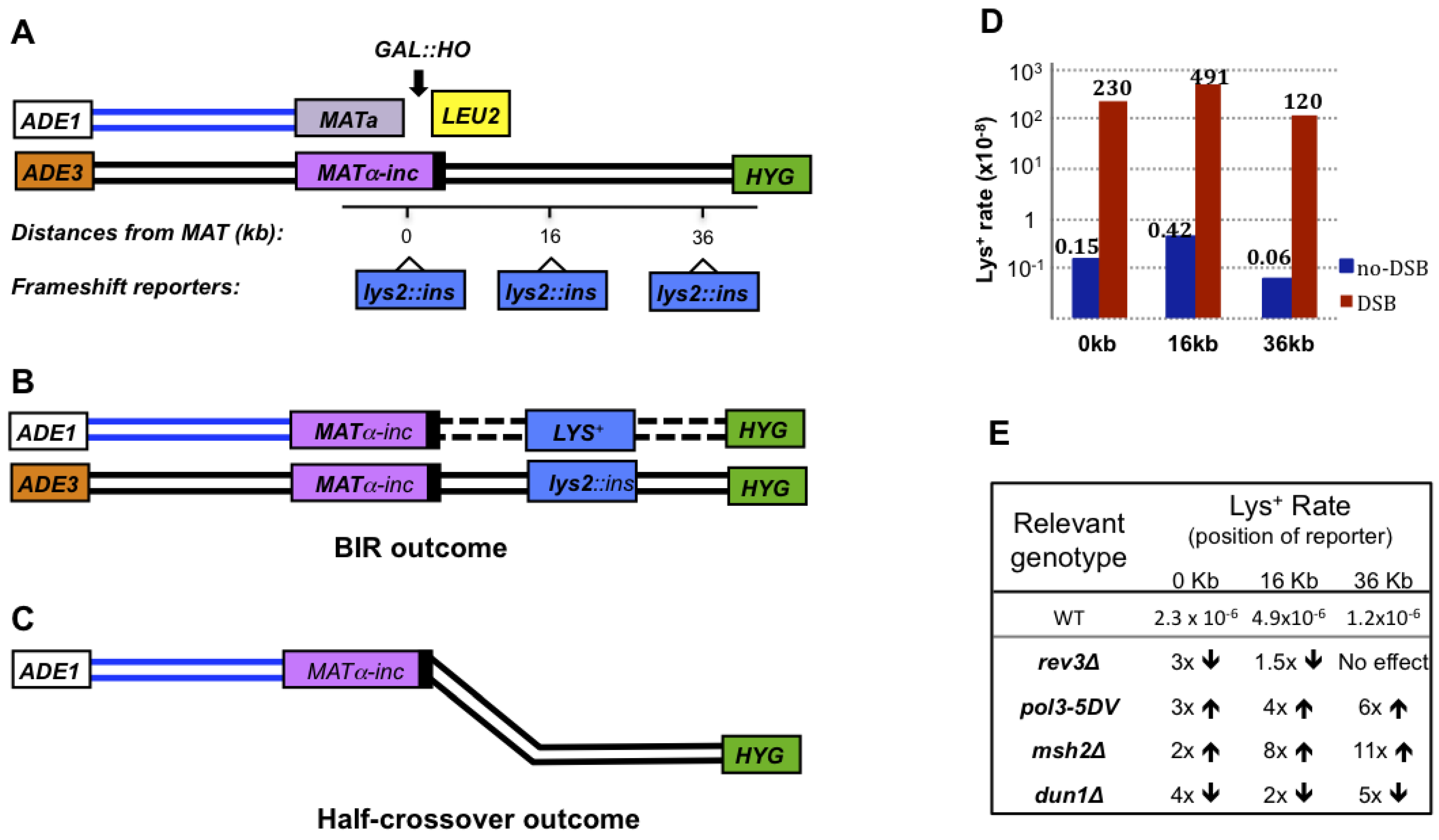
7. Chromosomal Rearrangements Associated with BIR
8. Microhomology-Mediated BIR (MMBIR)
9. Conclusions (Remaining Questions and Future Prospects)
Acknowledgments
Conflict of interest
References
- Felsher, D.W.; Bishop, J.M. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3940–3944. [Google Scholar] [CrossRef]
- Mai, S.; Fluri, M.; Siwarski, D.; Huppi, K. Genomic instability in MycER-activated Rat1A-MycER cells. Chromosome res. 1996, 4, 365–371. [Google Scholar] [CrossRef]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef]
- Woo, R.A.; Poon, R.Y. Activated oncogenes promote and cooperate with chromosomal instability for neoplastic transformation. Genes Dev. 2004, 18, 1317–1330. [Google Scholar] [CrossRef]
- Denko, N.C.; Giaccia, A.J.; Stringer, J.R.; Stambrook, P.J. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc. Natl. Acad. Sci. USA 1994, 91, 5124–5128. [Google Scholar]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; Kletsas, D.; Yoneta, A.; Herlyn, M.; Kittas, C.; Halazonetis, T.D. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell. Bio. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Kreuzer, K.N.; Brister, J.R. Initiation of bacteriophage T4 DNA replication and replication fork dynamics: a review in the Virology Journal series on bacteriophage T4 and its relatives. Virol. J. 2010, 7, 358. [Google Scholar] [CrossRef]
- Liu, J.; Morrical, S.W. Assembly and dynamics of the bacteriophage T4 homologous recombination machinery. Virol. J. 2010, 7, 357. [Google Scholar] [CrossRef]
- Maher, R.L.; Branagan, A.M.; Morrical, S.W. Coordination of DNA replication and recombination activities in the maintenance of genome stability. J. Cell Biochem. 2011, 112, 2672–2682. [Google Scholar] [CrossRef]
- Masai, H.; Tanaka, T.; Kohda, D. Stalled replication forks: making ends meet for recognition and stabilization. BioEssays 2010, 32, 687–697. [Google Scholar] [CrossRef]
- Gabbai, C.B.; Marians, K.J. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair 2010, 9, 202–209. [Google Scholar] [CrossRef]
- Heller, R.C.; Marians, K.J. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell. Biol. 2006, 7, 932–943. [Google Scholar] [CrossRef]
- Paques, F.; Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Moore, J.K.; Haber, J.E. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell Biol. 1996, 16, 2164–2173. [Google Scholar]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef]
- Paques, F.; Leung, W.Y.; Haber, J.E. Expansions and contractions in a tandem repeat induced by double-strand break repair. Mol. Cell Biol. 1998, 18, 2045–2054. [Google Scholar]
- Strathern, J.N.; Shafer, B.K.; McGill, C.B. DNA synthesis errors associated with double-strand-break repair. Genetics 1995, 140, 965–972. [Google Scholar]
- Hicks, W.M.; Kim, M.; Haber, J.E. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 2010, 329, 82–85. [Google Scholar]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef]
- Llorente, B.; Smith, C.E.; Symington, L.S. Break-induced replication: what is it and what is it for? Cell Cycle 2008, 7, 859–864. [Google Scholar] [CrossRef]
- McEachern, M.J.; Haber, J.E. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef]
- Deem, A.; Keszthelyi, A.; Blackgrove, T.; Vayl, A.; Coffey, B.; Mathur, R.; Chabes, A.; Malkova, A. Break-induced replication is highly inaccurate. PLoS Biol. 2011, 9, e1000594. [Google Scholar] [CrossRef]
- Bosco, G.; Haber, J.E. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 1998, 150, 1037–1047. [Google Scholar]
- Deem, A.; Barker, K.; Vanhulle, K.; Downing, B.; Vayl, A.; Malkova, A. Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics 2008, 179, 1845–1860. [Google Scholar] [CrossRef]
- Vanhulle, K.; Lemoine, F.J.; Narayanan, V.; Downing, B.; Hull, K.; McCullough, C.; Bellinger, M.; Lobachev, K.; Petes, T.D.; Malkova, A. Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol. Cell Biol. 2007, 7, 2601–2614. [Google Scholar]
- Smith, C.E.; Lam, A.F.; Symington, L.S. Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol. Cell Biol. 2009, 29, 1432–1441. [Google Scholar] [CrossRef]
- Smith, C.E.; Llorente, B.; Symington, L.S. Template switching during break-induced replication. Nature 2007, 447, 102–105. [Google Scholar] [CrossRef]
- Jain, S.; Sugawara, N.; Lydeard, J.; Vaze, M.; Tanguy Le Gac, N.; Haber, J.E. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 2009, 23, 291–303. [Google Scholar] [CrossRef]
- Ruiz, J.F.; Gomez-Gonzalez, B.; Aguilera, A. Chromosomal translocations caused by either Pol32-dependent or Pol32-independent triparental break-induced replication. Mol. Cell Biol. 2009, 29, 5441–5454. [Google Scholar] [CrossRef]
- Davis, A.P.; Symington, L.S. RAD51-dependent break-induced replication in yeast. Mol. Cell Biol. 2004, 24, 2344–2351. [Google Scholar] [CrossRef]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef]
- Tan, F.J.; Hoang, M.L.; Koshland, D. DNA resection at chromosome breaks promotes genome stability by constraining non-allelic homologous recombination. PLoS Genet. 2012, 8, e1002633. [Google Scholar] [CrossRef]
- Hoang, M.L.; Tan, F.J.; Lai, D.C.; Celniker, S.E.; Hoskins, R.A.; Dunham, M.J.; Zheng, Y.; Koshland, D. Competitive repair by naturally dispersed repetitive DNA during non-allelic homologous recombination. PLoS Genet. 2010, 6, e1001228. [Google Scholar] [CrossRef]
- Fishman-Lobell, J.; Rudin, N.; Haber, J.E. Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell Biol. 1992, 12, 1292–1303. [Google Scholar]
- Morrow, D.M.; Connelly, C.; Hieter, P. Break copy" duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 1997, 147, 371–382. [Google Scholar]
- Malkova, A.; Naylor, M.L.; Yamaguchi, M.; Ira, G.; Haber, J.E. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol. Cell Biol. 2005, 25, 933–944. [Google Scholar] [CrossRef]
- Luder, A.; Mosig, G. Two alternative mechanisms for initiation of DNA replication forks in bacteriophage T4: priming by RNA polymerase and by recombination. Proc. Natl. Acad. Sci. the USA 1982, 79, 1101–1105. [Google Scholar] [CrossRef]
- Mosig, G. Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu. Rev. Genet. 1998, 32, 379–413. [Google Scholar] [CrossRef]
- Kreuzer, K.N.; Saunders, M.; Weislo, L.J.; Kreuzer, H.W. Recombination-dependent DNA replication stimulated by double-strand breaks in bacteriophage T4. J. Bacteriol. 1995, 177, 6844–6853. [Google Scholar]
- George, J.W.; Kreuzer, K.N. Repair of double-strand breaks in bacteriophage T4 by a mechanism that involves extensive DNA replication. Genetics 1996, 143, 1507–1520. [Google Scholar]
- Mueller, J.E.; Clyman, J.; Huang, Y.J.; Parker, M.M.; Belfort, M. Intron mobility in phage T4 occurs in the context of recombination-dependent DNA replication by way of multiple pathways. Genes Dev. 1996, 10, 351–364. [Google Scholar] [CrossRef]
- Kreuzer, K.N. Recombination-dependent DNA replication in phage T4. Trends Biochem. Sci. 2000, 25, 165–173. [Google Scholar] [CrossRef]
- Asai, T.; Bates, D.B.; Kogoma, T. DNA replication triggered by double-stranded breaks in E. coli: dependence on homologous recombination functions. Cell 1994, 78, 1051–1061. [Google Scholar] [CrossRef]
- Asai, T.; Sommer, S.; Bailone, A.; Kogoma, T. Homologous recombination-dependent initiation of DNA replication from DNA damage-inducible origins in Escherichia coli. EMBO J. 1993, 12, 3287–3295. [Google Scholar]
- Kogoma, T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 1997, 61, 212–238. [Google Scholar]
- Kuzminov, A.; Stahl, F.W. Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes Dev. 1999, 13, 345–356. [Google Scholar] [CrossRef]
- Cairns, J.; Foster, P.L. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 1991, 128, 695–701. [Google Scholar]
- Harris, R.S.; Longerich, S.; Rosenberg, S.M. Recombination in adaptive mutation. Science 1994, 264, 258–260. [Google Scholar]
- Formosa, T.; Alberts, B.M. DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell 1986, 47, 793–806. [Google Scholar] [CrossRef]
- Lark, K.G.; Lark, C.A. RecA-dependent DNA replication in the absence of protein synthesis: characteristics of a dominant lethal replication mutation, DnaT, and requirement for RecA+ function. Cold Spring Harb Symp. Quant. Biol. 1979, 43 Pt 1, 537–549. [Google Scholar]
- Magee, T.R.; Kogoma, T. Requirement of RecBC enzyme and an elevated level of activated RecA for induced stable DNA replication in Escherichia coli. J. Bacteriol. 1990, 172, 1834–1839. [Google Scholar]
- Bleuit, J.S.; Xu, H.; Ma, Y.; Wang, T.; Liu, J.; Morrical, S.W. Mediator proteins orchestrate enzyme-ssDNA assembly during T4 recombination-dependent DNA replication and repair. Proc. Natl. Acad. Sci. USA 2001, 98, 8298–8305. [Google Scholar] [CrossRef]
- Marians, K.J. PriA: at the crossroads of DNA replication and recombination. Prog. Nucleic. Acid Res. Mol. Biol. 1999, 63, 39–67. [Google Scholar] [CrossRef]
- Cao, Y.; Kogoma, T. Requirement for the polymerization and 5'-->3' exonuclease activities of DNA polymerase I in initiation of DNA replication at oriK sites in the absence of RecA in Escherichia coli rnhA mutants. J. Bacteriol. 1993, 175, 7254–7259. [Google Scholar]
- Kogoma, T.; Lark, K.G. Characterization of the replication of Escherichia coli DNA in the absence of protein synthesis: stable DNA replication. J. Mol. Biol. 1975, 94, 243–256. [Google Scholar] [CrossRef]
- Marians, K.J. PriA-directed replication fork restart in Escherichia coli. Trends Biochem. Sci. 2000, 25, 185–189. [Google Scholar] [CrossRef]
- Hong, X.; Cadwell, G.W.; Kogoma, T. Escherichia coli RecG and RecA proteins in R-loop formation. EMBO J. 1995, 14, 2385–2392. [Google Scholar]
- Marrero, V.A.; Symington, L.S. Extensive DNA end processing by Exo1 and Sgs1 inhibits break-induced replication. PLoS Genet. 2010, 6, e1001007. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Puddu, F.; Costanzo, V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat. Struct. Mol. Biol. 2012, 19, 17–24. [Google Scholar]
- Richardson, C.; Jasin, M. Coupled homologous and nonhomologous repair of a double-strand break preserves genomic integrity in mammalian cells. Mol. Cell Biol. 2000, 20, 9068–9075. [Google Scholar] [CrossRef]
- Johnson, R.D.; Jasin, M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem. Soc. Trans. 2001, 29, 196–201. [Google Scholar] [CrossRef]
- Pierce, A.J.; Stark, J.M.; Araujo, F.D.; Moynahan, M.E.; Berwick, M.; Jasin, M. Double-strand breaks and tumorigenesis. Trends Cell Biol. 2001, 11, 52–59. [Google Scholar]
- Elliott, B.; Jasin, M. Double-strand breaks and translocations in cancer. Cell. Mol. Life Sci. : CMLS 2002, 59, 373–385. [Google Scholar] [CrossRef]
- Rowley, J.D. Chromosome translocations: dangerous liaisons revisited. Nat. Rev. Cancer 2001, 1, 245–250. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Ferguson, D.O.; Alt, F.W. DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene 2001, 20, 5572–5579. [Google Scholar] [CrossRef]
- Neumann, A.A.; Reddel, R.R. Telomere maintenance and cancer -- look, no telomerase. Nat. Rev. Cancer 2002, 2, 879–884. [Google Scholar] [CrossRef]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef]
- Durant, S.T. Telomerase-independent paths to immortality in predictable cancer subtypes. J. Cancer 2012, 3, 67–82. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Reddel, R.R.; Bryan, T.M.; Colgin, L.M.; Perrem, K.T.; Yeager, T.R. Alternative lengthening of telomeres in human cells. Radiat. Res. 2001, 155, 194–200. [Google Scholar] [CrossRef]
- Reddel, R.R. Alternative lengthening of telomeres, telomerase, and cancer. Cancer Lett. 2003, 194, 155–162. [Google Scholar] [CrossRef]
- Chung, W.H.; Zhu, Z.; Papusha, A.; Malkova, A.; Ira, G. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 2010, 6, e1000948. [Google Scholar] [CrossRef]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef]
- Signon, L.; Malkova, A.; Naylor, M.L.; Klein, H.; Haber, J.E. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol. Cell Biol. 2001, 21, 2048–2056. [Google Scholar] [CrossRef]
- Downing, B.; Morgan, R.; VanHulle, K.; Deem, A.; Malkova, A. Large inverted repeats in the vicinity of a single double-strand break strongly affect repair in yeast diploids lacking Rad51. Mutat. Res. 2008, 645, 9–18. [Google Scholar] [CrossRef]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef]
- Le, S.; Moore, J.K.; Haber, J.E.; Greider, C.W. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 1999, 152, 143–152. [Google Scholar]
- Teng, S.C.; Zakian, V.A. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol. Cell Biol. 1999, 19, 8083–8093. [Google Scholar]
- Teng, S.C.; Chang, J.; McCowan, B.; Zakian, V.A. Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol. Cell 2000, 6, 947–952. [Google Scholar] [CrossRef]
- Tsukamoto, Y.; Taggart, A.K.; Zakian, V.A. The role of the Mre11-Rad50-Xrs2 complex in telomerase- mediated lengthening of Saccharomyces cerevisiae telomeres. Curr. Biol. 2001, 11, 1328–1335. [Google Scholar] [CrossRef]
- Chen, Q.; Ijpma, A.; Greider, C.W. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol. Cell Biol. 2001, 21, 1819–1827. [Google Scholar] [CrossRef]
- Sugawara, N.; Ivanov, E.L.; Fishman-Lobell, J.; Ray, B.L.; Wu, X.; Haber, J.E. DNA structure-dependent requirements for yeast RAD genes in gene conversion. Nature 1995, 373, 84–86. [Google Scholar] [CrossRef]
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef]
- Wang, X.; Ira, G.; Tercero, J.A.; Holmes, A.M.; Diffley, J.F.; Haber, J.E. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell Biol. 2004, 24, 6891–6899. [Google Scholar] [CrossRef]
- Lydeard, J.R.; Lipkin-Moore, Z.; Sheu, Y.J.; Stillman, B.; Burgers, P.M.; Haber, J.E. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 2010, 24, 1133–1144. [Google Scholar] [CrossRef]
- Holmes, A.M.; Haber, J.E. Double-strand break repair in yeast requires both leading and lagging strand DNA polymerases. Cell 1999, 96, 415–424. [Google Scholar] [CrossRef]
- Schmidt, K.H.; Viebranz, E.; Doerfler, L.; Lester, C.; Rubenstein, A. Formation of complex and unstable chromosomal translocations in yeast. PLoS One 2010, 5, e12007. [Google Scholar]
- Yang, Y.; Sterling, J.; Storici, F.; Resnick, M.A.; Gordenin, D.A. Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLoS Genet. 2008, 4, e1000264. [Google Scholar] [CrossRef]
- Lemoine, F.J.; Degtyareva, N.P.; Lobachev, K.; Petes, T.D. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 2005, 120, 587–598. [Google Scholar] [CrossRef]
- Lemoine, F.J.; Degtyareva, N.P.; Kokoska, R.J.; Petes, T.D. Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol. Cell Biol. 2008, 28, 5359–5368. [Google Scholar] [CrossRef]
- Narayanan, V.; Mieczkowski, P.A.; Kim, H.M.; Petes, T.D.; Lobachev, K.S. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell 2006, 125, 1283–1296. [Google Scholar] [CrossRef]
- Schmidt, K.H.; Wu, J.; Kolodner, R.D. Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol. Cell Biol. 2006, 26, 5406–5420. [Google Scholar] [CrossRef]
- Putnam, C.D.; Hayes, T.K.; Kolodner, R.D. Specific pathways prevent duplication-mediated genome rearrangements. Nature 2009, 460, 984–989. [Google Scholar] [CrossRef]
- Sabatier, L.; Ricoul, M.; Pottier, G.; Murnane, J.P. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 2005, 3, 139–150. [Google Scholar] [CrossRef]
- Haber, J.E.; Hearn, M. Rad52-independent mitotic gene conversion in Saccharomyces cerevisiae frequently results in chromosomal loss. Genetics 1985, 111, 7–22. [Google Scholar]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef]
- Slack, A.; Thornton, P.C.; Magner, D.B.; Rosenberg, S.M.; Hastings, P.J. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006, 2, e48. [Google Scholar] [CrossRef]
- Payen, C.; Koszul, R.; Dujon, B.; Fischer, G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008, 4, e1000175. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; Cuppen, E. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 2011, 20, 1916–1924. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Liu, P.; Erez, A.; Nagamani, S.C.; Dhar, S.U.; Kolodziejska, K.E.; Dharmadhikari, A.V.; Cooper, M.L.; Wiszniewska, J.; Zhang, F.; Withers, M.A.; Bacino, C.A.; et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 2011, 146, 889–903. [Google Scholar] [CrossRef]
- Vissers, L.E.; Bhatt, S.S.; Janssen, I.M.; Xia, Z.; Lalani, S.R.; Pfundt, R.; Derwinska, K.; de Vries, B.B.; Gilissen, C.; Hoischen, A.; et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009, 18, 3579–3593. [Google Scholar] [CrossRef]
- Motobayashi, M.; Nishimura-Tadaki, A.; Inaba, Y.; Kosho, T.; Miyatake, S.; Niimi, T.; Nishimura, T.; Wakui, K.; Fukushima, Y.; Matsumoto, N.; Koike, K. Neurodevelopmental features in 2q23.1 microdeletion syndrome: report of a new patient with intractable seizures and review of literature. Am. J. Med. Genet. A. 2012, 158A, 861–868. [Google Scholar] [CrossRef]
- Bondurand, N.; Fouquet, V.; Baral, V.; Lecerf, L.; Loundon, N.; Goossens, M.; Duriez, B.; Labrune, P.; Pingault, V. Alu-mediated deletion of SOX10 regulatory elements in Waardenburg syndrome type 4. Eur. J. Hum. Genet. 2012.
- Zhang, F.; Khajavi, M.; Connolly, A.M.; Towne, C.F.; Batish, S.D.; Lupski, J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009, 41, 849–853. [Google Scholar]
- Lawson, A.R.; Hindley, G.F.; Forshew, T.; Tatevossian, R.G.; Jamie, G.A.; Kelly, G.P.; Neale, G.A.; Ma, J.; Jones, T.A.; Ellison, D.W.; Sheer, D. RAF gene fusion breakpoints in pediatric brain tumors are characterized by significant enrichment of sequence microhomology. Genome Res. 2011, 21, 505–514. [Google Scholar] [CrossRef]
- Marechal, A.; Parent, J.S.; Veronneau-Lafortune, F.; Joyeux, A.; Lang, B.F.; Brisson, N. Whirly proteins maintain plastid genome stability in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 14693–14698. [Google Scholar]
- McConnell Smith, A.; Takeuchi, R.; Pellenz, S.; Davis, L.; Maizels, N.; Monnat, R.J., Jr.; Stoddard, B.L. Generation of a nicking enzyme that stimulates site-specific gene conversion from the I-AniI LAGLIDADG homing endonuclease. Proc. Natl. Acad. Sci. USA 2009, 106, 5099–5104. [Google Scholar]
- Nielsen, I.; Bentsen, I.B.; Lisby, M.; Hansen, S.; Mundbjerg, K.; Andersen, A.H.; Bjergbaek, L. A Flp-nick system to study repair of a single protein-bound nick in vivo. Nat. Methods 2009, 6, 753–757. [Google Scholar]
- Munoz-Galvan, S.; Tous, C.; Blanco, M.G.; Schwartz, E.K.; Ehmsen, K.T.; West, S.C.; Heyer, W.D.; Aguilera, A. Distinct roles of Mus81, Yen1, Slx1-Slx4 and Rad1 nucleases in the repair of replication-born double strand breaks by sister chromatid exchange. Mol. Cell Biol. 2012, 9, 1592–1603. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sakofsky, C.J.; Ayyar, S.; Malkova, A. Break-Induced Replication and Genome Stability. Biomolecules 2012, 2, 483-504. https://doi.org/10.3390/biom2040483
Sakofsky CJ, Ayyar S, Malkova A. Break-Induced Replication and Genome Stability. Biomolecules. 2012; 2(4):483-504. https://doi.org/10.3390/biom2040483
Chicago/Turabian StyleSakofsky, Cynthia J., Sandeep Ayyar, and Anna Malkova. 2012. "Break-Induced Replication and Genome Stability" Biomolecules 2, no. 4: 483-504. https://doi.org/10.3390/biom2040483
APA StyleSakofsky, C. J., Ayyar, S., & Malkova, A. (2012). Break-Induced Replication and Genome Stability. Biomolecules, 2(4), 483-504. https://doi.org/10.3390/biom2040483