Reliable Metabolic Flux Estimation in Escherichia coli Central Carbon Metabolism Using Intracellular Free Amino Acids

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Continuous Culture of E. coli MG1655
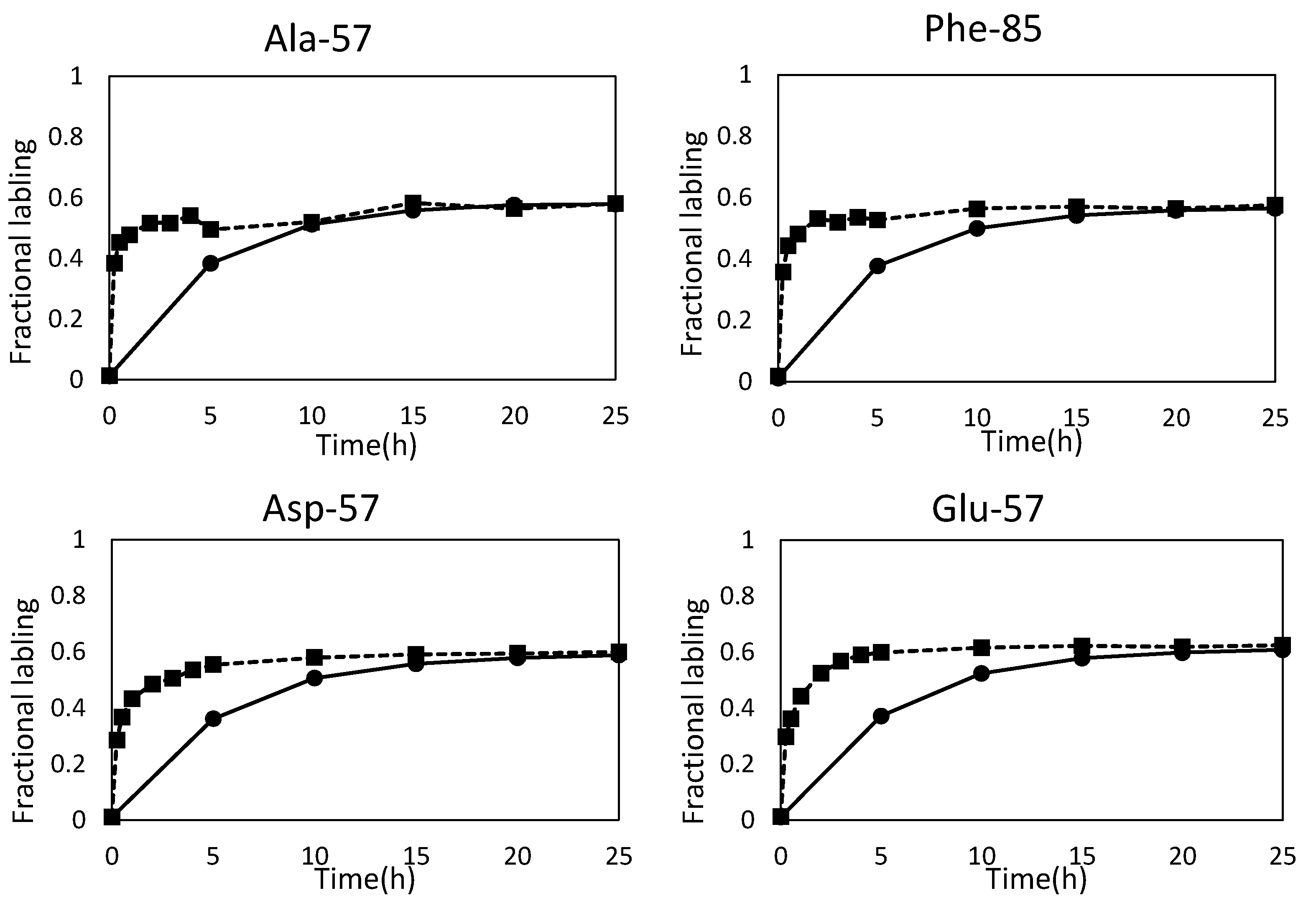
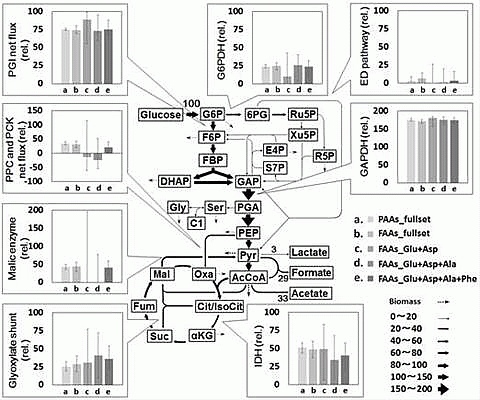
2.2. Metabolic Flux Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
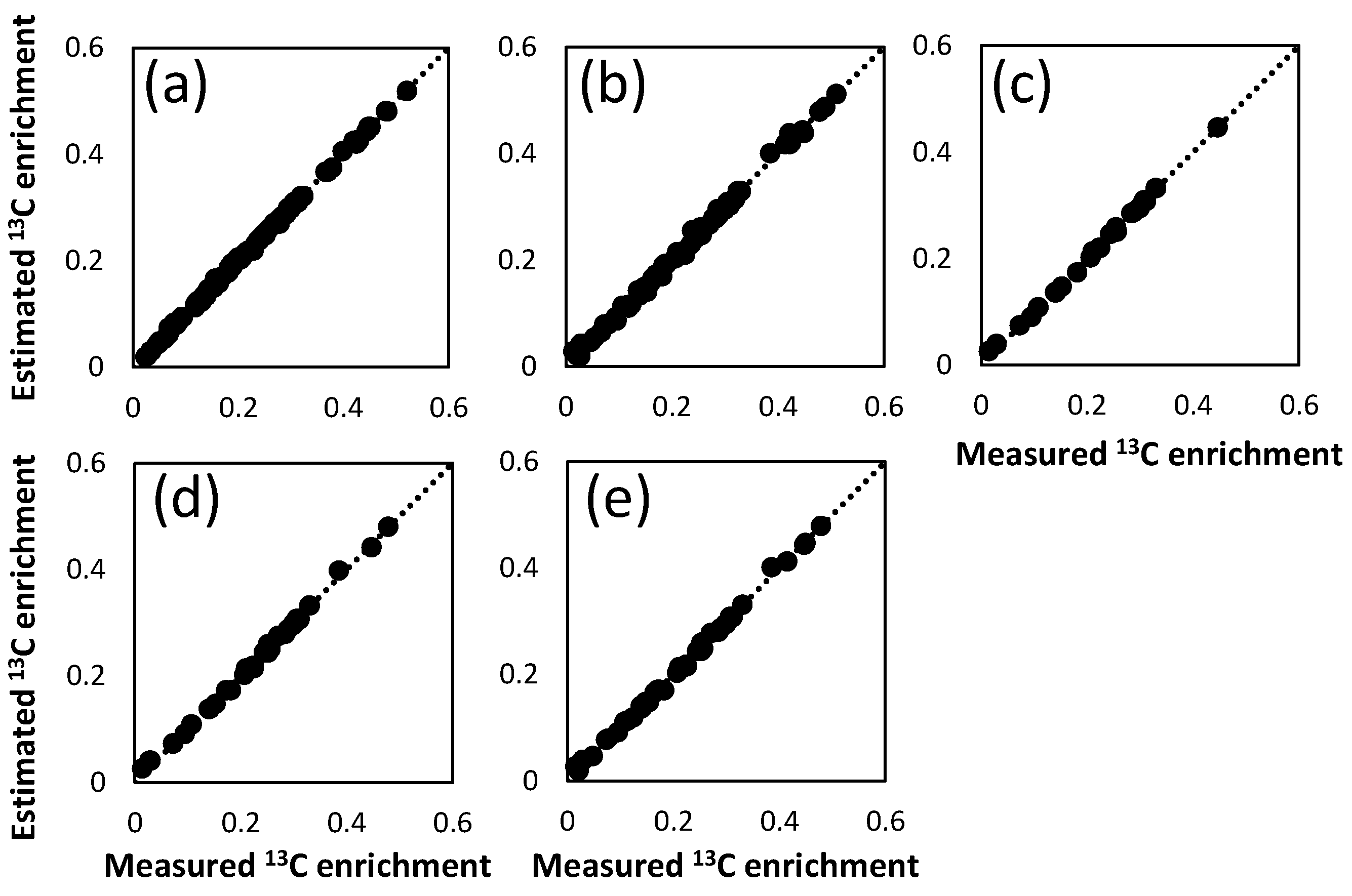
| Number of independent measurements (=n) | Number of metabolites used for fitting | Number of fragments used for fitting | n-p 1 | RSS | RSS/(n-p) | |
|---|---|---|---|---|---|---|
| PAAs_fullset | 92 | 11 | 25 | 71 | 0.0013 | 0.00002 |
| FAAs_fullset | 66 | 9 | 19 | 45 | 0.0037 | 0.00008 |
| FAAs_Glu+Asp | 25 | 2 | 7 | 9 | 0.0007 | 0.00017 |
| FAAs_Glu+Asp+Ala | 30 | 3 | 9 | 19 | 0.0012 | 0.00014 |
| FAAs_Glu+Asp+Ala+Phe | 40 | 4 | 11 | 4 | 0.0015 | 0.00008 |
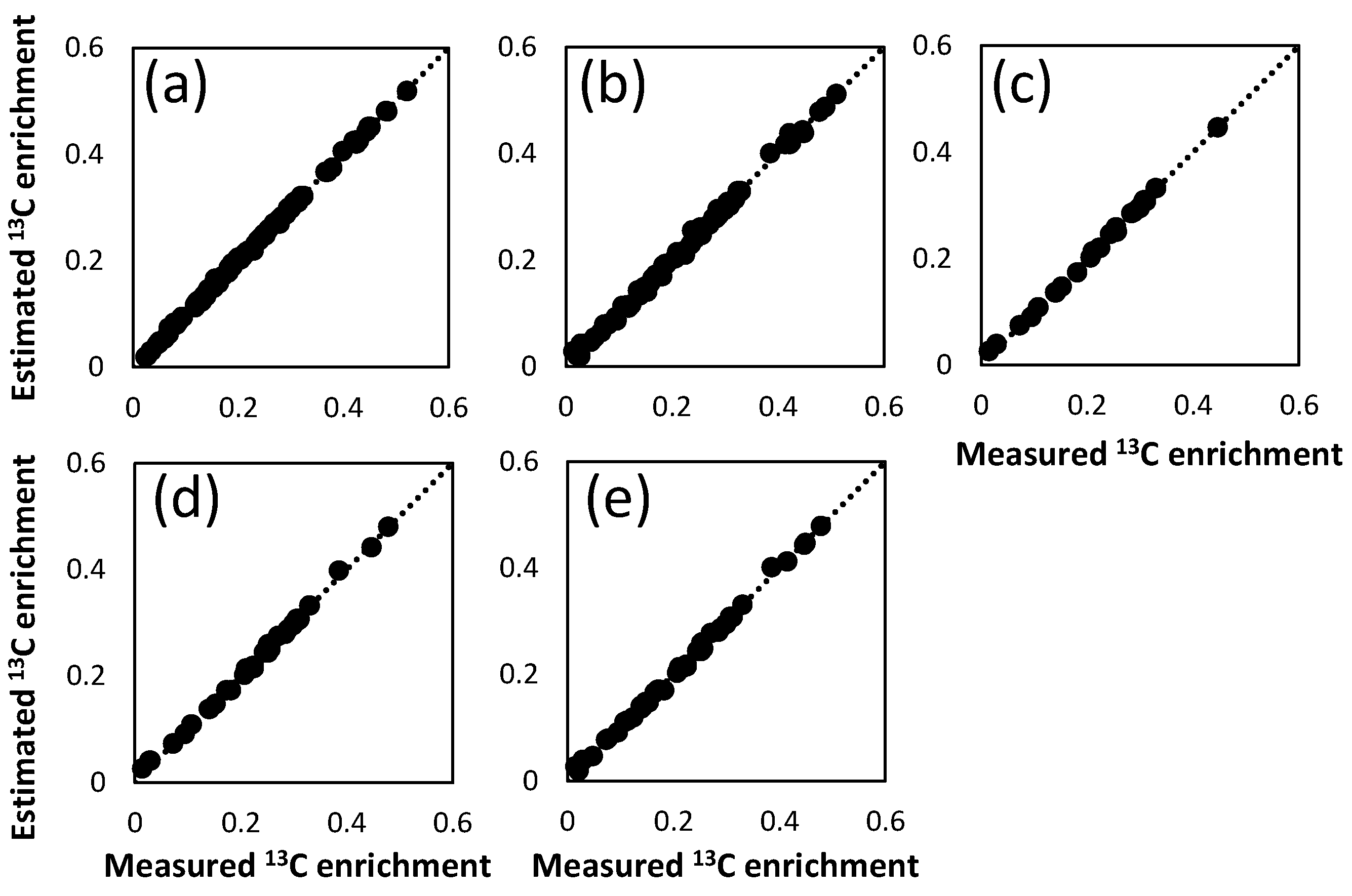

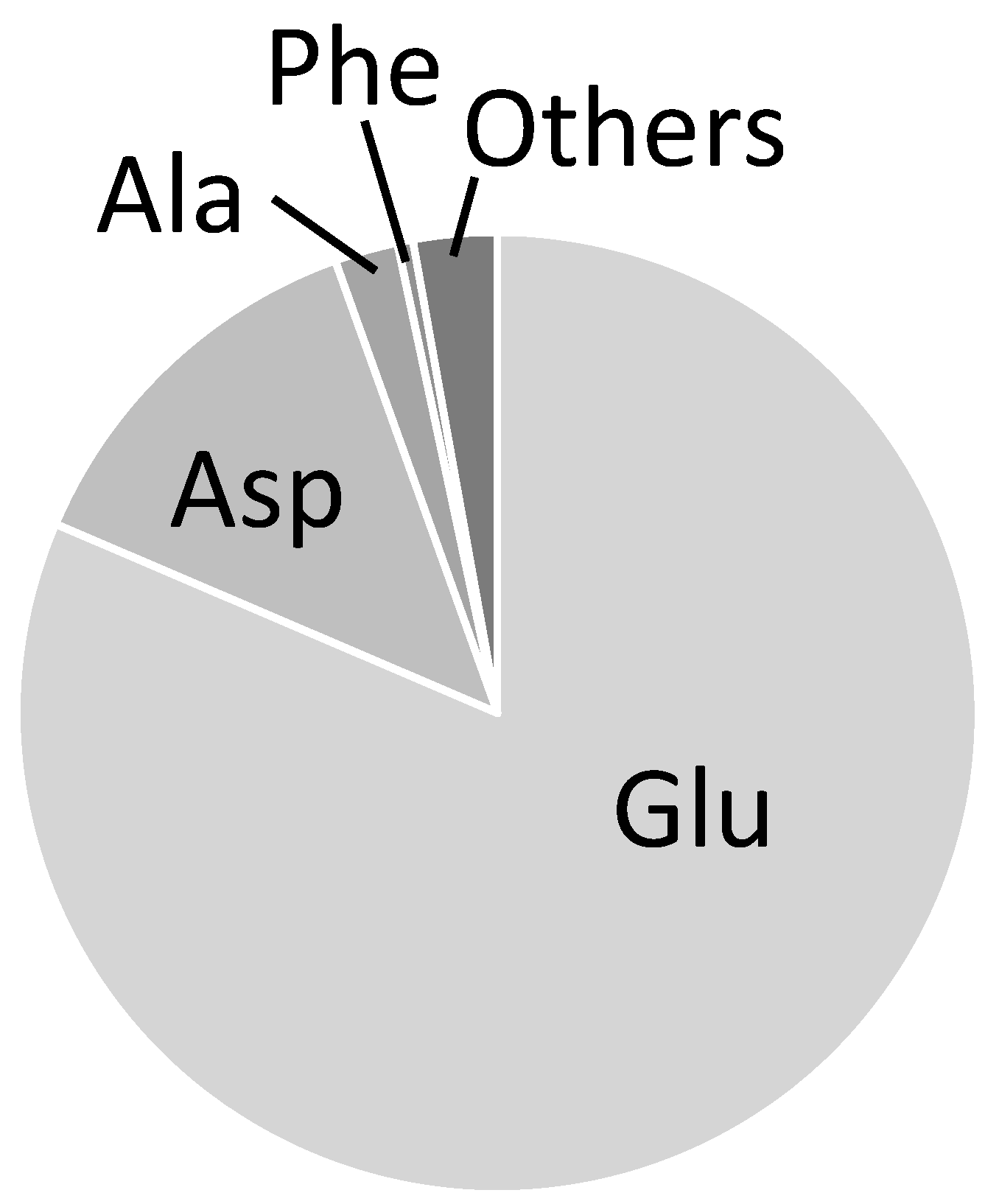
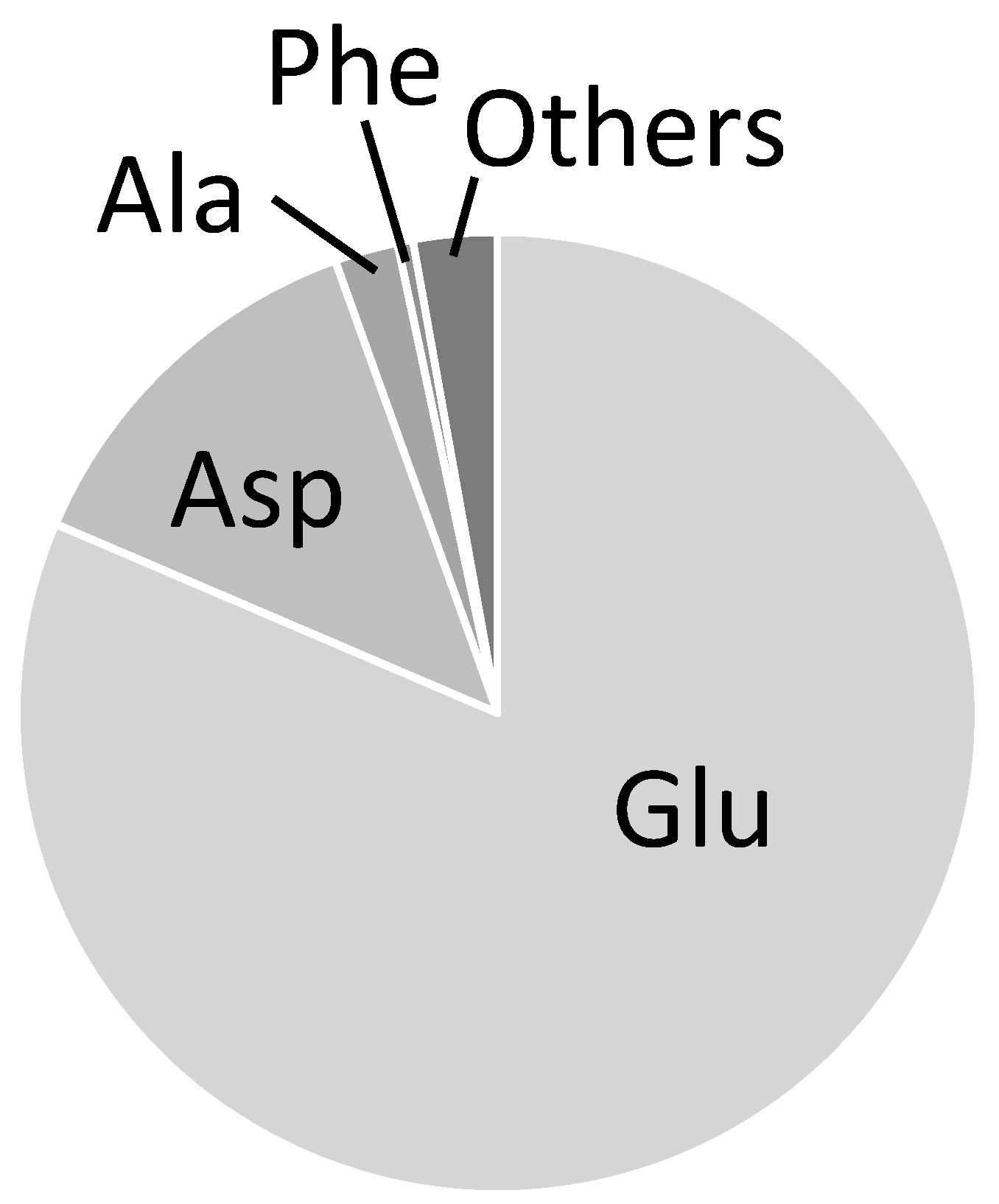
2.3. Combination of Amino Acids for Reliable FAAs-Based MFA
| This study | Mori et al. (2011) [17] | Toya et al. (2010) [19] | Iwatani et al. (2007) [14] | |
|---|---|---|---|---|
| Experimental conditions | ||||
| Analysis | GC-MS | GC-MS | CE-TOFMS | LC-MS/MS |
| Culture | Chemostat culture | Chemostat culture | Batch culture | Fed-batch culture |
| Amino acids | ||||
| Alanine | + | + | + | + |
| Valine | + | - | + | + |
| Leucine | + | + | + | - |
| Isoleucine | - | - | + | - |
| Lysine | - | - | + | - |
| Aspartate | + | + | + | + |
| Asparagine | - | - | - | + |
| Threonine | + | - | + | + |
| Methionine | - | - | - | - |
| Glutamate | + | + | + | + |
| Glutamine | - | - | - | + |
| Arginine | - | - | + | - |
| Proline | - | - | + | - |
| Glycine | + | - | + | + |
| Serine | - | + | + | + |
| Cysteine | - | - | - | - |
| Histidine | - | - | + | - |
| Tyrosine | + | - | + | + |
| Phenylalanine | + | + | + | + |
| Tryptophan | - | - | - | - |
3. Experimental Section
3.1. Strain and Medium
3.2. Culture Condition
3.3. Off-Line Measurements
3.4. Sample Preparation for GC-MS Analysis
3.5. GC-MS Analysis of PAAs and FAAs
3.6. Metabolic Flux Analysis
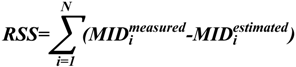
 is the mass isotopomer distribution (MID) of the ith measured metabolite,
is the mass isotopomer distribution (MID) of the ith measured metabolite, 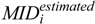 is the estimated MID of the corresponding metabolite, and N is the number of metabolites used for flux estimation. Optimization was started from 20 sets of random flux distributions. Confidence intervals were calculated by a grid search method as described previously [30,31,32]. The metabolic flux of reaction r is fixed to vopt,r + d and the objective function is re-optimized. Here, vopt,r is the optimized metabolic flux of reaction r and d is the perturbation level. The procedure is iterated with increased or decreased d. The range of fixed metabolic flux whose RSS is less than the threshold level is the confidence interval. The threshold level is determined by:
is the estimated MID of the corresponding metabolite, and N is the number of metabolites used for flux estimation. Optimization was started from 20 sets of random flux distributions. Confidence intervals were calculated by a grid search method as described previously [30,31,32]. The metabolic flux of reaction r is fixed to vopt,r + d and the objective function is re-optimized. Here, vopt,r is the optimized metabolic flux of reaction r and d is the perturbation level. The procedure is iterated with increased or decreased d. The range of fixed metabolic flux whose RSS is less than the threshold level is the confidence interval. The threshold level is determined by:
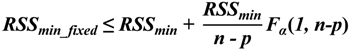
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shirai, T.; Fujimura, K.; Furusawa, C.; Nagahisa, K.; Shioya, S.; Shimizu, H. Study on roles of anaplerotic pathways in glutamate overproduction of Corynebacterium glutamicum by metabolic flux analysis. Microb. Cell Fact. 2007, 6, 19. [Google Scholar] [CrossRef]
- Nielsen, J.; Jewett, M.C. Impact of systems biology on metabolic engineering of Saccharomyces cerevisiae. FEMS Yeast Res. 2008, 8, 122–131. [Google Scholar] [CrossRef]
- Becker, J.; Zelder, O.; Häfner, S.; Schröder, H.; Wittmann, C. From zero to hero—Design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab. Eng. 2011, 13, 159–168. [Google Scholar] [CrossRef]
- Yim, H.; Haselbeck, R.; Niu, W.; Pujol-Baxley, C.; Burgard, A.; Boldt, J.; Khandurina, J.; Trawick, J.D.; Osterhout, R.E.; Stephan, R.; et al. Metabolic engineering of Escherichia coli for direct production of 1, 4-butanediol. Nat. Chem. Biol. 2011, 7, 445–452. [Google Scholar] [CrossRef]
- Toya, Y.; Shimizu, H. Flux analysis and metabolomics for systematic metabolic engineering of microorganisms. Biotechnol. Adv. 2013, 31, 818–826. [Google Scholar] [CrossRef]
- Stephanopoulos, G. Metabolic fluxes and metabolic engineering. Metab. Eng. 1999, 1, 1–11. [Google Scholar] [CrossRef]
- Wiechert, W. 13C metabolic flux analysis. Metab. Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef]
- Wittmann, C. Fluxome analysis using GC-MS. Microb. Cell Fact. 2007, 6, 6. [Google Scholar] [CrossRef]
- Zamboni, N.; Fendt, S.-M.; Rühl, M.; Sauer, U. 13C-based metabolic flux analysis. Nat. Protoc. 2009, 4, 878–892. [Google Scholar] [CrossRef]
- Christensen, B.; Nielsen, J. Isotopomer analysis using GC-MS. Metab. Eng. 1999, 1, 282–290. [Google Scholar] [CrossRef]
- Szyperski, T. Biosynthetically directed fractional 13C-labeling of proteinogenic amino acids. An efficient analytical tool to investigate intermediary metabolism. Eur. J. Biochem. 1995, 232, 433–448. [Google Scholar] [CrossRef]
- Krömer, J.O.; Sorgenfrei, O.; Klopprogge, K.; Heinzle, E.; Wittmann, C. In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J. Bacteriol. 2004, 186, 1769–1784. [Google Scholar] [CrossRef]
- Rühl, M.; Coq, D.L.; Aymerich, S.; Sauer, U. 13C-flux analysis reveals NADPH-balancing transhydrogenation cycles in stationary phase of nitrogen-starving Bacillus subtilis. J. Biol. Chem. 2012, 287, 27959–27970. [Google Scholar] [CrossRef]
- Iwatani, S.; Dien, S.V.; Shimbo, K.; Kubota, K.; Kageyama, N.; Iwahata, D.; Miyano, H.; Hirayama, K.; Usuda, Y.; Shimizu, K.; et al. Determination of metabolic flux changes during fed-batch cultivation from measurements of intracellular amino acids by LC-MS/MS. J. Biotechnol. 2007, 128, 93–111. [Google Scholar] [CrossRef]
- Rühl, M.; Zamboni, N.; Sauer, U. Dynamic flux responses in riboflavin overproducing Bacillus subtilis to increasing glucose limitation in fed-batch culture. Biotechnol. Bioeng. 2010, 105, 795–804. [Google Scholar]
- Wittmann, C.; Hans, M.; Heinzle, E. In vivo analysis of intracellular amino acid labelings by GC/MS. Anal. Biochem. 2002, 307, 379–382. [Google Scholar] [CrossRef]
- Mori, E.; Furusawa, C.; Kajihata, S.; Shirai, T.; Shimizu, H. Evaluating 13C enrichment data of free amino acids for precise metabolic flux analysis. Biotechnol. J. 2011, 6, 1377–1387. [Google Scholar] [CrossRef]
- Nöh, K.; Wiechert, W. The benefits of being transient: Isotope-based metabolic flux analysis at the short time scale. Appl. Microbiol. Biotechnol. 2011, 91, 1247–1265. [Google Scholar] [CrossRef]
- Toya, Y.; Ishii, N.; Nakahigashi, K.; Hirasawa, T.; Soga, T.; Tomita, M.; Shimizu, K. 13C-metabolic flux analysis for batch culture of Escherichia coli and its pyk and pgi gene knockout mutants based on mass isotopomer distribution of intracellular metabolites. Biotechnol. Prog. 2010, 26, 975–992. [Google Scholar]
- Ishii, N.; Nakahigashi, K.; Baba, T.; Robert, M.; Soga, T.; Kanai, A.; Hirasawa, T.; Naba, M.; Hirai, K.; Hoque, A.; et al. Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 2007, 316, 593–597. [Google Scholar] [CrossRef]
- Fischer, E.; Sauer, U. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 2003, 270, 880–891. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kraynie, D.F.; Laffend, L.A.; Gonzales-Lergier, J.; Kelleher, J.K.; Stephanopoulos, G. Metabolic flux analysis in a nonstationary system: Fed-batch fermentation of a high yielding strain of E. coli producing 1,3-propanediol. Metab. Eng. 2007, 9, 277–292. [Google Scholar] [CrossRef]
- Wittmann, C.; Krömer, J.O.; Kiefer, P.; Binz, T.; Heinzle, E. Impact of the cold shock phenomenon on quantification of intracellular metabolites in bacteria. Anal. Biochem. 2004, 327, 135–139. [Google Scholar] [CrossRef]
- Bolten, C.J.; Kiefer, P.; Letisse, F.; Portais, J.-C.; Wittmann, C. Sampling for metabolome analysis of microorganisms. Anal. Chem. 2007, 79, 3843–3849. [Google Scholar] [CrossRef]
- Ingraham, J.L.; Maaløe, O.; Neidhardt, F.C. Growth of the Bacterial Cell; Sinauer Associates Inc.: Sunderland, MA, USA, 1983; p. 128. [Google Scholar]
- Van Winden, W.A.; Wittmann, C.; Heinzle, E.; Heijnen, J.J. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 2002, 80, 477–479. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Accurate assessment of amino acid mass isotopomer distributions for metabolic flux analysis. Anal. Chem. 2007, 79, 7554–7559. [Google Scholar] [CrossRef]
- Kajihata, S.; Furusawa, C.; Matsuda, F.; Shimizu, H. OpenMebius: An open source software for isotopically nonstationary 13C-based metabolic flux analysis. BioMed Res. Int. 2014, in press. [Google Scholar]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Elementary metabolite units (EMU): A novel framework for modeling isotopic distributions. Metab. Eng. 2007, 9, 68–86. [Google Scholar] [CrossRef]
- Kleijn, R.J.; van Winden, W.A.; Ras, C.; van Gulic, W.M.; Schipper, D.; Heijnen, J.J. 13C-labeled gluconate tracing as a direct and accurate method for determining the pentose phosphate pathway split ratio in Penicillium chrysogenum. Appl. Environ. Microbiol. 2006, 72, 4743–4754. [Google Scholar] [CrossRef]
- Costenoble, R.; Müller, D.; Barl, T.; van Gulic, W.M.; van Winden, W.A.; Reuss, M.; Heijnen, J.J. 13C-Labeled metabolic flux analysis of a fed-batch culture of elutriated Saccharomyces cerevisiae. FEMS Yeast Res. 2007, 7, 511–526. [Google Scholar] [CrossRef]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab. Eng. 2006, 8, 324–337. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Okahashi, N.; Kajihata, S.; Furusawa, C.; Shimizu, H. Reliable Metabolic Flux Estimation in Escherichia coli Central Carbon Metabolism Using Intracellular Free Amino Acids. Metabolites 2014, 4, 408-420. https://doi.org/10.3390/metabo4020408
Okahashi N, Kajihata S, Furusawa C, Shimizu H. Reliable Metabolic Flux Estimation in Escherichia coli Central Carbon Metabolism Using Intracellular Free Amino Acids. Metabolites. 2014; 4(2):408-420. https://doi.org/10.3390/metabo4020408
Chicago/Turabian StyleOkahashi, Nobuyuki, Shuichi Kajihata, Chikara Furusawa, and Hiroshi Shimizu. 2014. "Reliable Metabolic Flux Estimation in Escherichia coli Central Carbon Metabolism Using Intracellular Free Amino Acids" Metabolites 4, no. 2: 408-420. https://doi.org/10.3390/metabo4020408
APA StyleOkahashi, N., Kajihata, S., Furusawa, C., & Shimizu, H. (2014). Reliable Metabolic Flux Estimation in Escherichia coli Central Carbon Metabolism Using Intracellular Free Amino Acids. Metabolites, 4(2), 408-420. https://doi.org/10.3390/metabo4020408