Cellulose Digestion and Metabolism Induced Biocatalytic Transitions in Anaerobic Microbial Ecosystems


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
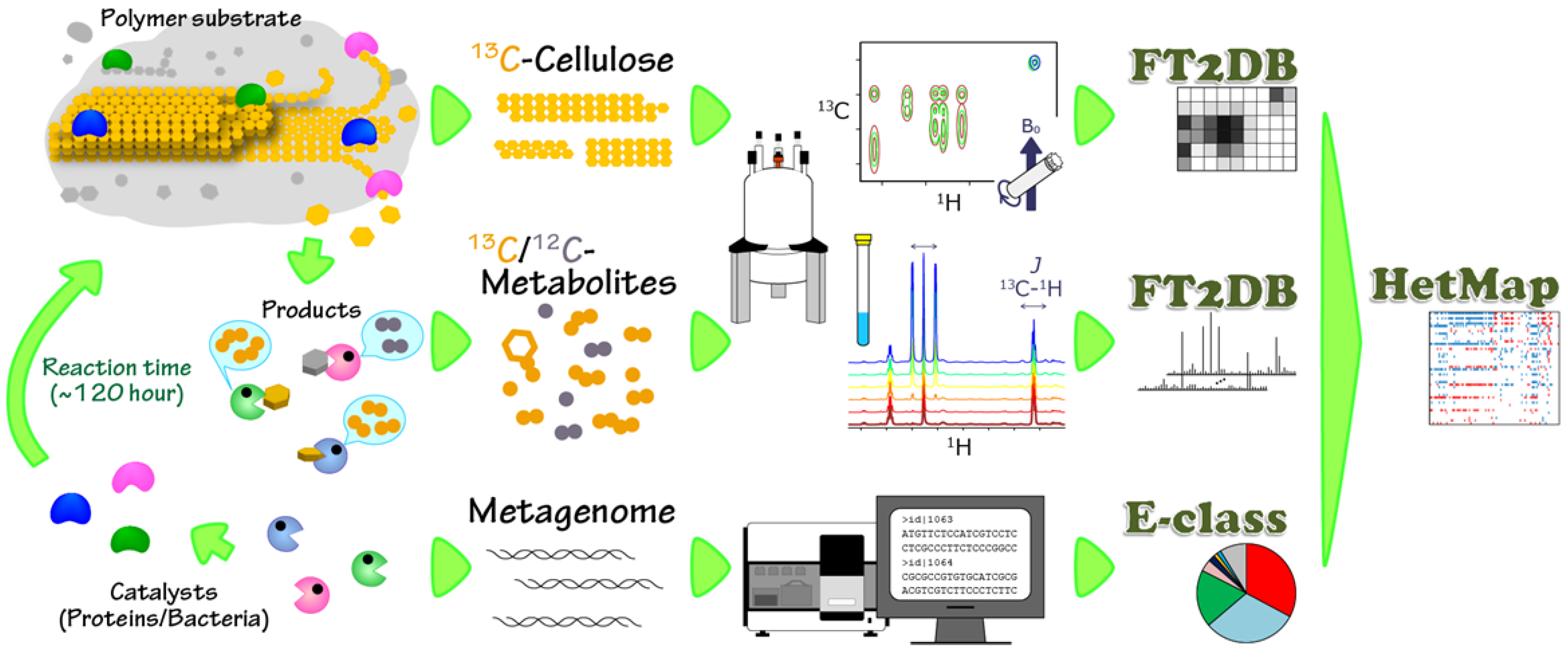
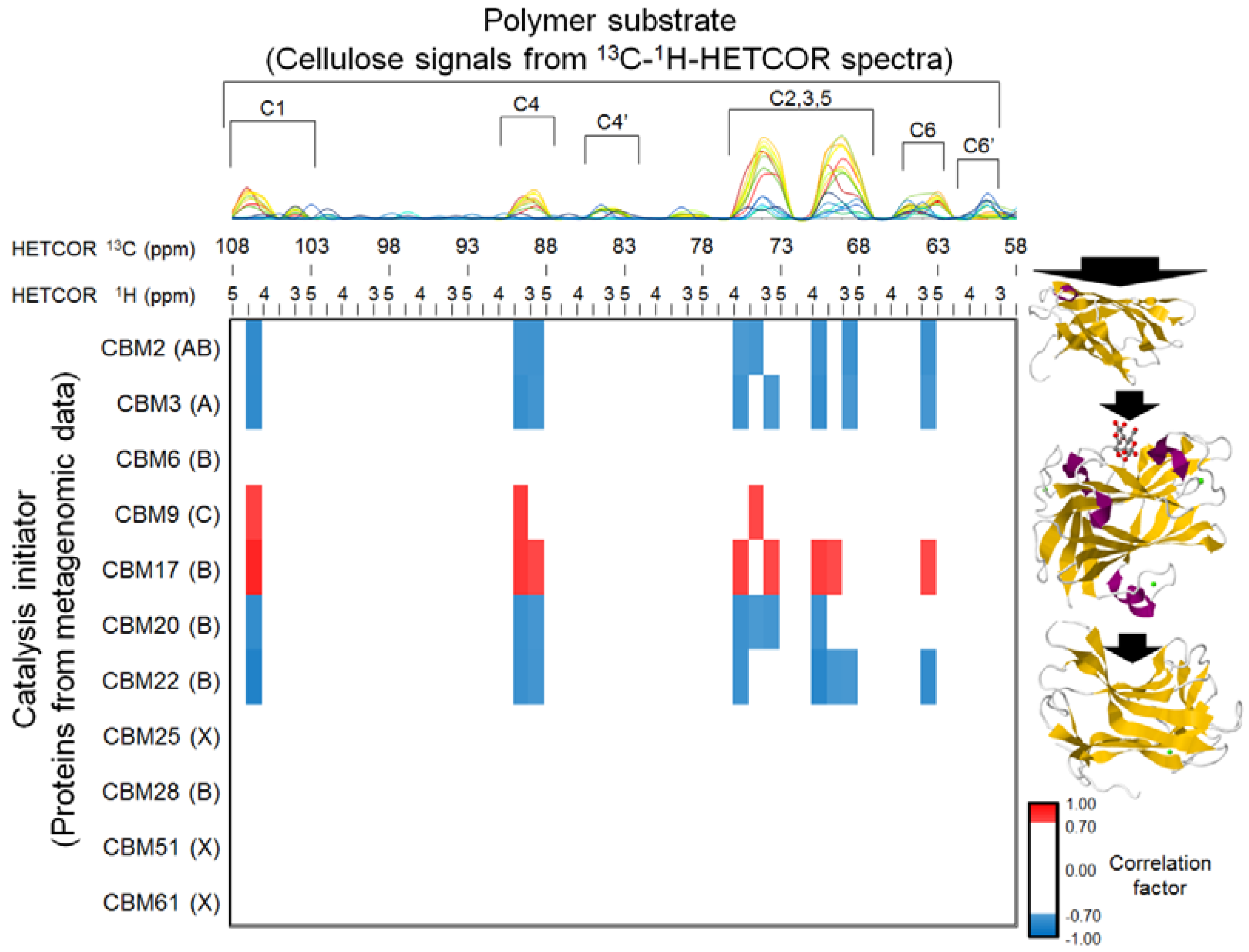
2.1. Characterization of Microbial BC Degradation by Using Solid-State 2D HETCOR NMR
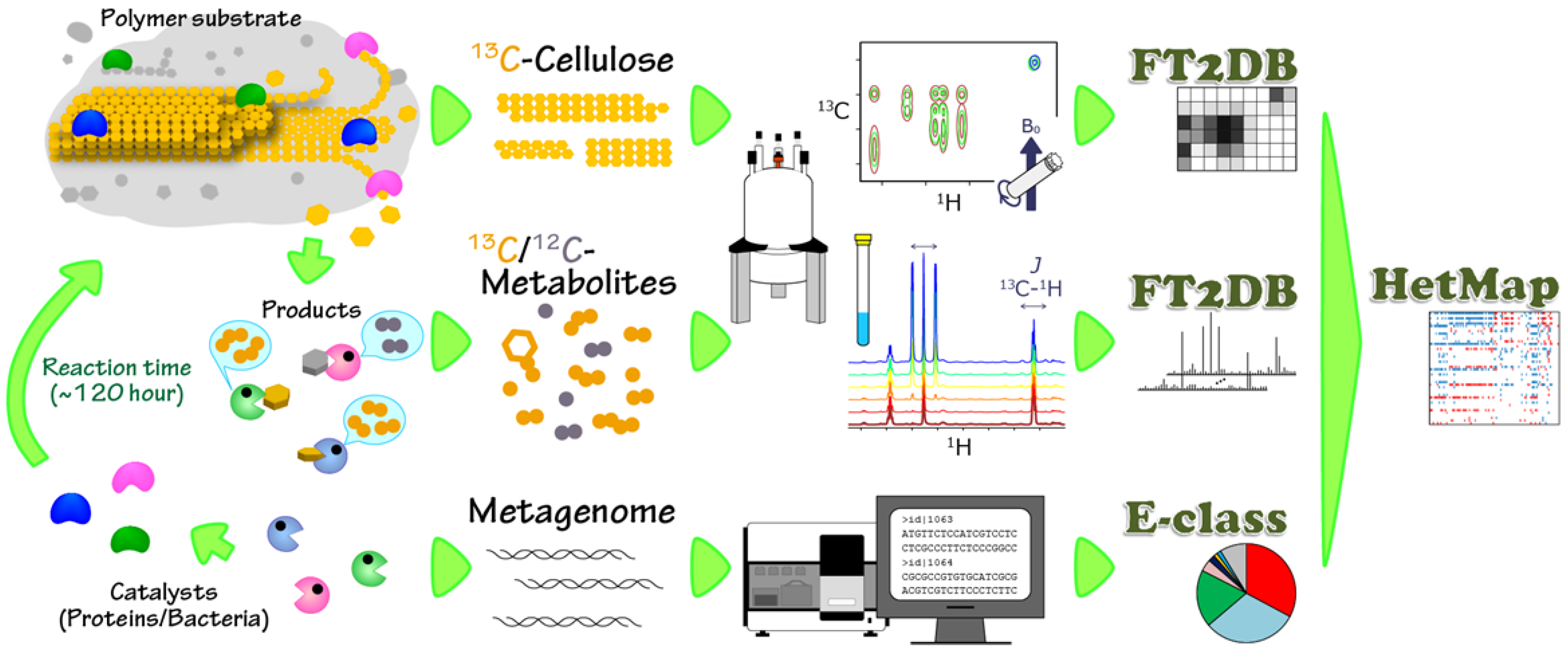
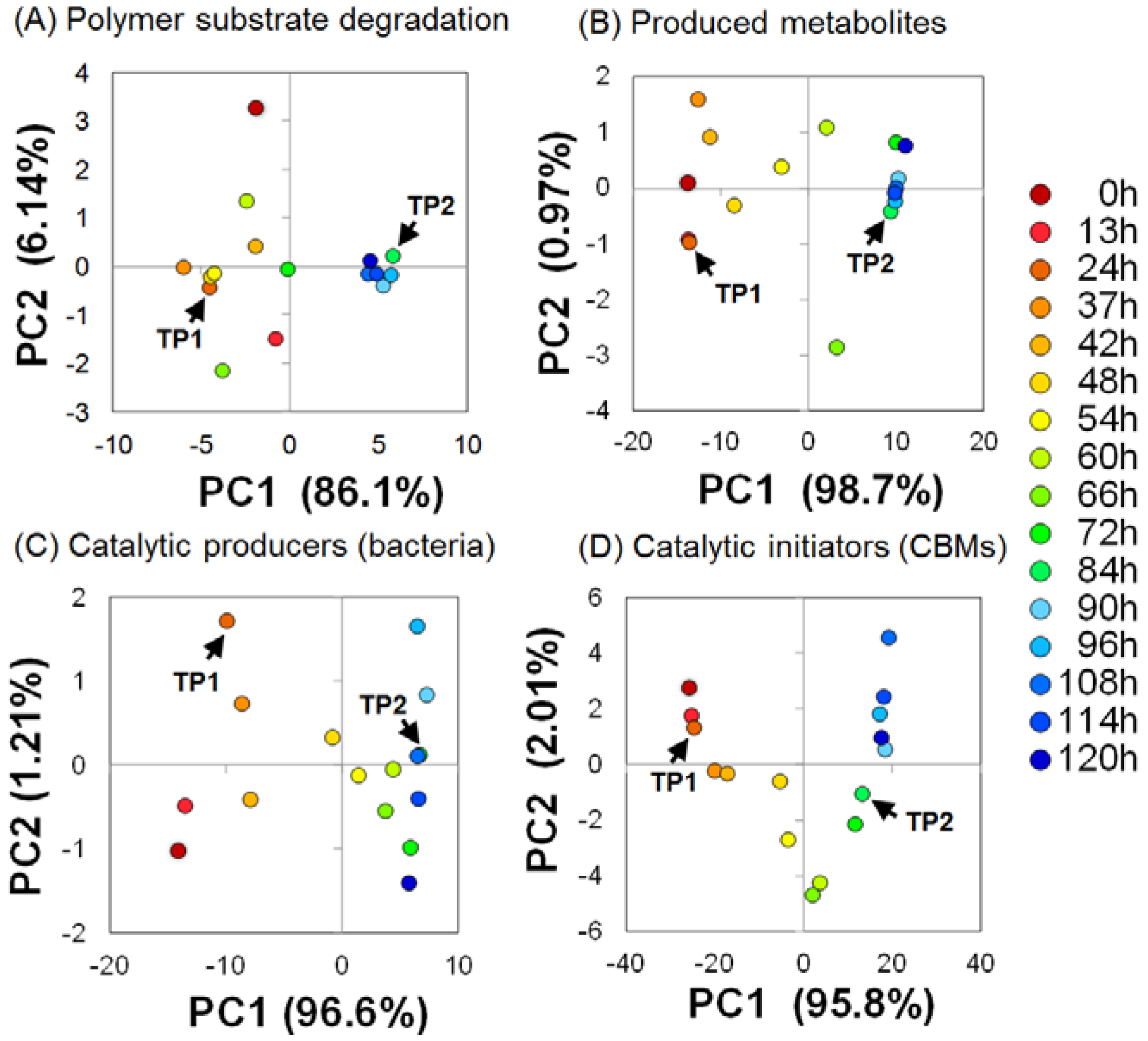
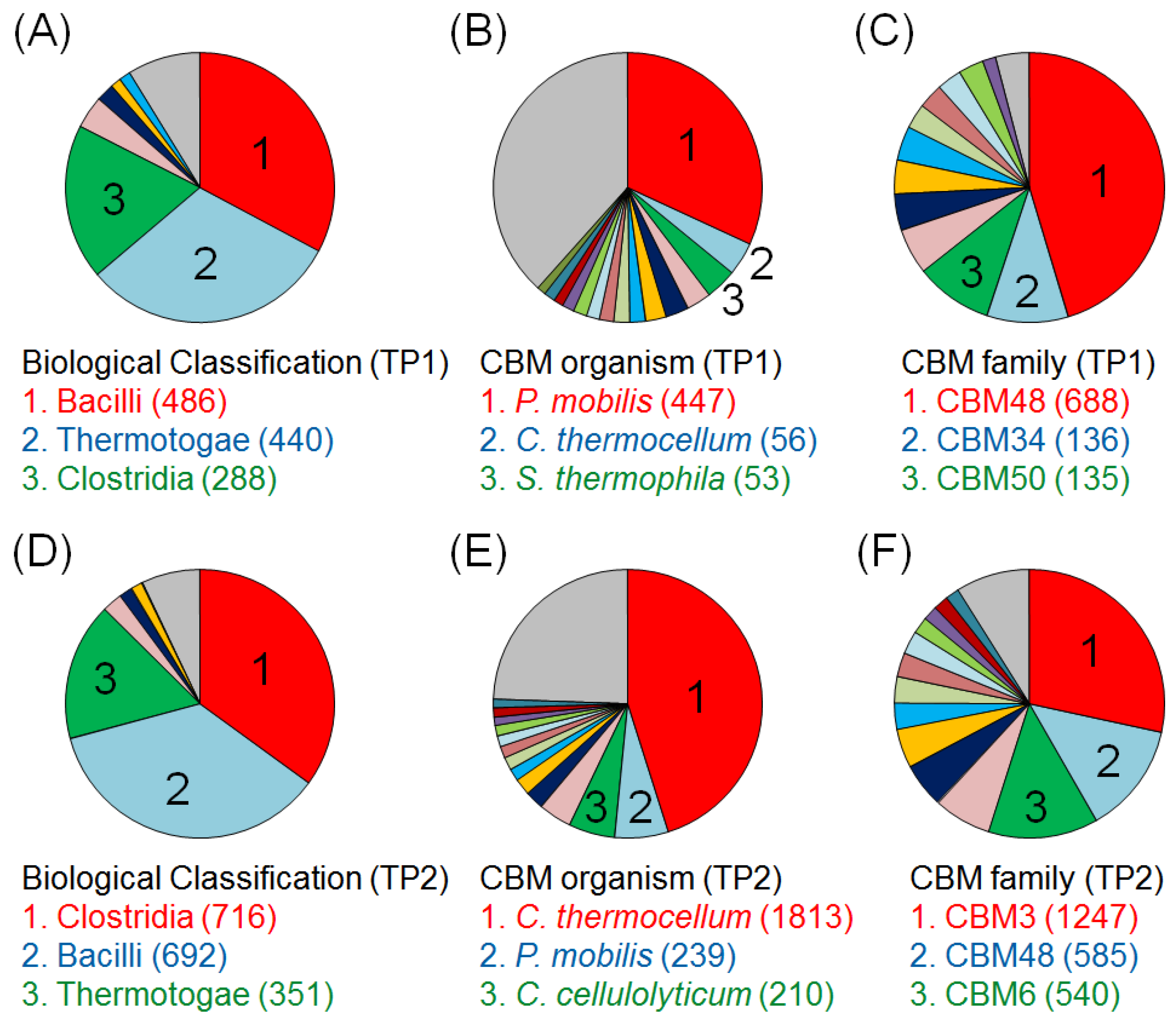
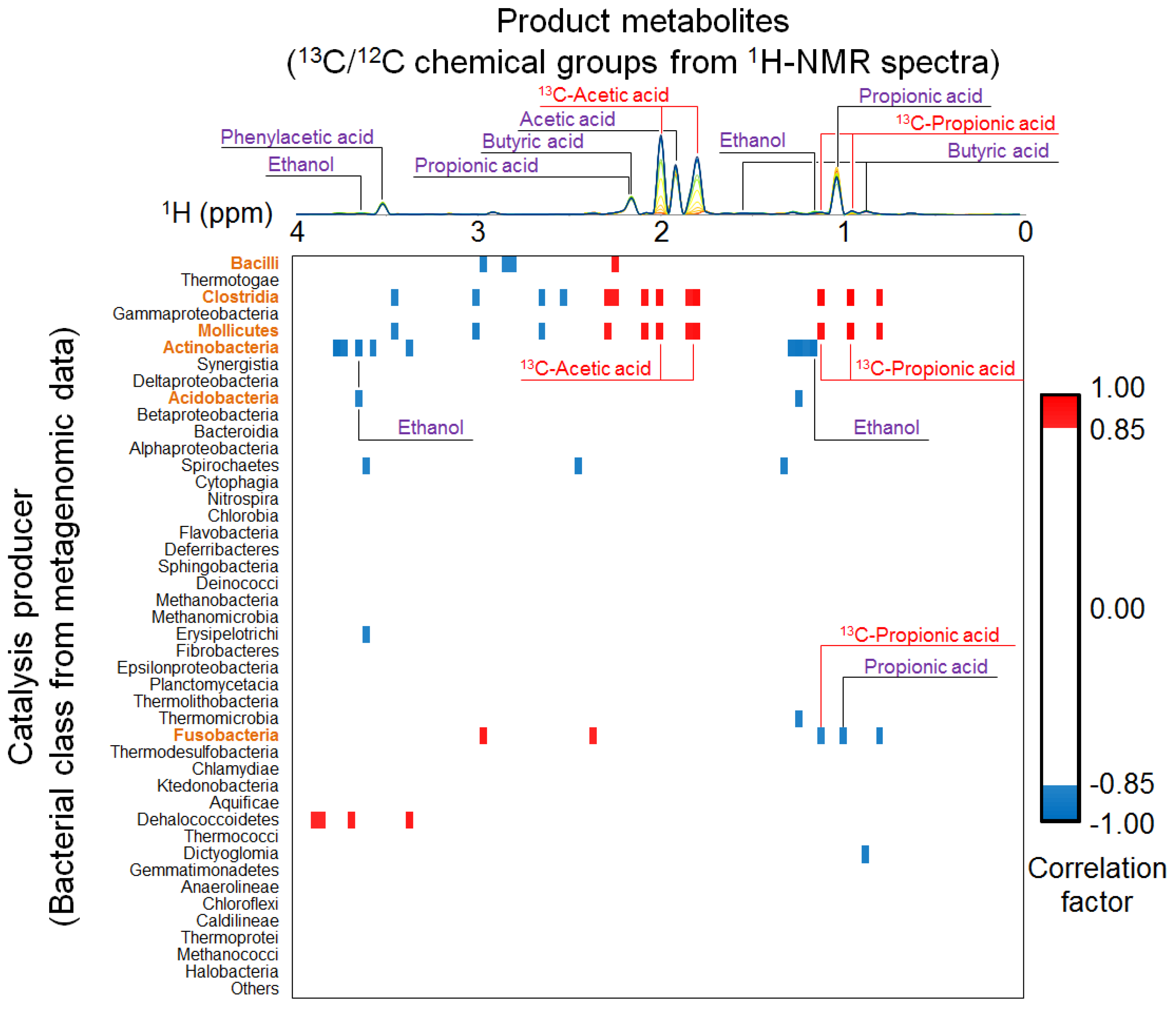
2.2. Time-Course Variations in Metabolites, Microbial Community Composition and CBMs
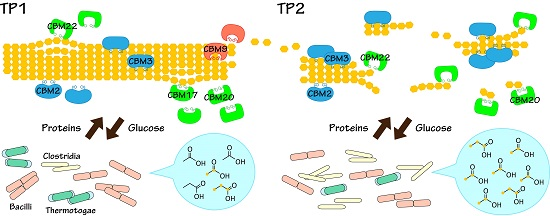
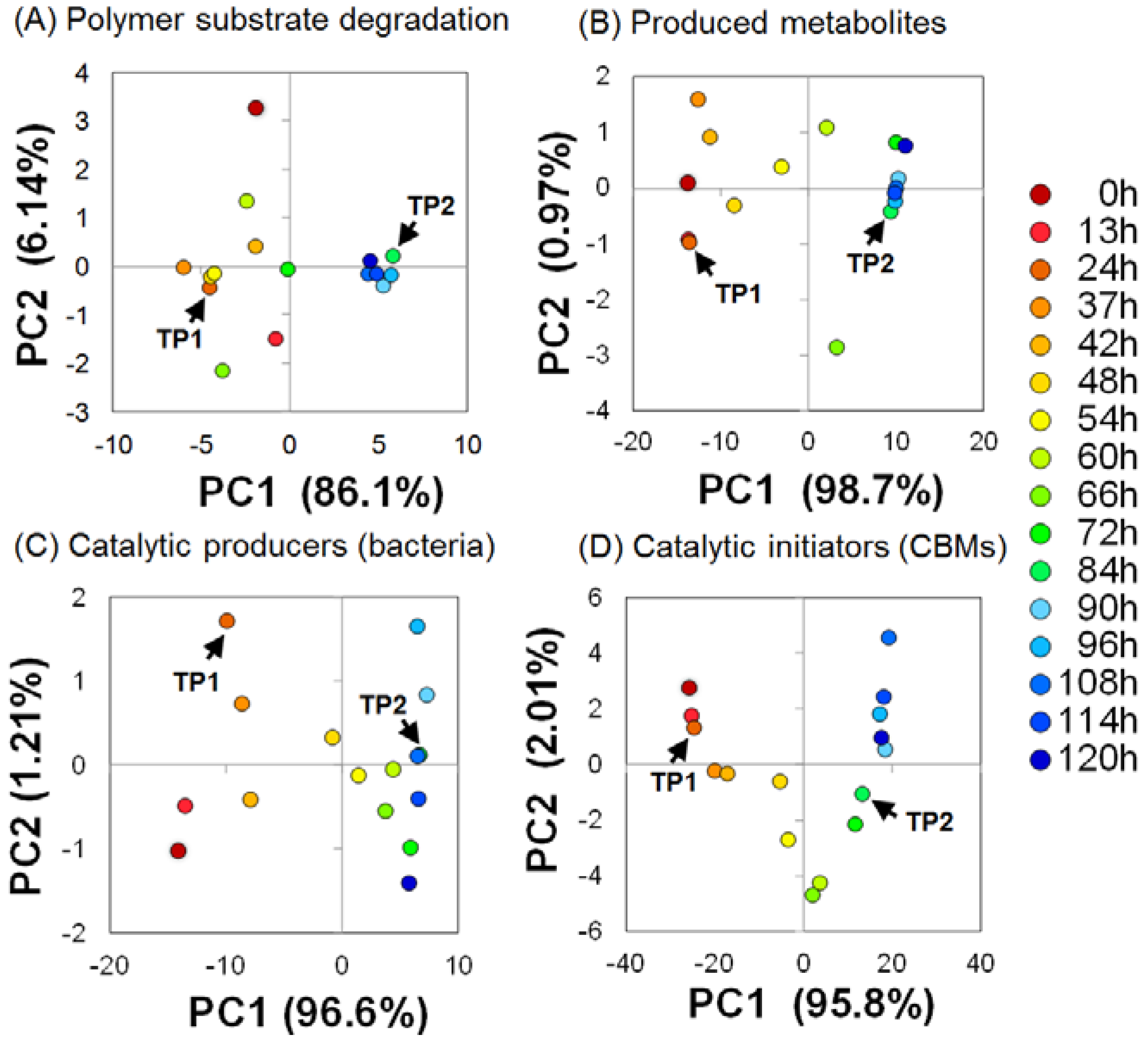
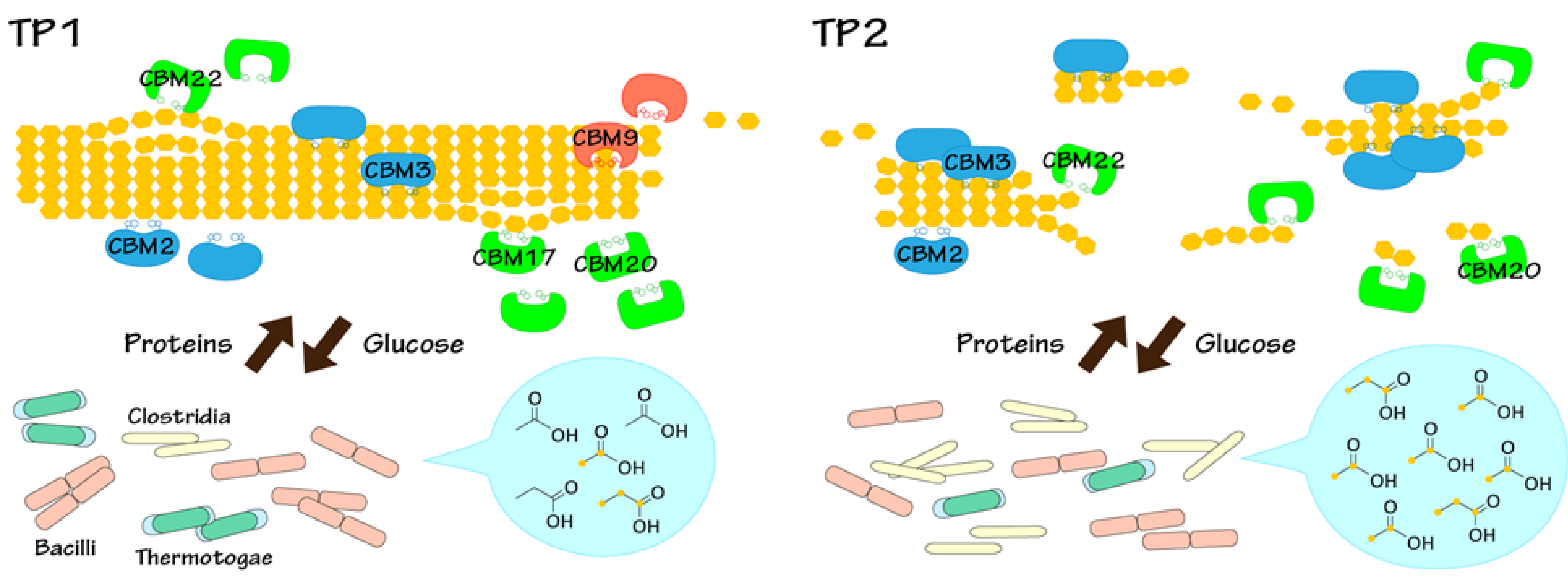
2.3. Key Components of Biocatalytic Transitions in BC Digestion Process

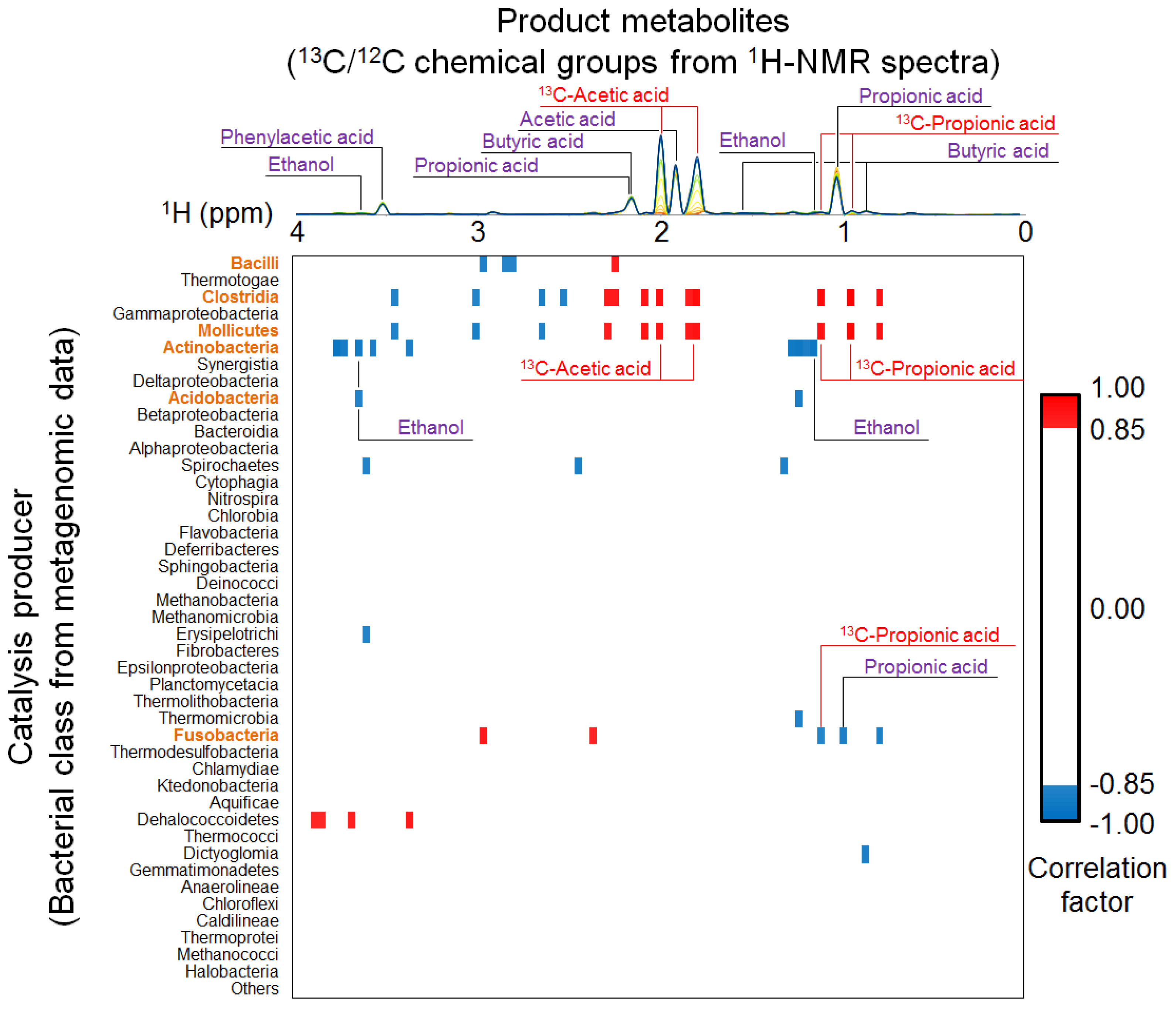
2.4. Relationships between Metabolic Dynamics and Biocatalytic Transitions of Microbial Community
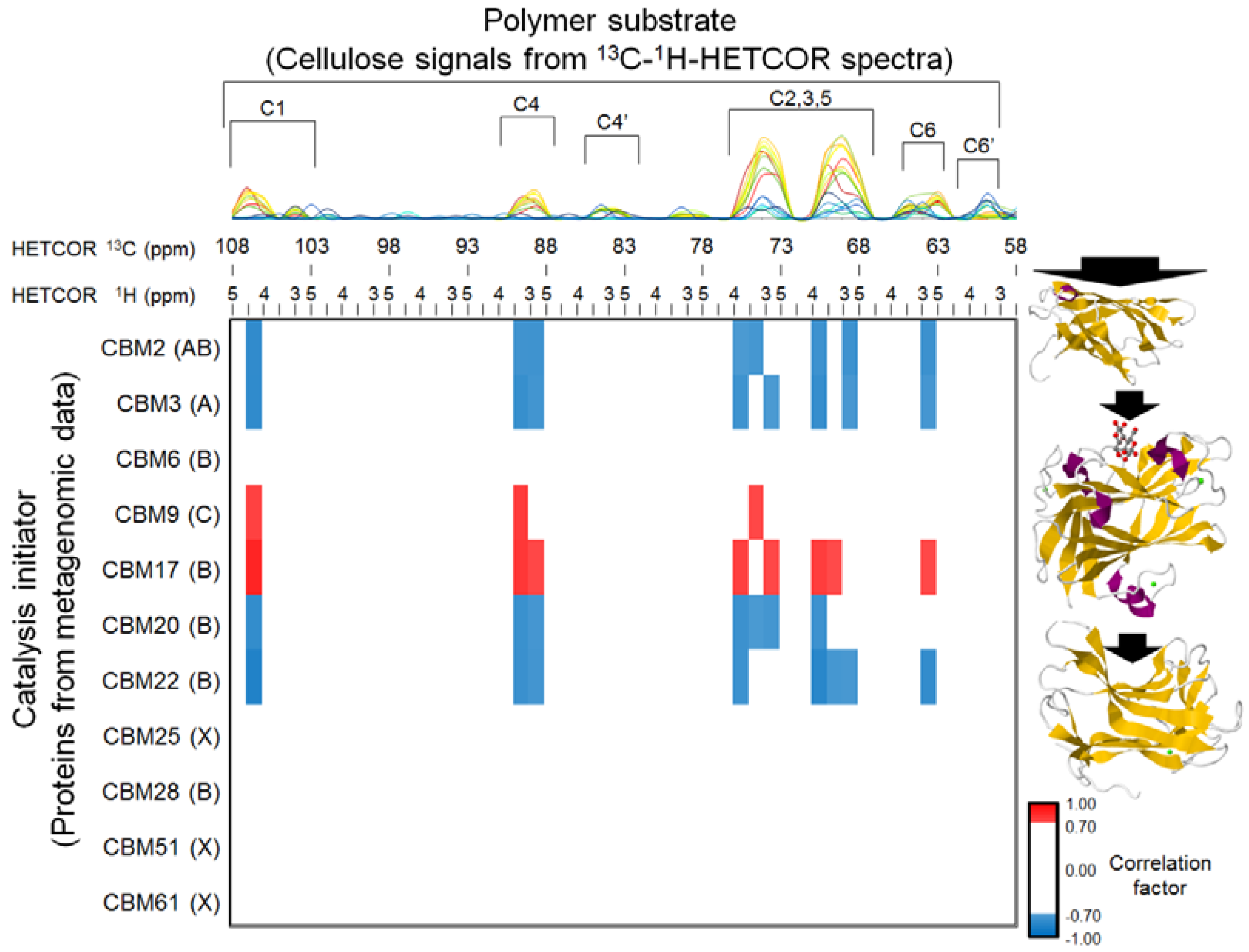
2.5. Relationships between BC Digestion and Biocatalytic Transitions of Proteins

3. Experimental
3.1. General
3.2. NMR Spectroscopy
3.3. DNA Extraction and Metagenomic Dataset Preparation
3.4. Classification of the Microbial Community and CBMs Using the E-Class Web Tool
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Conrad, R. The global methane cycle: Recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 2009, 1, 285–292. [Google Scholar]
- Chynoweth, D.P.; Owens, J.M.; Legrand, R. Renewable methane from anaerobic digestion of biomass. Renew. Energ. 2001, 22, 1–8. [Google Scholar] [CrossRef]
- Schink, B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 1997, 61, 262–280. [Google Scholar]
- Date, Y.; Iikura, T.; Yamazawa, A.; Moriya, S.; Kikuchi, J. Metabolic sequences of anaerobic fermentation on glucose-based feeding substrates based on correlation analyses of microbial and metabolite profiling. J. Proteome Res. 2012, 11, 5602–5610. [Google Scholar]
- Yamazawa, A.; Iikura, T.; Shino, A.; Date, Y.; Kikuchi, J. Solid-, solution-, and gas-state NMR monitoring of 13C-cellulose degradation in an anaerobic microbial ecosystem. Molecules 2013, 18, 9021–9033. [Google Scholar] [CrossRef]
- Kikuchi, J.; Asakura, T. Use of 13C conformation-dependent chemical shifts to elucidate the local structure of a large protein with homologous domains in solution and solid state. J. Biochem. Biophys. Methods 1999, 38, 203–208. [Google Scholar] [CrossRef]
- Mao, J.D.; Holtman, K.M.; Franqui-Villanueva, D. Chemical structures of corn stover and its residue after dilute acid prehydrolysis and enzymatic hydrolysis: Insight into factors limiting enzymatic hydrolysis. J. Agric. Food Chem. 2010, 58, 11680–11687. [Google Scholar] [CrossRef]
- Mao, J.D.; Schmidt-Rohr, K. Accurate quantification of aromaticity and nonprotonated aromatic carbon fraction in natural organic matter by 13C solid-state nuclear magnetic resonance. Environ. Sci. Technol. 2004, 38, 2680–2684. [Google Scholar] [CrossRef]
- Mori, T.; Chikayama, E.; Tsuboi, Y.; Ishida, N.; Shisa, N.; Noritake, Y.; Moriya, S.; Kikuchi, J. Exploring the conformational space of amorphous cellulose using NMR chemical shifts. Carbohydr. Polym. 2012, 90, 1197–1203. [Google Scholar] [CrossRef]
- Ogura, T.; Date, Y.; Kikuchi, J. Differences in cellulosic supramolecular structure of compositionally similar rice straw affect biomass metabolism by paddy soil microbiota. PLoS One 2013, 8, e66919. [Google Scholar] [CrossRef]
- Okushita, K.; Chikayama, E.; Kikuchi, J. Solubilization mechanism and characterization of the structural change of bacterial cellulose in regenerated states through ionic liquid treatment. Biomacromolecules 2012, 13, 1323–1330. [Google Scholar] [CrossRef]
- Okushita, K.; Komatsu, T.; Chikayama, E.; Kikuchi, J. Statistical approach for solid-state NMR spectra of cellulose derived from a series of variable parameters. Polym. J. 2012, 44, 895–900. [Google Scholar] [CrossRef]
- Komatsu, T.; Kikuchi, J. Selective signal detection in solid-state NMR using rotor-synchronized dipolar dephasing for the analysis of hemicellulose in lignocellulosic biomass. J. Phys. Chem. Lett. 2013, 4, 2279–2283. [Google Scholar] [CrossRef]
- Earl, W.L.; Vanderhart, D.L. Observations by high-resolution C-13 nuclear magnetic-resonance of cellulose-I related to morphology and crystal-structure. Macromolecules 1981, 14, 570–574. [Google Scholar] [CrossRef]
- Vanderhart, D.L.; Atalla, R.H. Studies of microstructure in native celluloses using solid-state carbon-13 NMR. Macromolecules 1984, 17, 1465–1472. [Google Scholar] [CrossRef]
- Everroad, R.C.; Yoshida, S.; Tsuboi, Y.; Date, Y.; Kikuchi, J.; Moriya, S. Concentration of metabolites from low-density planktonic communities for environmental metabolomics using nuclear magnetic resonance spectroscopy. J. Vis. Exp. 2012, 62, e3163. [Google Scholar]
- Fukuda, S.; Nakanishi, Y.; Chikayama, E.; Ohno, H.; Hino, T.; Kikuchi, J. Evaluation and characterization of bacterial metabolic dynamics with a novel profiling technique, real-time metabolotyping. PLoS One 2009, 4, e4893. [Google Scholar]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect host from enteropathgenic infection through production acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Fukuda, S.; Chikayama, E.; Kimura, Y.; Ohno, H.; Kikuchi, J. Dynamic omics approach identifies nutrition-mediated microbial interactions. J. Proteome Res. 2010, 10, 824–836. [Google Scholar]
- Date, Y.; Nakanishi, Y.; Fukuda, S.; Kato, T.; Tsuneda, S.; Ohno, H.; Kikuchi, J. New monitoring approach for metabolic dynamics in microbial ecosystems using stable-isotope-labeling technologies. J. Biosci. Bioeng. 2010, 110, 87–93. [Google Scholar] [CrossRef]
- McHardy, A.C.; Rigoutsos, I. What’s in the mix: Phylogenetic classification of metagenome sequence samples. Curr. Opin. Microbiol. 2007, 10, 499–503. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Demain, A.L.; Newcomb, M.; Wu, J.H. Cellulase, clostridia, and ethanol. Microbiol. Mol. Biol. Rev. 2005, 69, 124–154. [Google Scholar] [CrossRef]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280, 309–316. [Google Scholar]
- Boraston, A.B.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Carbohydrate-binding modules: Fine-tuning polysaccharide recognition. Biochem. J. 2004, 382, 769–781. [Google Scholar] [CrossRef]
- E-class, ECOMICS: Web tools for environmental and metabolic systems. Available online: https://database.riken.jp/ecomics/eclass/ (accessed on 20 December 2013).
- ECOMICS, ECOMICS: Web tools for environmental and metabolic systems. Available online: https://database.riken.jp/ecomics/ (accessed on 20 December 2013).
- Ogata, Y.; Chikayama, E.; Morioka, Y.; Everroad, R.C.; Shino, A.; Matsushima, A.; Haruna, H.; Moriya, S.; Toyoda, T.; Kikuchi, J. ECOMICS: A web-based toolkit for investigating the biomolecular web in ecosystems using a trans-omics approach. PLoS One 2012, 7, e30263. [Google Scholar] [CrossRef]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef]
- FT2DB, ECOMICS: Web tools for environmental and metabolic systems. Available online: https://database.riken.jp/ecomics/chika/index2.html (accessed on 20 December 2013).
- Sekiyama, Y.; Chikayama, E.; Kikuchi, J. Profiling polar and semipolar plant metabolites throughout extraction processes using a combined solution-state and high-resolution magic angle spinning NMR approach. Anal. Chem. 2010, 82, 1643–1652. [Google Scholar] [CrossRef]
- Solution structure of a cellulose-binding domain from Cellulomonas fimi by nuclear magnetic resonance spectroscopy. Available online: http://www.rcsb.org/pdb/explore.do?structureId=1EXG (accessed on 20 December 2013).
- The role of conserved amino acids in the cleft of the C-terminal family 22 carbohydrate binding module of Clostridium Theermocellum XYN10B in ligand binding. Available online: http://www.rcsb.org/pdb/explore.do?structureId=1H6X (accessed on 20 December 2013).
- Crystal structures of the family 9 carbohydrate-binding module from Thermotoga maritima xylanase 10A in native and ligand-bound forms. Available online: http://www.rcsb.org/pdb/explore.do?structureId=1I82 (accessed on 20 December 2013).
- Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 20 December 2013).
- Gilbert, H.J. The biochemistry and structural biology of plant cell wall deconstruction. Plant Physiol. 2010, 153, 444–455. [Google Scholar] [CrossRef]
- Jervis, E.J.; Haynes, C.A.; Kilburn, D.G. Surface diffusion of cellulases and their isolated binding domains on cellulose. J. Biol. Chem. 1997, 272, 24016–24023. [Google Scholar] [CrossRef]
- Kellett, L.E.; Poole, D.M.; Ferreira, L.M.; Durrant, A.J.; Hazlewood, G.P.; Gilbert, H.J. Xylanase B and an arabinofuranosidase from Pseudomonas fluorescens subsp. cellulosa contain identical cellulose-binding domains and are encoded by adjacent genes. Biochem. J. 1990, 272, 369–376. [Google Scholar]
- Blake, A.W.; McCartney, L.; Flint, J.E.; Bolam, D.N.; Boraston, A.B.; Gilbert, H.J.; Knox, J.P. Understanding the biological rationale for the diversity of cellulose-directed carbohydrate-binding modules in prokaryotic enzymes. J. Biol. Chem. 2006, 281, 29321–29329. [Google Scholar]
- Shoseyov, O.; Shani, Z.; Levy, I. Carbohydrate binding modules: Biochemical properties and novel applications. Microbiol. Mol. Biol. Rev. 2006, 70, 283–295. [Google Scholar] [CrossRef]
- Kono, H.; Erata, T.; Takai, M. Complete assignment of the CP/MAS C-13 NMR spectrum of cellulose IIII. Macromolecules 2003, 36, 3589–3592. [Google Scholar] [CrossRef]
- Kono, H.; Erata, T.; Takai, M. Determination of the through-bond carbon-carbon and carbon-proton connectivities of the native celluloses in the solid state. Macromolecules 2003, 36, 5131–5138. [Google Scholar] [CrossRef]
- Larsson, P.T.; Hult, E.L.; Wickholm, K.; Pettersson, E.; Iversen, T. CP/MAS C-13-NMR spectroscopy applied to structure and interaction studies on cellulose I. Solid State Nuclear Magn. Reson. 1999, 15, 31–40. [Google Scholar] [CrossRef]
- Larsson, P.T.; Westermark, U.; Iversen, T. Determination of the cellulose I alpha allomorph content in a tunicate cellulose by CP/MAS C-13-NMR spectroscopy. Carbohydr. Res. 1995, 278, 339–343. [Google Scholar] [CrossRef]
- Larsson, P.T.; Wickholm, K.; Iversen, T. A CP/MAS C-13 NMR investigation of molecular ordering in celluloses. Carbohydr. Res. 1997, 302, 19–25. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. Nmrpipe—A multidimensional spectral processing system based on unix pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar]
- Harris, D.M.; Corbin, K.; Wang, T.; Gutierrez, R.; Bertolo, A.L.; Petti, C.; Smilgies, D.M.; Estevez, J.M.; Bonetta, D.; Urbanowicz, B.R.; et al. Cellulose microfibril crystallinity is reduced by mutating C-terminal transmembrane region residues CESA1(A903V) and CESA3(T942I) of cellulose synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 4098–4103. [Google Scholar]
- Date, Y.; Sakata, K.; Kikuchi, J. Chemical profiling of complex biochemical mixtures from various seaweeds. Polymer J. 2012, 44, 888–894. [Google Scholar] [CrossRef]
- HetMap, ECOMICS: Web tools for environmental and metabolic systems. Available online: https://database.riken.jp/ecomics/chika/ (accessed on 20 December 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yamazawa, A.; Iikura, T.; Morioka, Y.; Shino, A.; Ogata, Y.; Date, Y.; Kikuchi, J. Cellulose Digestion and Metabolism Induced Biocatalytic Transitions in Anaerobic Microbial Ecosystems. Metabolites 2014, 4, 36-52. https://doi.org/10.3390/metabo4010036
Yamazawa A, Iikura T, Morioka Y, Shino A, Ogata Y, Date Y, Kikuchi J. Cellulose Digestion and Metabolism Induced Biocatalytic Transitions in Anaerobic Microbial Ecosystems. Metabolites. 2014; 4(1):36-52. https://doi.org/10.3390/metabo4010036
Chicago/Turabian StyleYamazawa, Akira, Tomohiro Iikura, Yusuke Morioka, Amiu Shino, Yoshiyuki Ogata, Yasuhiro Date, and Jun Kikuchi. 2014. "Cellulose Digestion and Metabolism Induced Biocatalytic Transitions in Anaerobic Microbial Ecosystems" Metabolites 4, no. 1: 36-52. https://doi.org/10.3390/metabo4010036