Miniaturized Bioaffinity Assessment Coupled to Mass Spectrometry for Guided Purification of Bioactives from Toad and Cone Snail

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Chemical and Biological Reagents
2.2. Instrumentation
2.2.1. Microfluidic Confocal Fluorescence Detection System
2.2.2. Nano-LC and Solvent Delivery System
2.2.3. Microfluidic Chip
2.2.4. Microfluidic LED Based LIF Detector
2.2.5. Nano-LC-MS
2.2.6. MS-Guided Fractionation
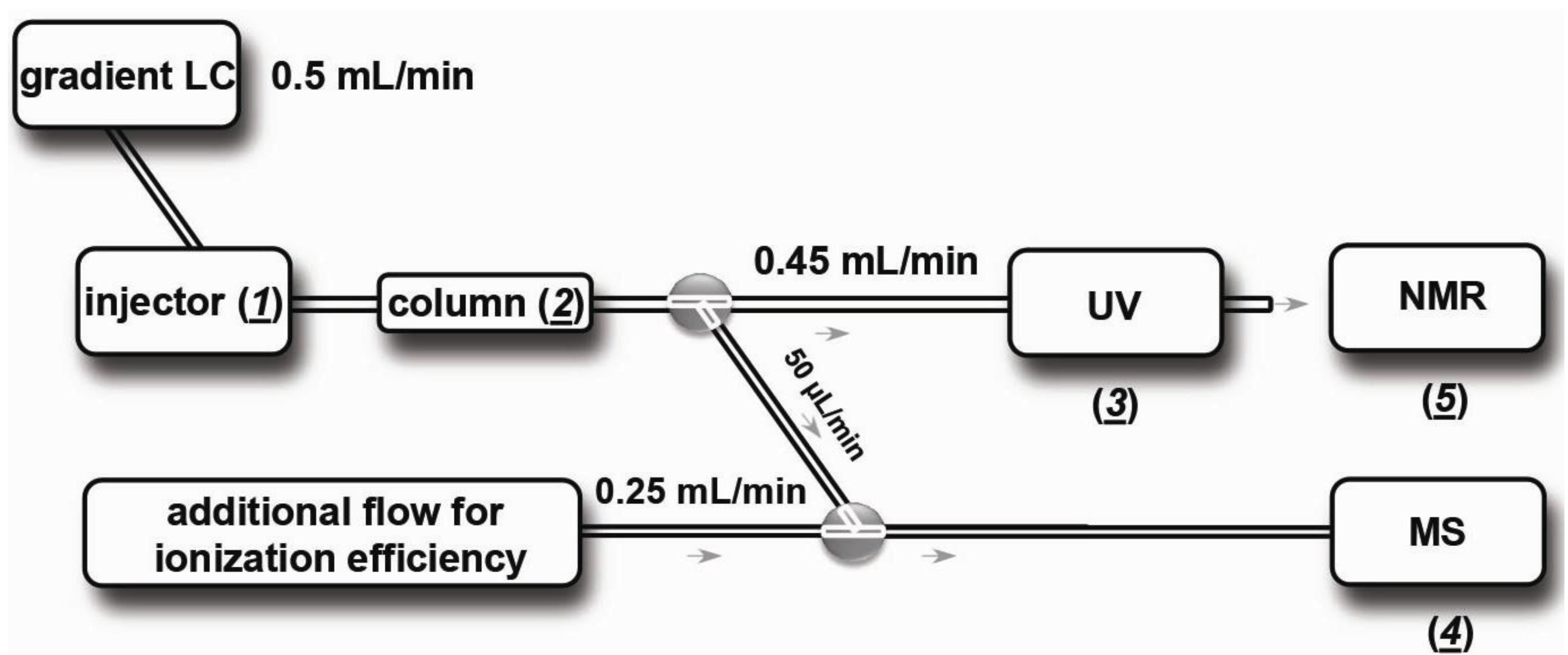
2.2.7. Off-Line NMR
2.3. Biochemical Assays and Samples
2.3.1. Biochemical Assay
2.3.2. Radioligand Binding Assay
2.3.3. Conus textile Venom
2.3.4. Toad Skin Excretion Extracts
3. Results and Discussion
3.1. Method Evaluation for Cone Snail Venoms
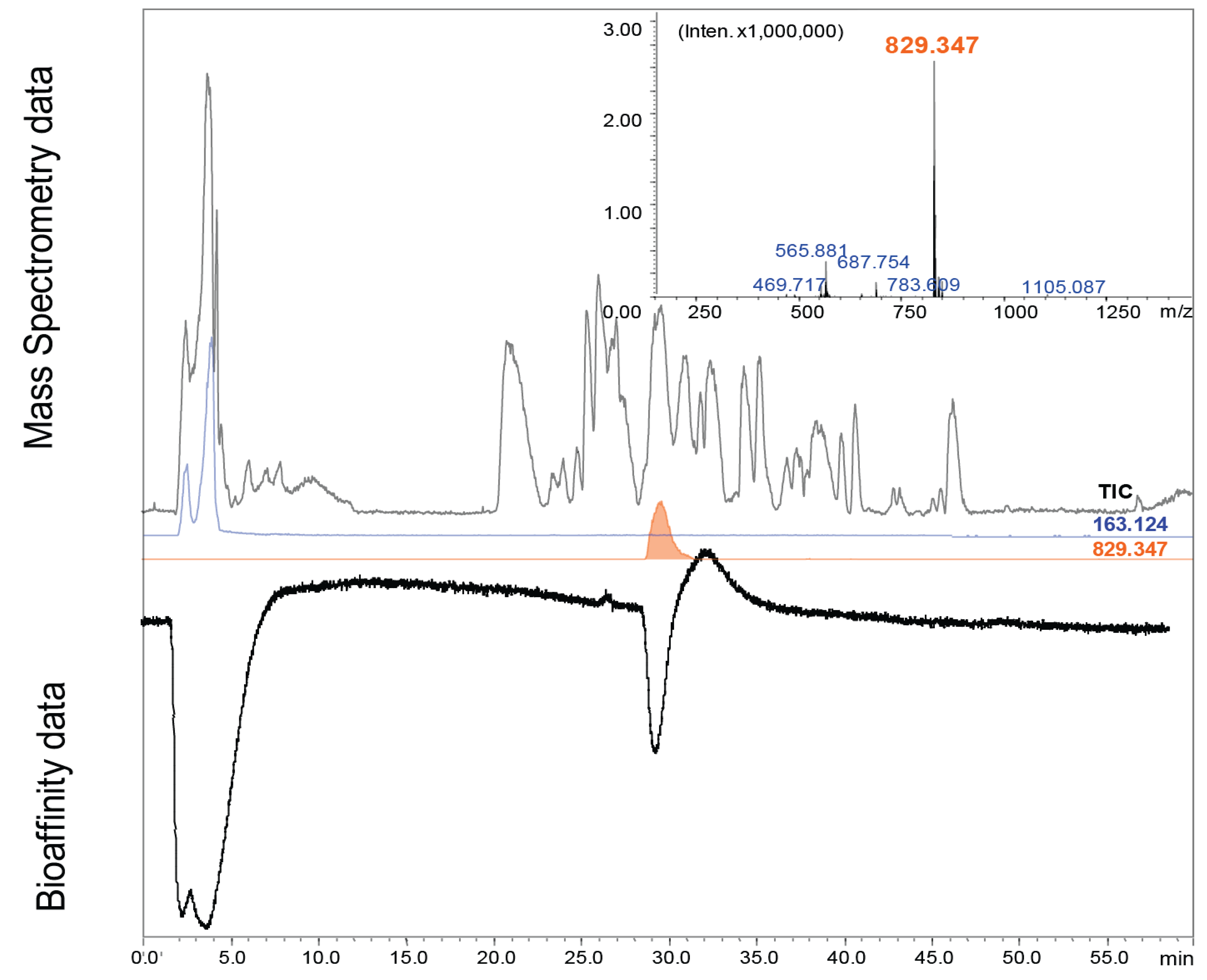
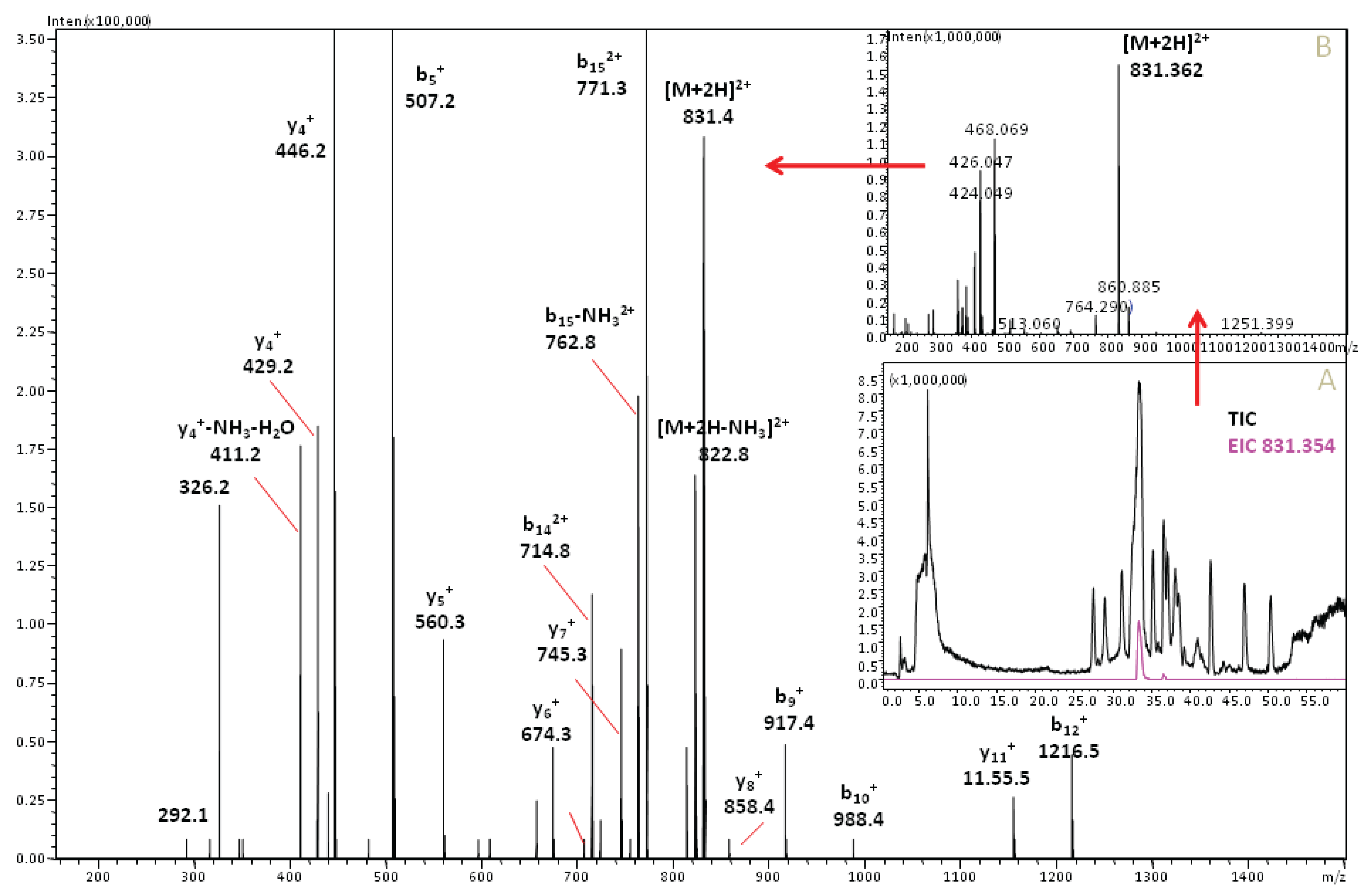
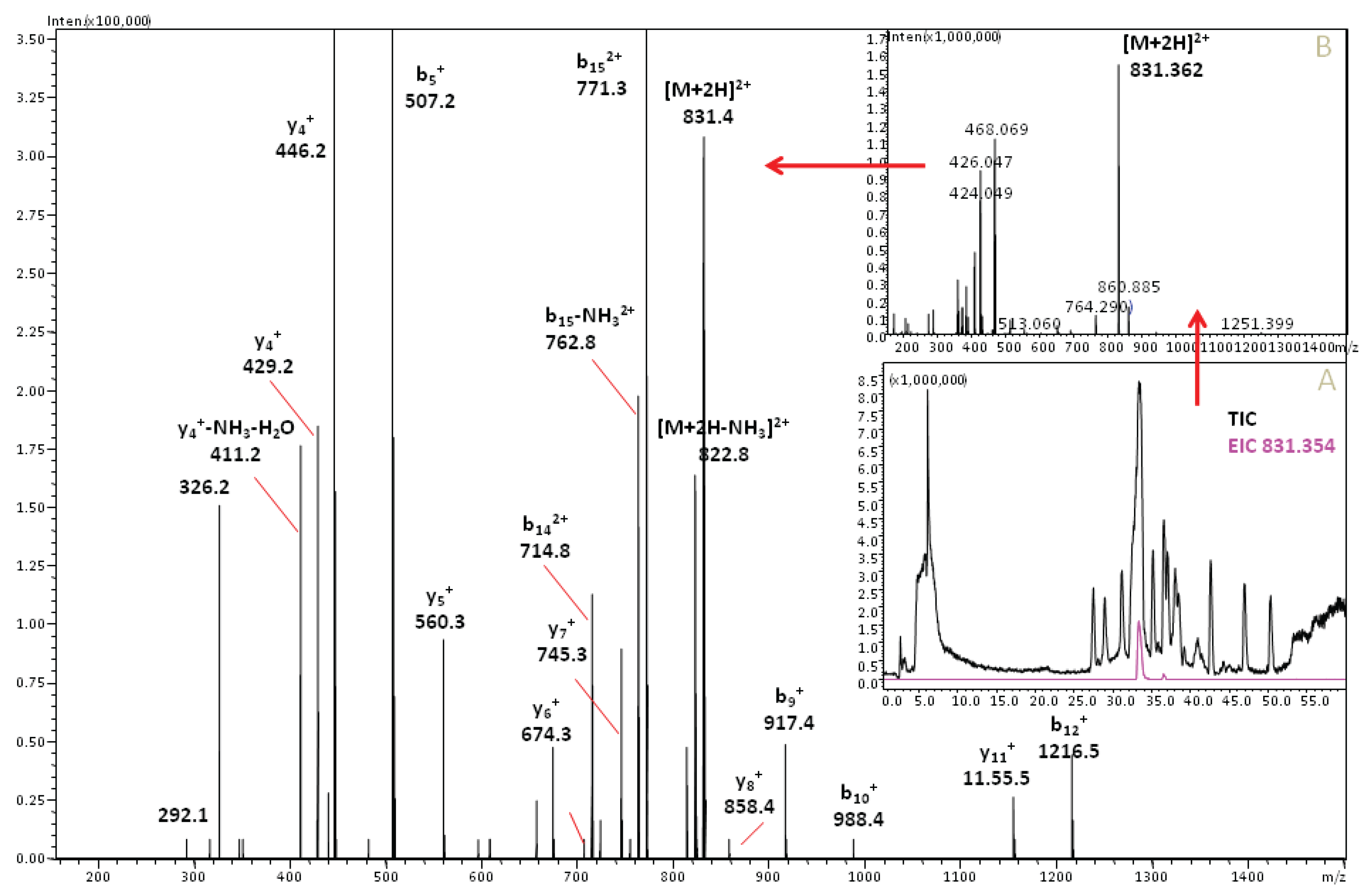
3.2. Cone Snail Toxin Screening
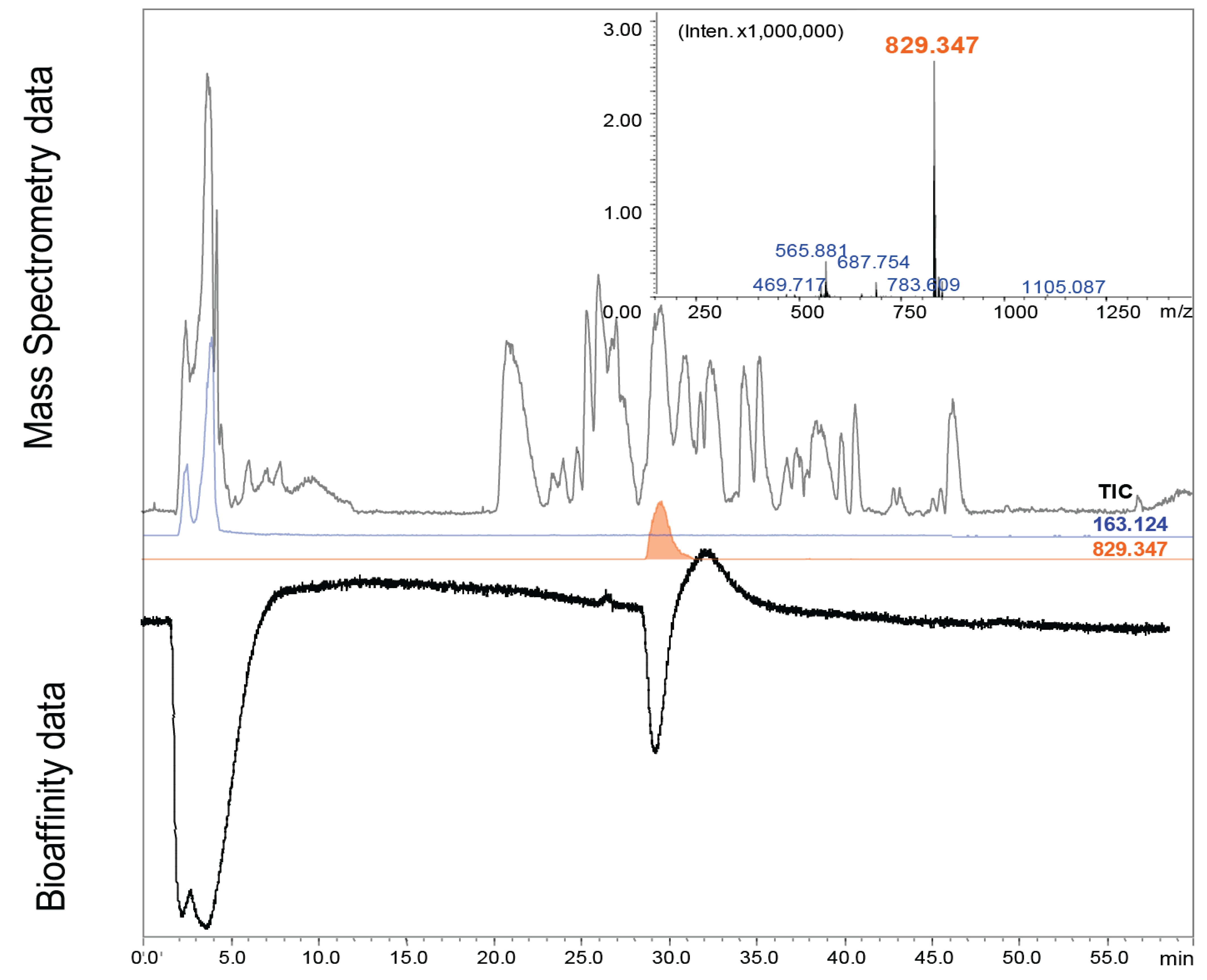
3.3. Screening of Toad Skin Excretions


3.4. NMR Data on Toad Fractions
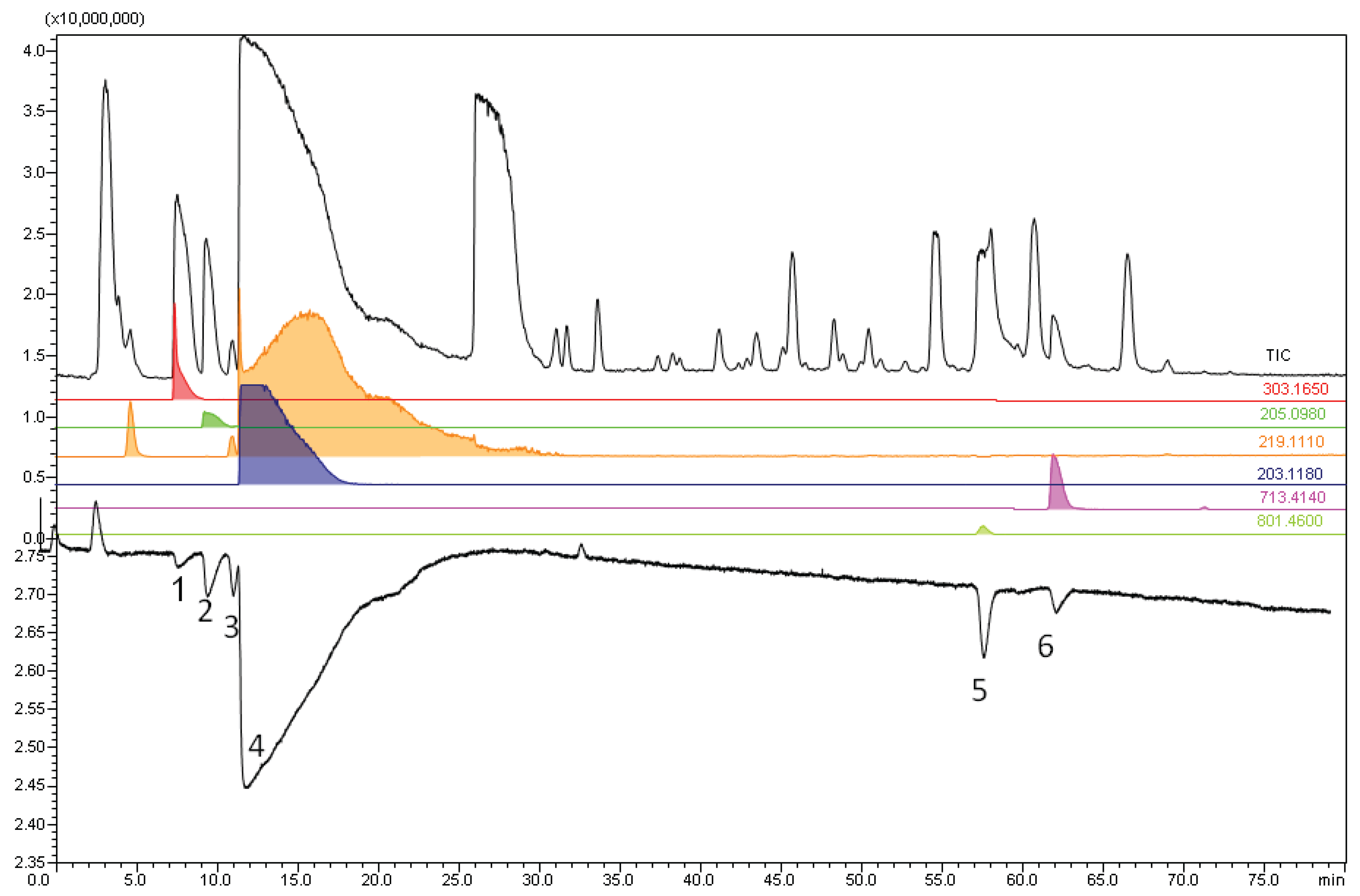
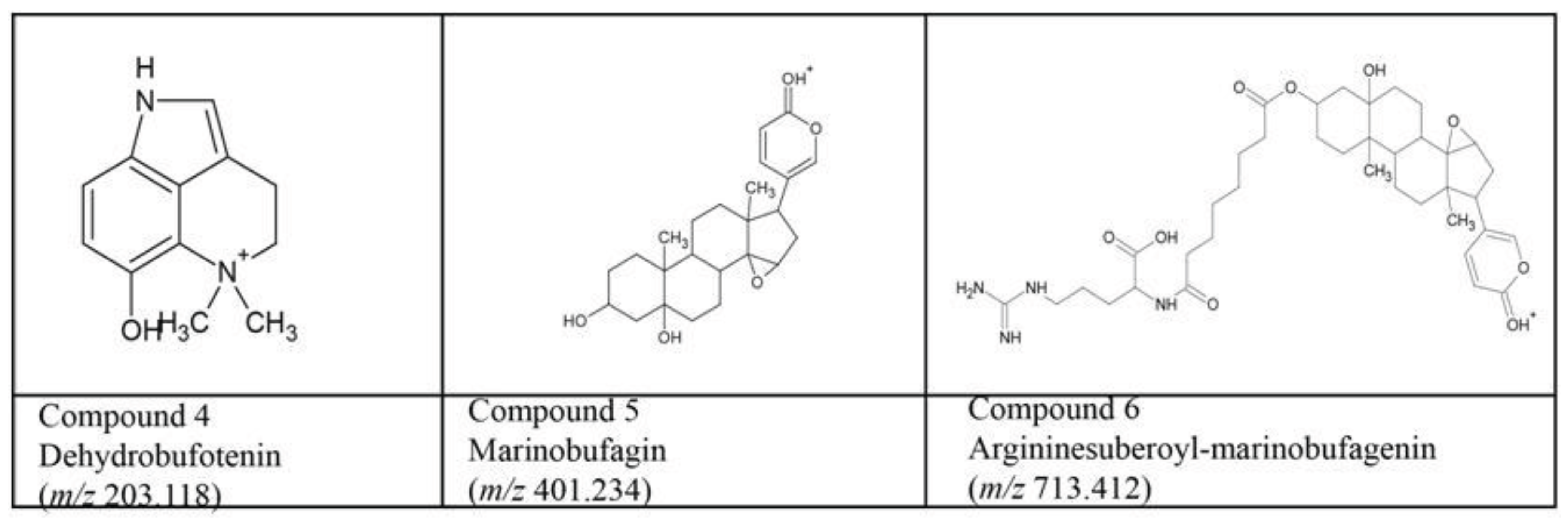
3.5. Bufo alvarius Bioaffinity Assessment
4. Conclusions
Abbreviations
| α7-nAChR | α7-nicotinic acetylcholine receptor |
| 3H-MLA | 3H-methyllycaconitine |
| AChBP | Acetylcholine binding protein |
| DAHBA | (E)-3-(3-(4-diethylamino-2-hydroxybenzylidene)3,4,5,6-tetrahydropyridin-2-yl)pyridine |
| DTT | DL-dithiothreitol |
| ESI | Electrospray Ionization |
| EICs | Extracted Ion Chromatograms |
| FA | Formic Acid |
| HRS | High-Resolution Screening |
| i.d. | Internal diameter |
| IT TOF | Ion-Trap-Time-of-Flight |
| Ls | Lymnaea stagnalis |
| LC | Liquid Chromatography |
| MeOH | Methanol |
| RBA | Radioligand Binding Assay |
| TIC | Total Ion Chromatogram |
| TFA | Trifluoroacetic acid |
Supplementary Files
Acknowledgments
Author contributions
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef]
- Norton, R.S.; Olivera, B.M. Conotoxins down under. Toxicon 2006, 48, 780–798. [Google Scholar] [CrossRef]
- Olivera, B.M.; Teichert, R.W. Diversity of the neurotoxic conus peptides: A model for concerted pharmacological discovery. Mol. Interv. 2007, 7, 251–260. [Google Scholar] [CrossRef]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. 2003, 2, 790–802. [Google Scholar]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- Tsetlin, V.I.; Kasheverov, I.E.; Zhmak, M.N.; Utkin, Y.N.; Vulfius, C.A.; Smit, A.B.; Bertrand, D. Alpha-conotoxin analogs with enhanced affinity for nicotinic receptors and acetylcholine-binding proteins. J. Mol. Neurosci. 2006, 30, 77–78. [Google Scholar] [CrossRef]
- Livett, B.G.; Sandall, D.W.; Keays, D.; Down, J.; Gayler, K.R.; Satkunanathan, N.; Khalil, Z. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon 2006, 48, 810–829. [Google Scholar] [CrossRef]
- Nasiripourdori, A.; Taly, V.; Grutter, T.; Taly, A. From toxins targeting ligand gated ion channels to therapeutic molecules. Toxins 2011, 3, 260–293. [Google Scholar] [CrossRef]
- Loughnan, M.; Nicke, A.; Jones, A.; Schroeder, C.I.; Nevin, S.T.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Identification of a novel class of nicotinic receptor antagonists: Dimeric conotoxins vxxiia, vxxiib, and vxxiic from conus vexillum. J. Biol. Chem. 2006, 281, 24745–24755. [Google Scholar] [CrossRef]
- Romeo, C.; di Francesco, L.; Oliverio, M.; Palazzo, P.; Massilia, G.R.; Ascenzi, P.; Polticelli, F.; Schinina, M.E. Conus ventricosus venom peptides profiling by HPLC-MS: A new insight in the intraspecific variation. J. Sep. Sci. 2008, 31, 488–498. [Google Scholar] [CrossRef]
- Mandal, A.K.; Ramasamy, M.R.; Sabareesh, V.; Openshaw, M.E.; Krishnan, K.S.; Balaram, P. Sequencing of t-superfamily conotoxins from conus virgo: Pyroglutamic acid identification and disulfide arrangement by maldi mass spectrometry. J. Am. Soc. Mass Spectrom. 2007, 18, 1396–1404. [Google Scholar] [CrossRef]
- Favreau, P.; Menin, L.; Michalet, S.; Perret, F.; Cheneval, O.; Stocklin, M.; Bulet, P.; Stocklin, R. Mass spectrometry strategies for venom mapping and peptide sequencing from crude venoms: Case applications with single arthropod specimen. Toxicon 2006, 47, 676–687. [Google Scholar] [CrossRef]
- Lenaerts, C.; Demeyer, M.; Gerbaux, P.; Blankert, B. Analytical aspects of marinobufagenin. Clin. Chim. Acta 2013, 421, 193–201. [Google Scholar] [CrossRef]
- Erspamer, V.; Vitali, T.; Roseghini, M.; Cei, J.M. 5-Methoxy- and 5-hydroxyindoles in the skin of bufo alvarius. Biochem. Pharmacol. 1967, 16, 1149–1164. [Google Scholar] [CrossRef]
- Weil, A.T.; Davis, W. Bufo alvarius: A potent hallucinogen of animal origin. J. Ethnopharmacol. 1994, 41, 1–8. [Google Scholar] [CrossRef]
- Dutertre, S.; Croker, D.; Daly, N.L.; Andersson, A.; Muttenthaler, M.; Lumsden, N.G.; Craik, D.J.; Alewood, P.F.; Guillon, G.; Lewis, R.J. Conopressin-t from conus tulipa reveals an antagonist switch in vasopressin-like peptides. J. Biol. Chem. 2008, 283, 7100–7108. [Google Scholar] [CrossRef]
- Schroeder, C.I.; Ekberg, J.; Nielsen, K.J.; Adams, D.; Loughnan, M.L.; Thomas, L.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Neuronally micro-conotoxins from conus striatus utilize an alpha-helical motif to target mammalian sodium channels. J. Biol. Chem. 2008, 283, 21621–21628. [Google Scholar] [CrossRef]
- Loughnan, M.L.; Nicke, A.; Jones, A.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Chemical and functional identification and characterization of novel sulfated alpha-conotoxins from the cone snail conus anemone. J. Med. Chem. 2004, 47, 1234–1241. [Google Scholar] [CrossRef]
- Mishra, K.P.; Ganju, L.; Sairam, M.; Banerjee, P.K.; Sawhney, R.C. A review of high throughput technology for the screening of natural products. Biomed. Pharmacother. 2008, 62, 94–98. [Google Scholar] [CrossRef]
- Lam, K.S. New aspects of natural products in drug discovery. Trends Microbiol. 2007, 15, 279–289. [Google Scholar] [CrossRef]
- Liu, S.; Yi, L.Z.; Liang, Y.Z. Traditional chinese medicine and separation science. J. Sep. Sci. 2008, 31, 2113–2137. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. 2005, 4, 206–220. [Google Scholar]
- Potterat, O.; Hamburger, M. Concepts and technologies for tracking bioactive compounds in natural product extracts: Generation of libraries, and hyphenation of analytical processes with bioassays. Nat. Prod. Rep. 2013, 30, 546–564. [Google Scholar] [CrossRef]
- Mouhat, S.; Andreotti, N.; Jouirou, B.; Sabatier, J.M. Animal toxins acting on voltage-gated potassium channels. Curr. Pharm. Des. 2008, 14, 2503–2518. [Google Scholar] [CrossRef]
- Annis, D.A.; Nickbarg, E.; Yang, X.; Ziebell, M.R.; Whitehurst, C.E. Affinity selection-mass spectrometry screening techniques for small molecule drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 518–526. [Google Scholar] [CrossRef]
- Johnson, B.M.; Nikolic, D.; van Breemen, R.B. Applications of pulsed ultrafiltration-mass spectrometry. Mass Spectrom. Rev. 2002, 21, 76–86. [Google Scholar] [CrossRef]
- Kool, J.; Eggink, M.; van Rossum, H.; van Liempd, S.M.; van Elswijk, D.A.; Irth, H.; Commandeur, J.N.; Meerman, J.H.; Vermeulen, N.P. Online biochemical detection of glutathione-s-transferase p1-specific inhibitors in complex mixtures. J. Biomol. Screen. 2007, 12, 396–405. [Google Scholar] [CrossRef]
- Kool, J.; Giera, M.; Irth, H.; Niessen, W.M. Advances in mass spectrometry-based post-column bioaffinity profiling of mixtures. Anal. Bioanal. Chem. 2011, 399, 2655–2668. [Google Scholar] [CrossRef]
- Kool, J.; van Liempd, S.M.; Harmsen, S.; Beckman, J.; van Elswijk, D.; Commandeur, J.N.; Irth, H.; Vermeulen, N.P. Cytochrome p450 bio-affinity detection coupled to gradient HPLC: On-line screening of affinities to cytochrome p4501a2 and 2d6. J. Chromatogr. 2007, 858, 49–58. [Google Scholar] [CrossRef]
- Kool, J.; van Liempd, S.M.; Harmsen, S.; Schenk, T.; Irth, H.; Commandeur, J.N.; Vermeulen, N.P. An on-line post-column detection system for the detection of reactive-oxygen-species-producing compounds and antioxidants in mixtures. Anal. Bioanal. Chem. 2007, 388, 871–879. [Google Scholar] [CrossRef]
- Schebb, N.H.; Faber, H.; Maul, R.; Heus, F.; Kool, J.; Irth, H.; Karst, U. Analysis of glutathione adducts of patulin by means of liquid chromatography (HPLC) with biochemical detection (BCD) and electrospray ionization tandem mass spectrometry (ESI-MS/MS). Anal. Bioanal. Chem. 2009, 394, 1361–1373. [Google Scholar] [CrossRef]
- Edink, E.; Rucktooa, P.; Retra, K.; Akdemir, A.; Nahar, T.; Zuiderveld, O.; van Elk, R.; Janssen, E.; van Nierop, P.; van Muijlwijk-Koezen, J.; et al. Fragment growing induces conformational changes in acetylcholine-binding protein: A structural and thermodynamic analysis. J. Am. Chem. Soc. 2011, 133, 5363–5371. [Google Scholar] [CrossRef]
- De Kloe, G.E.; Retra, K.; Geitmann, M.; Kallblad, P.; Nahar, T.; van Elk, R.; Smit, A.B.; van Muijlwijk-Koezen, J.E.; Leurs, R.; Irth, H.; et al. Surface plasmon resonance biosensor based fragment screening using acetylcholine binding protein identifies ligand efficiency hot spots (le hot spots) by deconstruction of nicotinic acetylcholine receptor alpha7 ligands. J. Med. Chem. 2010, 53, 7192–7201, Same as ref 40. [Google Scholar] [CrossRef]
- Celie, P.H.; Kasheverov, I.E.; Mordvintsev, D.Y.; Hogg, R.C.; van Nierop, P.; van Elk, R.; van Rossum-Fikkert, S.E.; Zhmak, M.N.; Bertrand, D.; Tsetlin, V.; et al. Crystal structure of nicotinic acetylcholine receptor homolog achbp in complex with an alpha-conotoxin pnia variant. Nat. Struct. Mol. Biol. 2005, 12, 582–588. [Google Scholar] [CrossRef]
- Smit, A.B.; Brejc, K.; Syed, N.; Sixma, T.K. Structure and function of achbp, homologue of the ligand-binding domain of the nicotinic acetylcholine receptor. Ann. NY Acad. Sci. 2003, 998, 81–92. [Google Scholar] [CrossRef]
- Kool, J.; de Kloe, G.E.; Bruyneel, B.; de Vlieger, J.S.; Retra, K.; Wijtmans, M.; van Elk, R.; Smit, A.B.; Leurs, R.; Lingeman, H.; et al. Online fluorescence enhancement assay for the acetylcholine binding protein with parallel mass spectrometric identification. J. Med. Chem. 2010, 53, 4720–4730. [Google Scholar] [CrossRef]
- Heus, F.; Giera, M.; de Kloe, G.E.; van Iperen, D.; Buijs, J.; Nahar, T.T.; Smit, A.B.; Lingeman, H.; de Esch, I.J.; Niessen, W.M.; et al. Development of a microfluidic confocal fluorescence detection system for the hyphenation of nano-lc to on-line biochemical assays. Anal. Bioanal. Chem. 2010, 398, 3023–3032. [Google Scholar] [CrossRef]
- Heus, F.; Vonk, F.; Otvos, R.A.; Bruyneel, B.; Smit, A.B.; Lingeman, H.; Richardson, M.; Niessen, W.M.; Kool, J. An efficient analytical platform for on-line microfluidic profiling of neuroactive snake venoms towards nicotinic receptor affinity. Toxicon 2013, 61, 112–124. [Google Scholar] [CrossRef]
- Ulens, C.; Akdemir, A.; Jongejan, A.; van Elk, R.; Bertrand, S.; Perrakis, A.; Leurs, R.; Smit, A.B.; Sixma, T.K.; Bertrand, D.; et al. Use of acetylcholine binding protein in the search for novel alpha7 nicotinic receptor ligands. In silico docking, pharmacological screening, and X-ray analysis. J. Med. Chem. 2009, 52, 2372–2383. [Google Scholar] [CrossRef]
- Dutertre, S.; Ulens, C.; Buttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; et al. Achbp-targeted alpha-conotoxin correlates distinct binding orientations with nachr subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar] [CrossRef]
- Mebs, D.; Wagner, M.G.; Pogoda, W.; Maneyro, R.; Kwet, A.; Kauert, G. Lack of bufadienolides in the skin secretion of red bellied toads, Melanophryniscus spp. (anura, bufonidae), from uruguay. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2007, 144, 398–402. [Google Scholar]
- Gao, H.; Zehl, M.; Leitner, A.; Wu, X.; Wang, Z.; Kopp, B. Comparison of toad venoms from different bufo species by hplc and LC-DAD-MS/MS. J. Ethnopharmacol. 2010, 131, 368–376. [Google Scholar] [CrossRef]
- Cunha Filho, G.A.; Schwartz, C.A.; Resck, I.S.; Murta, M.M.; Lemos, S.S.; Castro, M.S.; Kyaw, C.; Pires, O.R., Jr.; Leite, J.R.S.; Bloch, C., Jr.; et al. Antimicrobial activity of the bufadienolides marinobufagin and telocinobufagin isolated as major components from skin secretion of the toad bufo rubescens. Toxicon 2005, 45, 777–782. [Google Scholar] [CrossRef]
- Yoshika, M.; Komiyama, Y.; Konishi, M.; Akizawa, T.; Kobayashi, T.; Date, M.; Kobatake, S.; Masuda, M.; Masaki, H.; Takahashi, H. Novel digitalis-like factor, marinobufotoxin, isolated from cultured y-1 cells, and its hypertensive effect in rats. Hypertension 2007, 49, 209–214. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Heus, F.; Otvos, R.A.; Aspers, R.L.E.G.; Van Elk, R.; Halff, J.I.; Ehlers, A.W.; Dutertre, S.; Lewis, R.J.; Wijmenga, S.; Smit, A.B.; et al. Miniaturized Bioaffinity Assessment Coupled to Mass Spectrometry for Guided Purification of Bioactives from Toad and Cone Snail. Biology 2014, 3, 139-156. https://doi.org/10.3390/biology3010139
Heus F, Otvos RA, Aspers RLEG, Van Elk R, Halff JI, Ehlers AW, Dutertre S, Lewis RJ, Wijmenga S, Smit AB, et al. Miniaturized Bioaffinity Assessment Coupled to Mass Spectrometry for Guided Purification of Bioactives from Toad and Cone Snail. Biology. 2014; 3(1):139-156. https://doi.org/10.3390/biology3010139
Chicago/Turabian StyleHeus, Ferry, Reka A. Otvos, Ruud L. E. G. Aspers, Rene Van Elk, Jenny I. Halff, Andreas W. Ehlers, Sébastien Dutertre, Richard J. Lewis, Sybren Wijmenga, August B. Smit, and et al. 2014. "Miniaturized Bioaffinity Assessment Coupled to Mass Spectrometry for Guided Purification of Bioactives from Toad and Cone Snail" Biology 3, no. 1: 139-156. https://doi.org/10.3390/biology3010139