Abstract
Hsp90 has become the target of intensive investigation, as inhibition of its function has the ability to simultaneously incapacitate proteins that function in pathways that represent the six hallmarks of cancer. While a number of Hsp90 inhibitors have made it into clinical trials, a number of short-comings have been noted, such that the search continues for novel Hsp90 inhibitors with superior pharmacological properties. To identify new potential Hsp90 inhibitors, we have utilized a high-throughput assay based on measuring Hsp90-dependent refolding of thermally denatured luciferase to screen natural compound libraries. Over 4,000 compounds were screen with over 100 hits. Data mining of the literature indicated that 51 compounds had physiological effects that Hsp90 inhibitors also exhibit, and/or the ability to downregulate the expression levels of Hsp90-dependent proteins. Of these 51 compounds, seven were previously characterized as Hsp90 inhibitors. Four compounds, anthothecol, garcinol, piplartine, and rottlerin, were further characterized, and the ability of these compounds to inhibit the refolding of luciferase, and reduce the rate of growth of MCF7 breast cancer cells, correlated with their ability to suppress the Hsp90-dependent maturation of the heme-regulated eIF2α kinase, and deplete cultured cells of Hsp90-dependent client proteins. Thus, this screen has identified an additional 44 compounds with known beneficial pharmacological properties, but with unknown mechanisms of action as possible new inhibitors of the Hsp90 chaperone machine.
1. Introduction
During the past 20 years Hsp90 has emerged as a major target for the development of cancer therapeutics. The story began in 1994 with the report by Whitesell and co-workers that benzoquinone ansamycins, natural products isolated from the soil actinomycetes species Streptomyces hygroscopicus, were inhibitors of Hsp90 and not tyrosine kinases [1]. In 1997, the crystal structure of the Hsp90 N-terminal domain in complex with the benzoquinone ansamycin, geldanamycin was published, and later that year it was demonstrated that the geldanamycin-bindng site was responsible for the binding and hydrolysis of ATP (adenosine triphosphate) by Hsp90 [2]. Subsequently in 1998, radicicol, an antibiotic isolated from the mycoparasitic fungus Humicola fuscoatra, was found to bind similarly to the N-terminal domain of Hsp90 [3,4], and with the additional co-crystal structure in 1999, the road to rational drug design was paved [5].
Upon the discovery of Hsp90 inhibitors, insight into the cellular function of Hsp90 rapidly emerged. Basic research led to the identification of proteins termed “clients,” which relied upon Hsp90 not only for their activity and function, but also in many cases for their stability. Disruption of Hsp90’s function resulted in the depletion of Hsp90-dependent clients from cells either by destabilizing the client and accelerating their degradation via the ubiquitin/proteasome pathway [6], or by preventing the replenishment of mature client proteins as they turned over in the cell through the inhibition of their folding, which led to the degradation of newly synthesized clients. From these early studies it became apparent that many of the Hsp90 clients were components of signal transduction cascades, and Hsp90 became know as the “signal-transduction” chaperone [7]. Excitement over Hsp90 as a cancer therapeutic target grew when it became apparent that Hsp90-dependent clients were present in all six (now eight [8]) hallmarks of cancer, and that Hsp90 inhibitors could derail multiple cellular signaling pathways simultaneously [9].
Hsp90 inhibitors based on the benzoquinone ansamycin scaffold of geldanamycin, the resorcinol scaffold of radicicol, the purine scaffold of ATP, and other unique chemical scaffolds discovered to inhibit Hsp90, are currently being evaluated in over 80 ongoing or completed clinical trials [10]. While some of the results from these trials have been encouraging, reports of hepato-, cardio-, ocular-toxicity, peripheral neuropathy and in some cases difficult dosing schedules have tempered enthusiasm for clinical Hsp90 inhibition. However, due to Hsp90’s importance toward maintaining the viability of cancer cells, significant effort is still being dedicated to the discovery and synthesis of novel and more efficacious Hsp90 inhibitors.
Because Hsp90 inhibitors affect the function of such a diverse set of cellular pathways, the inhibitors have an unusual physiological signature. Distinctive properties of Hsp90 inhibitors include: (1) the ability to inhibit multiple, yet seemingly unrelated signal transduction pathways [9]; (2) the ability to inhibit the activities of proteins for which no rational expectation for a common binding specificity exists; (3) a high differential selectivity of the compound for cancer versus normal cells [11,12]; and (4) protection of cells from toxicity induced by the accumulation of protein aggregates [13,14,15] (i.e., aggregates of tau or Aβ amyloid).
These distinctive properties manifested by Hsp90 inhibitors have widespread and sometimes varied effects within a cell. Depending on the conditions, the physiological manifestations of this inhibition can also vary. For example, Hsp90 is highly involved in the inflammatory response, as several mediating proteins, such as IκB Kinase (IKK) [16,17] and nitric oxide synthase [18,19], are dependent upon Hsp90 for their function. Accordingly, Hsp90 inhibitors result in the down-regulation of these proteins and display anti-inflammatory activity, which makes them prominent in traditional medicine. In the case of cancer cells, accelerated growth and cell division is maintained by Hsp90-dependent clients, as is angiogenesis [20,21]. Thus, treatment with Hsp90 inhibitors results in the slowing of cell growth, inhibition of tumor vascularization, and potentially, induction of apoptosis. Similarly, the effects of Hsp90 inhibition can be seen in other medically relevant ways, including activity against viruses [22], bacteria, fungi [23], and parasites [24], specifically the causative agent of malaria, Plasmodium falciparum [25].
A number of high-throughput screening (HTS) assays based on a variety of techniques have been developed to screen large chemical libraries to identify new Hsp90 inhibitors (reviewed in [26]). Screens have been developed based on the ability of compounds to: (1) inhibit Hsp90 catalyzed ATP hydrolysis; (2) competitively inhibit the binding of ligand to Hsp90’s N-terminal ATP binding domain; (3) inhibit Hsp90-mediated refolding of denatured protein (e.g., luciferase); and (4) deplete cultured cells of Hsp90 client proteins. These assays have identified a large number of potential Hsp90 inhibitors. However, only a limited number of follow-up studies have been carried out to verify the mechanism of action of these compounds and to optimize their Hsp90-inhibitory activity.
As noted above, most HTS have been carried out on large chemical libraries. Here we focus on the use of a high-throughput assay to screen natural product libraries for novel inhibitors of the Hsp90 chaperone machine. The screening is based on the ability of Hsp90 inhibitors to block the refolding of thermally denatured firefly luciferase, which is catalyzed by the Hsp90 chaperone machinery present in rabbit reticulocyte lysate [27,28,29]. It was reasoned that natural products would be a fertile territory for identification of additional Hsp90 inhibitors, as it would be reasonable to expect that evolutionary pressure would give plant or other species, which have acquired pathways leading to the synthesis of secondary metabolites that inhibit Hsp90 a competitive advantage, because such compounds would be expected to inhibit the growth and development of insect and pathological pests. Furthermore, as noted in a recent review, the majority of drugs approved for use by the FDA during the past 30 to 50 years are natural products or derivatives thereof [30,31]. In addition, there is vast literature on active compounds that have been isolated from traditional folk medicines that allowed us to mine the literature for compounds identified in our screen that have properties of Hsp90 inhibitors that were discussed above.
2. Experimental Section
2.1. High-Throughput Screen of Natural Product Libraries
2.1.1. Rabbit Reticulocyte Lysate
Rabbit reticulocyte lysate, prepared by lysing one volume of packed reticulocytes in two volumes of deionized water, followed by centrifugation for twenty minutes at 15,000 × g, was purchased from Green Hectares (Oregon, WI, USA).
2.1.2. Denatured Luciferase
Recombinant luciferase from Promega was diluted to 0.5 mg/mL in buffer consisting of 25 mM Tricine–HCl (pH 7.8), 8 mM MgSO4, 0.1 mM EDTA, and 10 mg/mL acetylated BSA. Next, the solution was adjusted to include 10% glycerol and 1% Triton X-100. Finally, the luciferase solution was heated to ~41°. Once the activity of the luciferase reached ~1% of its initial value, the mixture was placed on ice, or flash frozen in liquid nitrogen and placed at −80° for storage.
To prepare the denatured luciferase for use in re-folding assays, 125 µL of the 0.5 mg/mL mixture was added into a 10 mL mixture containing 80 mM Tris HCl, pH 7.7, 8 mM Mg(OAc)2, 300 mM KCl, 12 mM ATP, and 20 mM creatine phosphate, and 0.8 mg/mL creatine phosphokinase.
2.1.3. Assay Buffer
The assay buffer, which contains the luciferase substrate luciferin, consisted of 75 mM Tricine-HCl, pH 7.8, 24 mM MgSO4, 300 µM EDTA, 2 mM DTT, 313 µM D-luciferin, 640 µM coenzyme A, 660 µM ATP, 150 mM KCl, 10% (v/v) Triton X-100, 20% (v/v) glycerol, and 3.5% DMSO.
2.1.4. Compounds Library Screen
The natural product libraries were purchased from the following companies: Microsource Spectrum Collection (University of Kansas High Throughput Screening Center, 720 compounds); TimTec (480 compounds); AnalytiCon Discovery(2,511 compounds); BioFocus (272 compounds); and; BioMol Life Sciences/ ENZO Life Sciences (596 compounds). Of the 2,608 compounds that were present in the Microsource, TimTec, BioFocus, and BioMol libraries, approximately 209 were duplicates. Differences in naming, salts of the compounds and chemical nomenclature (e.g., D- versus (+)-) in the Excel spreads makes this number an estimate. The Analyticon did not come with a spreadsheet that was exportable to Excel.
The libraries were screened for compounds that inhibited the refolding of thermally denatured luciferase using a high-throughput assay carried out with a slight modification of the method previously described [27,29]. Briefly, compounds were reconstituted in 100% DMSO. Stocks of compounds purchased from Microsource, TimTec, Biofocus and BioMol were reconstituted to a concentration of 1 mg/mL. The stocks were diluted 40-fold into nano-pure water with the compounds being used at a final concentration of 12.5 µg/mL for the assay. Analyticon compounds (0.2 µmol) were reconstituted to a concentration of 4 mM, and were used at a final concentration of 40 µM in the assay. Assays were performed in 96-well microplates. To each well was added 30 µL of the water/DMSO compound solution, 15 µL of the reticulocyte lysate preparation, and 15 µL of the luciferase reagent. The plates were agitated and then allowed to incubate at 25 °C for one to three hours. After the incubation, 60 µL of assay buffer containing luciferin was added to each well. The plates were then read in a Molecular Devices LMaxII384 microplate reader, and luminescence was measured in relative light units, with an integration time of 10 s. Compounds that inhibited luciferase refolding by approximately fifty percent or greater were then titrated into a refolding reaction containing native luciferase, to eliminate false positive hits that were direct inhibitors of luciferase as previously described [27,29]. Compounds were classified as a potential Hsp90 inhibitor if they inhibited luciferase refolding by 60% or greater and there was no inhibition of the activity of native luciferase at the compound’s IC50 (concentration of the compound that inhibits luciferase refolding by 50%).
To obtain an estimate of a compound’s IC50, the compounds were titrated into the assay mixture described above starting at a concentration of 25 µg/mL and a series of three-fold dilutions. The titrations were repeated twice. For a more accurate determination of the IC50s for the compounds studied in Section 3.2.1 of this manuscript, a two-fold deletion series of the compounds were used in triplicate, with the experiment being repeated three times.
2.1.5. Statistical Analysis
Z-factors were calculated for the Microsource, TimTec and Analyticon compounds screens as the plates contained an adequate number of positive and negative controls to make the calculation. Robust statistics [32], which minimizes the effects of outliers on the statistical analysis, were used to calculate the median, robust standard deviation (rSD) and the robust percent coefficient of variance (%rCV).
2.2. Antiproliferation Assay
MCF7 cells were grown in Gibco Dulbecco’s Modified Essential Medium, supplemented with non-essential amino acids, 2 mM glutamine, and 10% fetal bovine serum. Cells were seeded at 2,000 cells per well in clear 96-well plates containing 100 µL of media per well, and the cells were allowed to attach overnight. The next day, serial dilutions of compounds in DMSO or DMSO control was added to the wells. Cells were then incubated at 37 °C for 48 h. Cell viability [33] was determined using the Promega Cell Titer 96 Aqueous One Solution Cell Proliferation Assay, which makes use of a soluble tetrazolium compound that is converted into a chromophore by living cells. After incubation with compounds, 20 µL of the assay substrate solution were added to the wells, and the plate was incubated at 37 °C for an additional hour. The plate was then read at absorbance at 490 nm using a Molecular Devices Versamax plate reader. Values are expressed as percent DMSO control. All experiments were done in triplicate.
2.3. Hsp90-Dependent Maturation of the Heme-Regulated eIF2α Kinase [33]
[35S]-Labeled His-tagged HRI was translated by coupled transcription/translation for 20 min in TnT reticulocyte lysate at 30 °C. Drugs or DMSO vehicle control were then added. After an additional 10 min, the reactions (4 µL aliquots) were diluted into 28 µL volumes of heme-supplemented control lysate or heme-deficient lysate, containing the same respective drugs and DMSO vehicle control. The samples were allowed to incubate for another 40 min at 37 °C. The [35S]-Labeled His-tagged HRI was then adsorbed from the reactions with NTA-Ni2+ agarose resin for 1 h at 4 °C. The agarose resins were the washed four times total, in P50T, P100T 2X, and P50T again. SDS sample buffer was then added to the pellets, the samples boiled, and then separated on an 8% PAGE gel, and transferred to PVDF membrane. Membranes were dried and exposed to X-ray film for autoradiography.
2.4. Inhibitor-Dependent Depletion of Hsp90-Dependent Clients from MCF7 Cells
MCF7 cells were grown to confluence in Advanced DMEM/F12 (1:1; Gibco) supplemented with L-glutamine (2 mM), streptomycin (500 µg/mL), and penicillin (100 units/mL) and re-seeded at 0.4 × 106 cells/well/2 mL. Cells were incubated in a humidified atmosphere (37 °C, 5% CO2) for 24 h and treated with varying concentrations of compound or 0.5 µM geldanamycin in DMSO (0.25% DMSO final concentration), or vehicle (DMSO) for 24 h. Cells were harvested in cold PBS and lysed using MPER (Thermo Scientific/Pierce, USA) supplemented with protease and phosphatase inhibitors (Roche Applied Science, USA) according to manufacturer’s directions. Lysates were clarified at 14,000 g for 15 min at 4 °C. Protein concentrations were determined using the Pierce BCA protein assay kit per the manufacturer’s instructions. Equal amounts of protein (15 μg) were electrophoresed under reducing conditions (10% acrylamide gels), transferred to PVDF, and immunoblotted with the corresponding antibody (Anti-pAkt, -Her2, and -Cdk6 antibodies were from Cell Signaling; anti-actin was from Santa Cruz Biotechnology; and anti-Hsp90 and -Hsp70 were from Enzo Lifesciences). Membranes were incubated with an appropriate horseradish peroxidase-labeled secondary antibody, developed with a chemiluminescent substrate, and visualized.
3. Results and Discussion
3.1. Natural Product Library Screen
An assay for chaperone-mediated protein renaturation was developed in 1994 using rabbit reticulocyte lysate (RRL) to refold thermally denatured firefly luciferase [34]. RRL had been used for decades for in vitro protein synthesis and was known to contain abundant quantities of the heat shock proteins. After the discovery that geldanamycin was an Hsp90 inhibitor, it was also demonstrated to inhibit the refolding of luciferase in RRL, confirming that luciferase renaturation was an Hsp90-dependent process [28]. The assay was demonstrated to be sensitive to compounds that bound and inhibited Hsp70, indicating that the assay could also detect compounds that interacted with other critical components of the Hsp90 chaperone machinery [27]. Subsequently, it was demonstrated that inhibitors that bind to the C-terminal domain of Hsp90 (e.g., novobiocin) also inhibited luciferase refolding in RRL [29]. The assay was then miniaturized and developed as a high-throughput screen for inhibitors of the Hsp90 chaperone machine [29]. Thus, while we generically refer to the compounds discussed below as Hsp90 inhibitors, the molecular target of the potential inhibitor could be any component of the Hsp90 chaperone machinery that is required for the refolding of luciferase.
Natural product libraries proved to be a fertile ground for identifying inhibitors of the Hsp90 chaperone machine. A scatter plot of percent inhibition of luciferase refolding versus 3,859 compounds screened from the TimTec, Biofocus, BioMol and Analyticon libraries is shown in Figure 1 (median = −0.041 ± 13 rSD). The Microsource compounds were part of a previously published screen [29] that had a Z-factor of 0.62 ± 0.09. The Z-factors for the TimTec and Analyticon screens were 0.77 ± 0.18 and 0.64 ± 0.14, respectively. The %rCV for the screens of the TimTec, Biofocus, BioMol and Analyticon screens were 8.9 ± 3.9, 20 ± 6.4, 18 ± 5.0 and 13 ± 6.2, respectively, indicating that the assay had a good signal-to-noise ration.
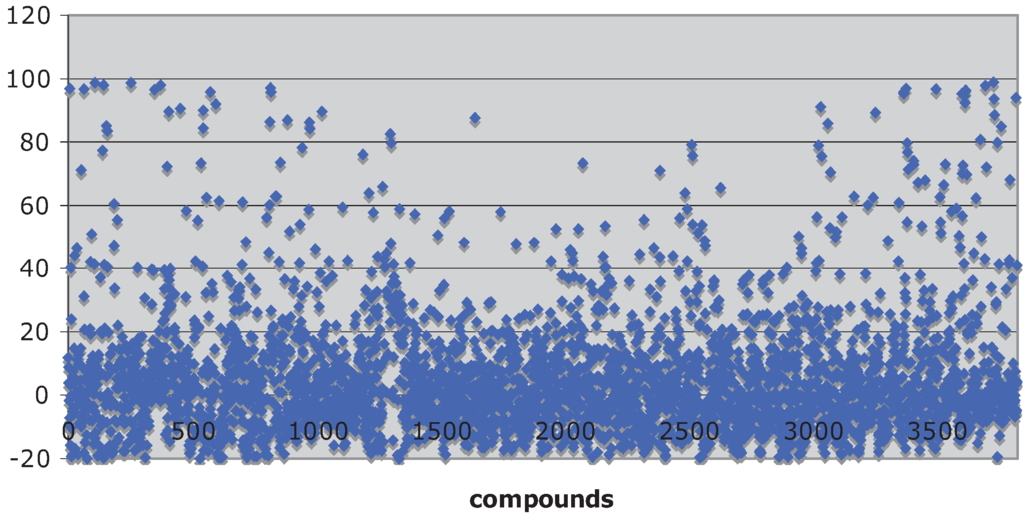
Figure 1.
Scatter plot showing the activities of 3,859 of the compounds screened.
The compounds, whose structures are shown in Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11 and are listed in Table 1, Table 2, Table 3, Table 4, Table 5, Table 6, Table 7, Table 8, Table 9, and Table 10, inhibited Hsp90-dependent refolding of luciferase by 60% or greater in the initial screen (a value greater than 4 rSD from the median), without inhibiting luciferase itself, with the exception of the screening of the Microsource compounds which used a cut-off of 70% inhibition [29]. The compounds have been sorted largely by structural classification, although some do not fit well into any particular structural group. Some compounds were present in more than one library. Curcumin (2 screens), gambogic acid (3 screens) and plumbagin (3 screens) were consistently identified as hits. Of the compounds that were present in more than one library, but were not identified as hits, six were present in the Microsource screen that used a cut-off of 70% inhibition, and four compounds failed to be identified as hits because they inhibited luciferase refolding less than the 60% cut-off used for the other libraries. The lone exception was luteolin, which exhibited no inhibitory activity in one of the screens, indicating that its identification as a hit should be viewed with some skepticism.
Because of Hsp90’s role in modulating proteins associated with signal transduction and protein shuttling, its inhibition has widespread and sometimes varied effects within a cell. Depending on the conditions, the physiological manifestations of this inhibition can also vary. Thus, Hsp90 inhibitors display not only anti-proliferative and anti-tumor properties, but they can also display anti-inflammatory, anti-metastatic [20,21,35], immunosuppressant, anti-viral [22], anti-bacterial, anti-fungal [23] and/or anti-malarial [24,25] properties depending on the system investigated. Another property of inhibitors that bind to Hsp90’s N-terminal ATP-binding domain is the induction of a robust heat shock response [10].
The significance of this screen, is that novel compounds not known to have any activity against Hsp90 or its co-chaperones can be implicated to exhibit multiple, seemingly unrelated, medically relevant biological activities. Subsequent to the screen, the literature was mined to identify reports on the physiological effect of potential compounds. As shown below, some of the compounds identified have been specifically shown to inhibit the activities of proteins known to be dependent upon Hsp90 for their function [9,10,20]. These proteins include Akt, STAT-3, Her2 (ErbB2), Insulin-like Growth Factor Receptor (IGFR), Endothelial Growth Factor Receptor (EGFR), telomerase and others. Compounds reported to block the actions of these proteins, or their downstream signaling partners, such as NF-κB, are of special interest. Since Hsp90 is required for the activity of viral polymerases [22,36,37], anti-viral activity is another hallmark manifested by Hsp90 inhibitors [22]. VEGFR1 and 2 [38], and HIF1α [35] are also Hsp90-dependent clients, and as such Hsp90 inhibitors are accordingly anti-angiogenic [39]. Even though they have not been identified as Hsp90 inhibitors, many of the compounds identified from this screen belong to structural families that contain known Hsp90 inhibitors. Below are examples of the biological activities manifested that make each class of compounds, or specific compounds, potential candidates as Hsp90 inhibitors.
3.1.1. Sesquiterpene Lactones, Tetracyclic Sesterpenoids and Sesterpines
Some of the compound hits in this screen belong to the sesquiterpene lactone family of compounds (Figure 2, Table 1). These molecules are characterized by a fifteen-carbon skeleton formed by the union of three isoprene units that contain a lactone group. These compounds are present in many types of plants, and have long been used for various purposes in traditional medicine. Given their effectiveness in the treatment of a wide variety of ailments, and their observed activity on multiple cellular functions and molecular targets, these compounds represent promising candidates as Hsp90 inhibitors.
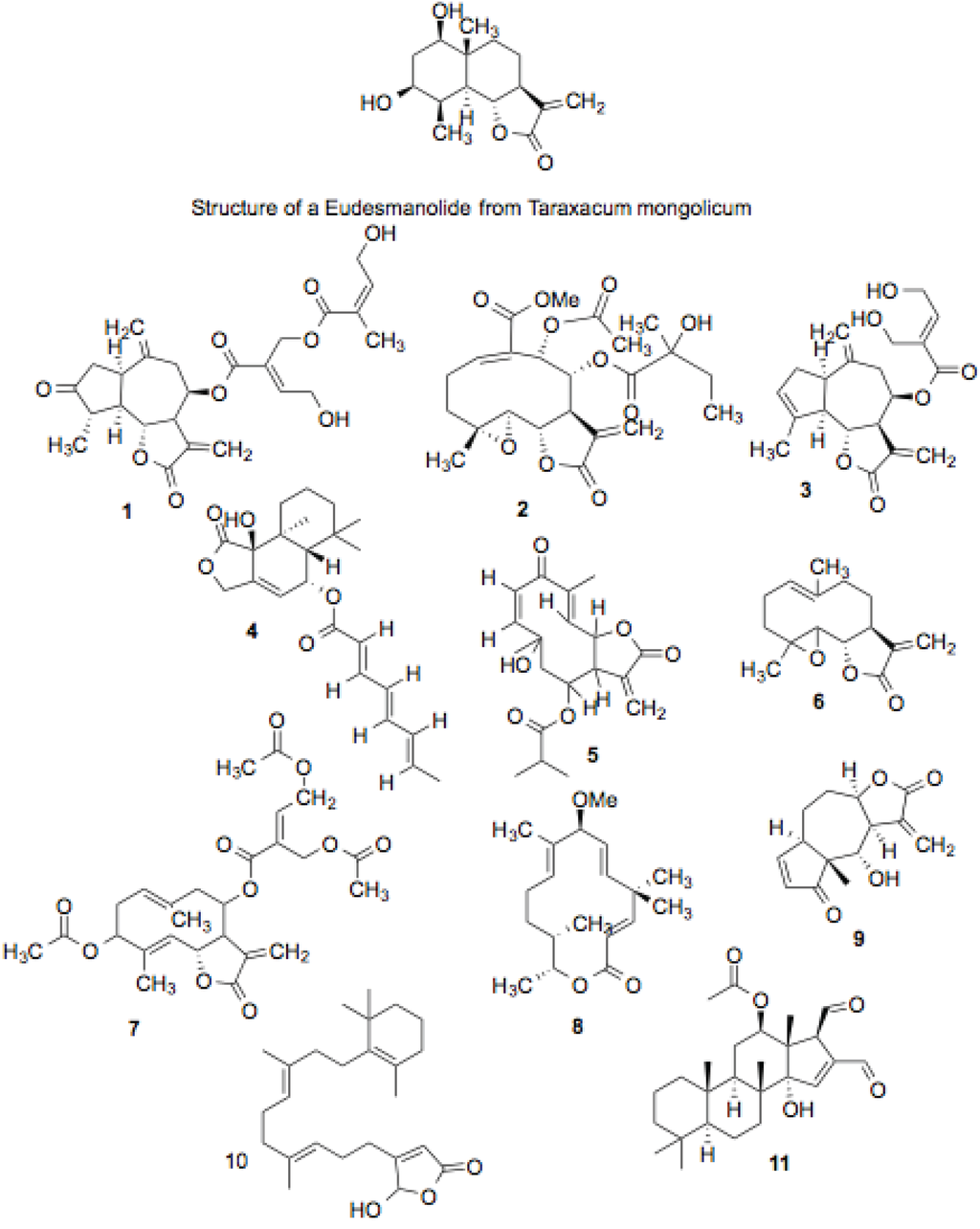
Figure 2.
Sesquiterpine lactones, tetracyclic sestorpenoids and sesterpines.
Sesquiterpene lactones have been grouped into seven general classes according to their structures. They are germacranolides, eudesmanolides, eremophilanolides, guaianolides, pseudoguaianolides, hypocretenolides, and iso-seco-tanapartholides. Compounds with reported biological activity come from all of the groups, although the germacranolides, guaianolides, and pseudoguaianolides appear most prominent [40].
Several sesquiterpene lactones with structures similar to those shown in Figure 1 have demonstrated biological activities. Isodeoxyelephantopin, and its nearly identical analog, deoxyelephantopin, were shown to inhibit the proliferation of mouse fibroblast tumor cells. The two compounds also inhibited DNA replication in both proliferating lymphocytes and tumor ascites [41]. Another pair of compounds fitting into this family, costunolide and eremanthin that have structures similar to compounds 6 and 7, were extracted from the ornamental plant Costus speciosus, and displayed anti-fungal activity similar to the standard anti-fungal, fluconazole, against two species of Trichophyton, and somewhat weaker activity against other fungi [42]. As nitric oxide is a mediator of inflammation, a compound’s effect on nitric oxide production is also an indicator of its anti-inflammatory potential, which is especially relevant to this study, as nitric oxide synthase is a well-known Hsp90 client. Eudesmanolides isolated from Taraxacum mongolicum inhibit nitric oxide production in RAW 264.7 mouse macrophages [43].
Compound 6, parthenolide, is from a different class of sesquiterpene lactones, but presents a similar structure. Additionally, it contains a methane group at the same location as the previously mentioned compound, a functional group that is the sole distinguishing feature from another, less active compound in the same study. Compound 4, an eudesmanolide (MEGxm0_000041), contains a similar moiety at its core, but it also contains an unsaturated eight-carbon fatty acid ester. Two compounds from the plant Eupatorium lindleyanum, eupalinolide A and eupalinolide B, are of the germacranolide sub-class. They induced expression of several heat shock proteins, including Hsp70 and Hsp90, in mouse squamous cell carcinoma and melanoma cells. The compounds were also shown to activate HSF1 [44], a potential indicator of Hsp90 inhibition.
Compound 6, parthenolide, has been identified as an anti-tumor and anti-inflammatory agent, and is currently in clinical trials along with several other sesquiterpene lactones for acute myeloid leukemia, acute lymphoblastic leukemia, and other types of blood and lymph node cancers [40]. Parthenolide’s anti-cancer and anti-inflammatory activities have been attributed to multiple mechanisms. It was shown to inhibit the activation of NF-κB by IKK, even when the kinase was constitutively active [45]. It was also able to sensitize TRAIL-resistant cancer cells by inhibiting STAT3 activation [46].
Compound 9, helenalin, suppresses NF-κB activation, promotes ROS generation, and induces apoptosis by bypassing Bcl-2 function [47]. Helenalin is cytotoxic against a number of cancer cells, and it also manifests immunosuppressant and anti-inflammatory activity [48]. In addition, helenalin is a potent inhibitor of telomerase [49], which further supports its potential as an Hsp90 inhibitor. Compound 3, also belongs to this class of compounds, and presents a similarity in the attached moieties.
Grouped with these compounds are two additional hits. Luffariellolide, a sesterterpene from a marine sponge, is cytotoxic to breast cancer cells, and inhibits the activation of the Hsp90-dependent protein HIF-1α [50]. Compound 11, 12-epi-scalardial, a tetracyclic sesterpenoid, inhibits EGFR-mediated activation of Akt [51].

Table 1.
Sesquiterpine lactones, tetracyclic sesterpenoids and sesterterpenes.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 1 | Guaianolide | ~60 | |
| 2 | Germacranolide | ~40 | |
| 3 | Guaianolide | ~40 | |
| 4 | Eudesmanolide: MEGxm0_000041 | ~60 | Cytotoxic to L5178Y lymphoblastic, PC12 neuroendrocrine, HeLa cervical cancer cells [52] |
| 5 | Tagitinin C | ~10 | Anti-malaria [53] |
| 6 | Parthenolide | >100 | Anti-tumor, anti-inflammatory [40,45,46] |
| 7 | 17-C4 | ~30 | |
| 8 | 16-H2 | ~10 | |
| 9 | Helenalin | ~80 | Inhibition of NF-κB and suppression of Bcl-2-mediated resistance to apoptosis [47]; anti-leukemic [54]; inhibition of telomerase [49]; induction of autophagy and cell cycle arrest [55] |
| 10 | Luffariellolide | ~10–20 | Cytotoxic, inhibits HIF-1α [50] |
| 11 | Scalaradial, 12-epi- | ~40–60 | Inhibits EGFR activation of Akt independent of PLA2 [51] |
3.1.2. Polyphenols
Polyphenols are defined as compounds that contain multiple phenolic moieties and often are poly-hydroxylated. This family of compounds is large, and contains multiple subtypes. Polyphenols generally exhibit anti-oxidant activity and can protect against ROS in vitro. The actual mechanisms behind these activities, however, have not been fully evaluated. Several of the compounds shown in Figure 3 and listed in Table 2 inhibit Hsp90-dependent proteins, and manifest cytotoxic, anti-proliferative, anti-inflammatory, and anti-viral activities, among others, that might be explained by their ability to inhibit Hsp90. Theaflavin (compound 15) has recently been reported to be an inhibitor of Hsp90 [56].
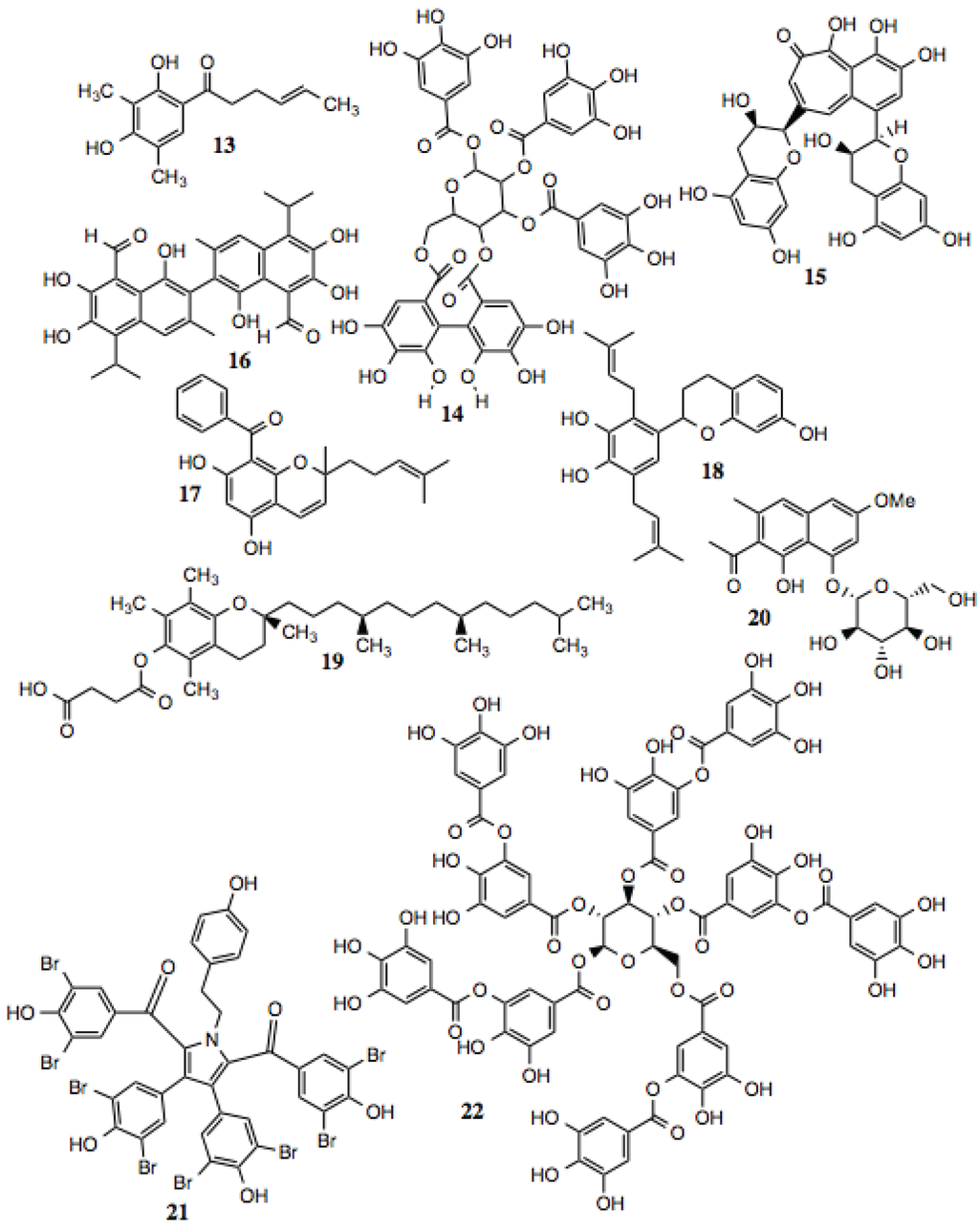
Figure 3.
Polyphenols and related compounds.
Table 2.
Polyphenols and related compounds.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 13 | 2'3'-dihydrosorbicillin | >100 | Moderate cytotoxic activity against cancer cell lines [57]. |
| 14 | Tellimagrandin II | ~70 | Anti-HIV [58]; suppression of sarcoma tumor cell growth [57]. |
| 15 | Theaflavin | ~25 | Anti-viral, anti-inflammatory [59]; anti-proliferative against leukemia cells via Akt down-regulation via Hsp90 inhibition [56]; inhibits NF-κB and MAPK signaling [60]; |
| 16 | Gossypol | ~50 | Anti-oxidant; broad anti-cancer activity; anti-viral; anti-protozoan; anti-bacterial; contraceptive [61]. |
| 17 | Similar to catechin (CID 10670714) | ~50 | |
| 18 | Flavan-3-ol (AC1MR5D9, CID 3512639) | ~60 | |
| 19 | (+)-α-Tocopherol acid succinate | ~50 | |
| 20 | Torachrysone 8-O-Glucoside (CID 11972479) | ~25 | Anti-oxidant [62]; inhibits LPS-induced NO production and NF-κB activation [63]. |
| 21 | Polycitone A | ~10–20 | |
| 22 | Tannic acid | ~5 |
3.1.3. Flavonoids
Flavonoids (Table 3, Figure 4) represent a large and diverse group of compounds that reside within the class of polyphenols. Flavonoids comprise approximately one half of all identified polyphenolic compounds. Aside from their aromaticity, the molecules have no unifying characteristic, except that they contain two or more six-membered rings, as well as at least one oxygen atom in the form of an ether or ketone. Many of these compounds contain multiple ketones and hydroxyl groups. This family of compounds is abundant in a number of substances used in traditional medicine. These substances have been shown to exhibit activities against allergy, inflammation, infection, tumors, diarrhea, and others. They have also been credited with wound healing and other beneficial properties. As ubiquitous as flavonoids are in plants, they are found in many foods. Examples are quercitin, epigallocatechin gallate (EGCG), resveratrol, and others.
Several hits from the screening belong to the flavonoid family (Table 3, Figure 4). Some of the compounds contain the typical bicyclic core along with a benzene ring fused to a pyran or pyrone, as well as a phenyl group attached to the flavan, isoflavan, or neoflavan. They also contain phenyl or aliphatic groups of varying saturation and oxygen incorporation. Additionally, some of the compounds fall into the subgroup of flavonoids known as chalcones (Table 4, Figure 5), which are metabolic precursors to flavonoids. These chalcones are characterized by two benzenes bridged by a 2-propen-1-one group.
Flavonoids are found throughout the plant kingdom. They have been used for the treatment of disease for centuries. Many flavonoids also demonstrate anti-microbial activity. Argentinian folk medicine has made use of a plant containing the glycosylated flavonol, quercetagetin-7-arabinosyl-galactoside, for the treatment of infectious diseases [64]. In another study, epigallocatechin gallate, a type of flavonoid found abundantly in green tea, demonstrated strong anti-bacterial activity, resulting from damage to the lipid bilayer [65]. However, EGCG has also been shown to bind Hsp90 and to induce degradation of Hsp90-dependent substrates [66].
Table 3.
Flavonoids.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 23 | Glabranine (CAS 41983-91-9) | >75 | Anti-viral [67]. |
| 24 | 5-Methoxyflavone | ~100 | |
| 25 | MolPort-005-945-561 (CID 4560115) | ~70 | |
| 26 | 2'-hydroxy-b-naphtho-flavone | ~350 | |
| 27 | 7,8-dihydroxy-2-(2-hydroxyphenyl)chromen-4-one | ~10 | |
| 28 | Biochanin A (5,7-dihydroxy-3-(4-methoxyphenyl)-chromen-4-one) | >90 | Anti-proliferative, anti-inflammatory, cytotoxic; inhibits iNOS expression, MAPK phosphorylation and NF-κB activation [68]. |
| 29 | 2',3',6-Trimethoxyflavone (2-(2,3-Dimethoxyphenyl)-6-methoxy-4H-chromen-4-one) | >90 | |
| 30 | 3',4'-Dimethoxy-3-hydroxy-6-methylflavone | ~30–80 | |
| 31 | Luteolin | >100 | Induction of unfolded protein response and apoptosis in neuroblastoma [69]; inhibits LPS-activated, Akt-mediated activation of NF-κB in macrophages [70]. Anti-tumor activity through EGFR pathway suppression in breast cancer cells [71]. Shown to inhibit Hsp90 [72]. |
| 32 | Mangostin [1,7-bis(3-methylbut-2-enyl)-3,6,8-trihydroxy-2-methoxy-xanthen-9-one] | ~60 | Xanthanoid–Induces cell cycle arrest and apoptosis in colon [73] and prostate cancer cells [74]. Blocks activation of MAPK and Akt pathways [75]. |
| 33 | MolPort-001-742-269 (CID 38356110) | ~65 | |
| 34 | 6-Hydroxy-7-methoxyflavone | ~45 | |
| 35 | Gambogic acid | ~2 | Demonstrated to inhibit Hsp90 [33,76]. |
| 36 | Tetrahydrogambogic acid | ~10 | |
| 37 | Dimethyl Gambogate | ~2 | |
| 38 | Derrubone | ~0.2 | Inhibitor of Hsp90 [77]. |
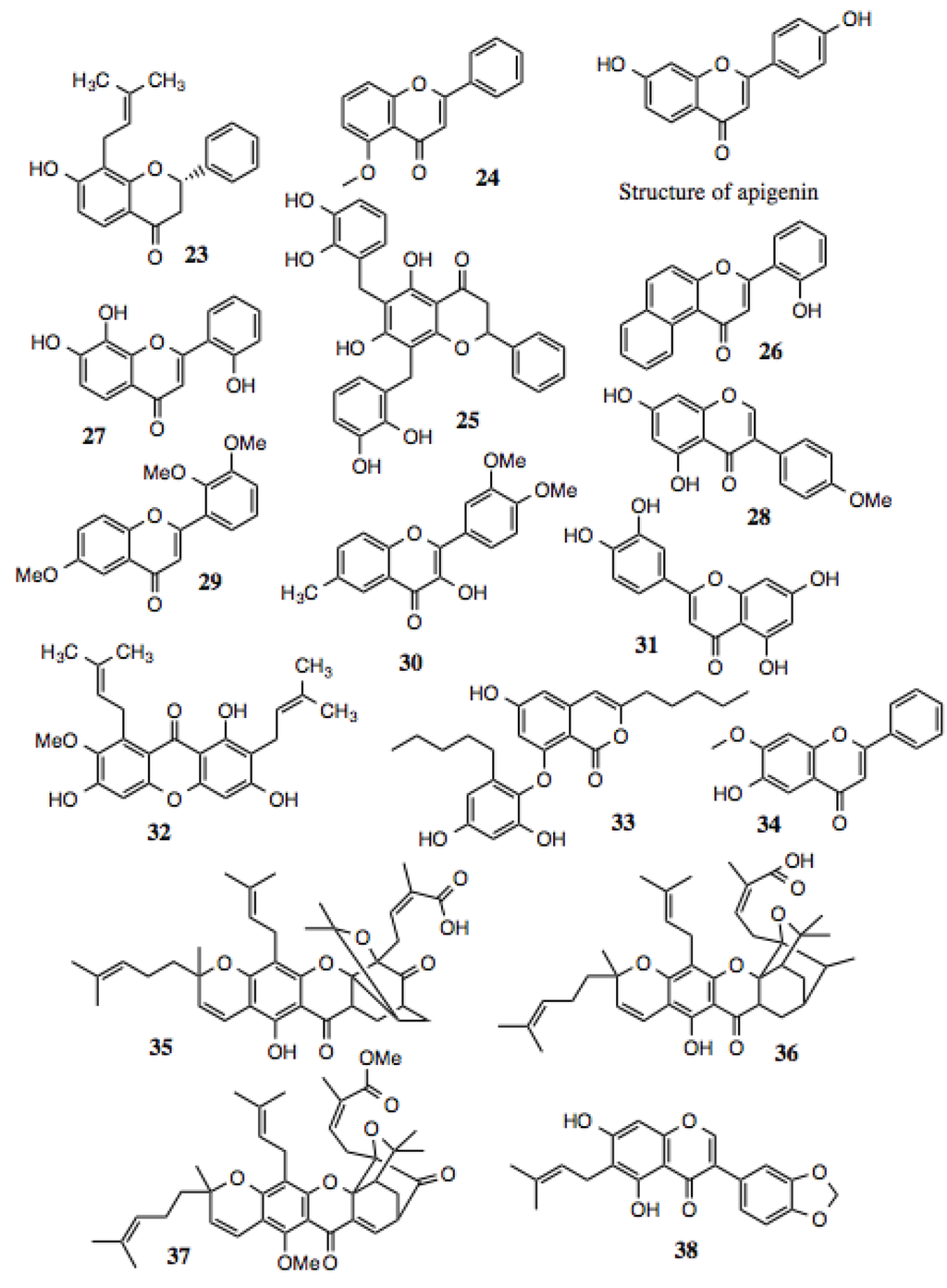
Figure 4.
Flavonoids and related compounds.
Over the course of twenty years, apigenin, a simple flavone, was assayed for its anti-bacterial activity, and was found active against more than fifteen types of pathogenic bacteria, including S. aureus, MRSA, E. coli, P. aeruginosa, and K. pneumonia [78]. In addition to anti-microbial activity, these compounds also demonstrate anti-cancer activity. For example, apigenin (Figure 3) exhibited strong in vitro anti-tumor and anti-angiogenic activity against human lung, prostate, and ovarian cancer cells. In each of these cases, the expression of VEGF and HIF-1alpha were suppressed [79,80], both of which are Hsp90-dependent clients.
Among the hits from this screen, compounds 23, 24, 27, 29, 30 and 34, contain the same flavone core as apigenin. Two contain a flavonone core that is nearly identical, except for saturation of the C-2 double bond found in flavones. The physiological effects of several of the flavonoid hits implicating them as possible Hsp90 inhibitors are noted in Table 3.
3.1.4.Chalcones
Chalcones are a structurally distinct subclass of flavonoids from which several hits were identified (Table 4, Figure 5). Chalcones share many of biochemical characteristics with other flavonoids, as they exhibit anti-fungal [81], anti-inflammatory [82], anti-tumorogenic [83], anti-HIV, and anti-plasmodial activities [84], amongst others (Table 4). A number of cellular proteins were identified as targets for the chalcones, many of which are known to be dependent upon Hsp90 (Table 4), including Akt, NF-κB, mTOR, STAT3, HIF-1α, iNOS, and others [85].
3.1.5. Pentacyclic Triterpenoids
Three pentacyclic triterpenoids were identified in the screening (Table 5, Figure 6), celastrol, its methyl ester and anthothecol. Celastrol is a well-established Hsp90 inhibitor [86]. Anthothecol is a limonoid related to degunin. Degunin has also been identified as an inhibitor of the Hsp90 machine [87,88].
Table 4.
Chalone compounds.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 39 | Phloretin | ~35–90 | Induction of apoptosis in breast cancer cells via Bcl-xL degradation [89]. |
| 40 | Curcumin | ~70 | Anti-proliferative, anti-tumor, anti-inflammatory via suppression of NF-κB activation [90]; reported Hsp90 inhibitor [91]. |
| 41 | 2',4-Dihydroxychalone | ~120 | |
| 42 | [6-methyl-5, 7-dihydroxy-2,2-dimethylchromen-8-yl]-3-phenylprop-2-en-1-one Similar to rotterlin and catechin | ~60 | |
| 43 | (CID 193568) Corylifolinin; isobacachalone | ~50 | Inhibition of LPS-induced NO production [92]. |
| 44 | Dimethyl cardamonin (CID 10424762) | ~70 | Antibacterial; anti-fungal [93,94]; anti-proliferative via cell cycle arrest; anti-inflammatory via blocking NF-κB activation [95,96,97,98]. |
| 45 | Nordihydroguaiaretic acid | ~35–80 | Phase II study for effect on prostate cancer. Increased doubling time of PSA. Thought to inhibit IGF1R and HER2 [99]. Repressed breast tumor growth via mTORC1 inhibition [100]. |
| 46 | Violastyrene | ~20 | |
| 47 | Rottlerin | ~60 | Anti-proliferative [101,102]; cytotoxic to pancreatic cancer cells via PI3K/Akt/mTOR inhibition [103]; inhibits NF-κB; STAT and amyloid aggregation [104]. |
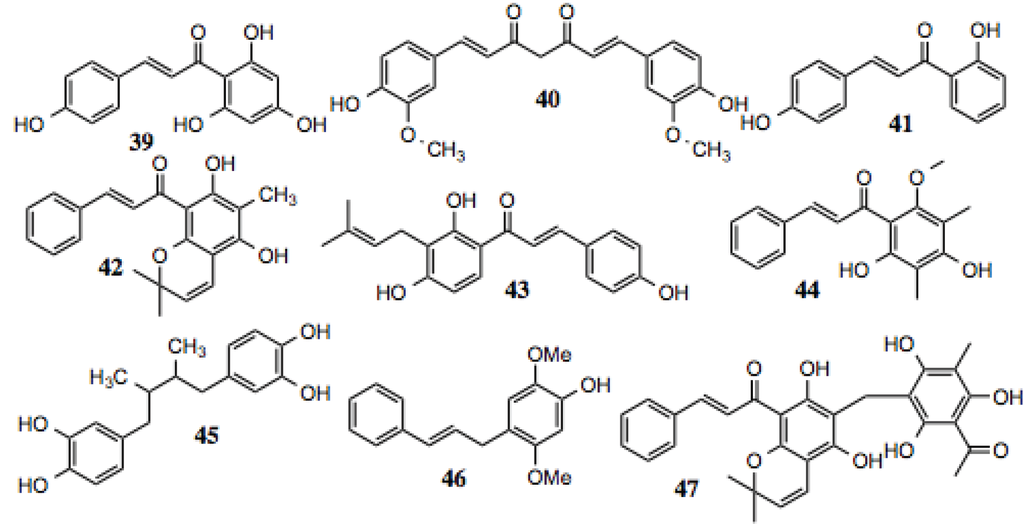
Figure 5.
Chalones and related compounds.
Table 5.
Pentacyclic triterpenoids.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 48 | Celastrol | ~2 | Hsp90 inhibitor [86]. |
| 49 | Celastrol methyl ester | ~2 | |
| 50 | Anthothecol | ~6 | Antimalarial [105]. |
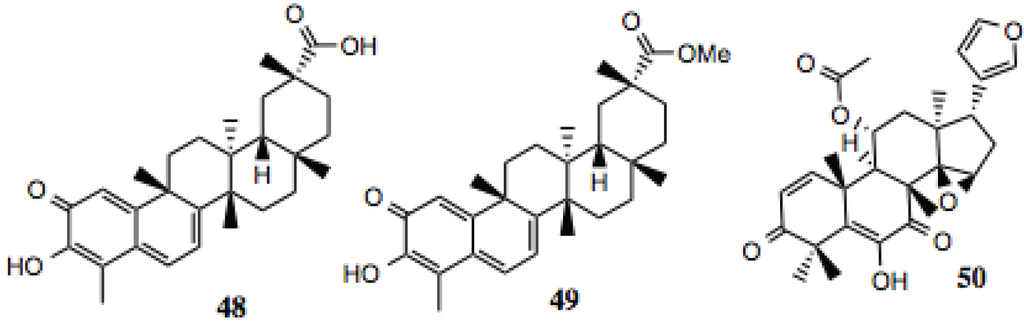
Figure 6.
Pentacyclic triterpenoids.
3.1.6. Alkaloids
Alkaloids are broadly defined as containing basic nitrogen atoms within their ring structures. As this definition includes a large number of potential compounds, the family is further broken down into smaller subdivisions. Regardless, the compounds within the family that have demonstrated biological activities are diverse, with no single structure or group of structures requisite for activity. For the purposes of this screen, alkaloids were regarded as molecules that contain a cyclic nitrogen atom. Similar to other groups identified in this screen, alkaloids (Table 6, Figure 7) demonstrate a wide range of medically relevant bioactivities, which include anti-tumor, anti-hypertensive, anti-depressant, anti-microbial, anti-inflammatory, and other activities [106], as well as inhibiting the function of some well known Hsp90-dependent proteins (Table 6). A well-known example of a medicinal alkaloid is quinine (Figure 7), isolated from the tropical medicinal plant Cinchona succirubra, which has been used to treat malaria for hundreds of years [53].
3.1.7. Benzylisoquinoline Alkaloids
Benzylisoquinoline alkaloids (Table 7, Figure 8) comprise a subset of alkaloid compounds characterized by an aromatic, tetracyclic skeleton, containing three benzene moieties, and a fourth cycle containing the alkaloid nitrogen. Most of the compounds shown in Figure 7 are aporphines that are highly similar, and differ only by the presence or location of a hydroxyl, methoxy, or keto group. Some, however, incorporate an additional ring, or exist as a dimer of two aporphine molecules. While there is little known about most of the aporphine compounds identified in this screen, some have no prior designation. One example is glaucine, which manifests a host of activities in vitro, including relaxation of bronchia via inhibition of its contraction, reduction in superoxide generation in stimulated polymorphonuclear leukocytes and eosinophils, reduction of elastase release, leukotriene production, and intracellular Ca2+ in PMN’s, platelet aggregation, and eosinophil peroxidase release. These effects make glaucine a likely candidate for the treatment of bronchiodilation and inflammation [107].
Table 6.
Alkaloid compounds.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 51 | Peganine (CID 72610) | ~130 | Modest anti-proliferative and cytotoxic activity [108]; anti-mycobacterial [109]; anti-leishmania [110,111]. |
| 52 | Gliotoxin acetate (CID 21127802) | ~15 | Anti-viral; anti-mycobacterial; inhibition of NF-κB; anti-leukemic; anti-tumor [112,113,114]. |
| 53 | 19-A7: (Z)-4-isopropyl-1-(2-methylpropylidene)-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione | ~60 | |
| 54 | (+)-Cinchonine | ~30 | Circumvention of P-glycoprotein mediated multi-drug resistance [115]. |
| 55 | (2S,3R,3aS,9aR)-2-(hydroxymethyl)-6-imino-2,3,3a,9a-tetrahydro-6H-furo[2',3':4,5]oxazolo[3,2-a] pyrimidin-3-ol | ~50 | |
| 56 | (−)-Eseroline | ~50–90 | |
| 57 | Metacycloprodigiosin; Streptorubin | ~10 | Cytoxic activity against cancer cell lines [116,117]. |
| 58 | Fredericamycin | ~3 | Cytotoxic, anti-bacterial, anti-fungal, anti-tumor activities; topoisomerase inhibitor; cell cycle arrest [118,119]. |
| 59 | Tryptanthrin | ~15 | Induced apoptosis in human leukemia cells; suppression of NO production [120]. |
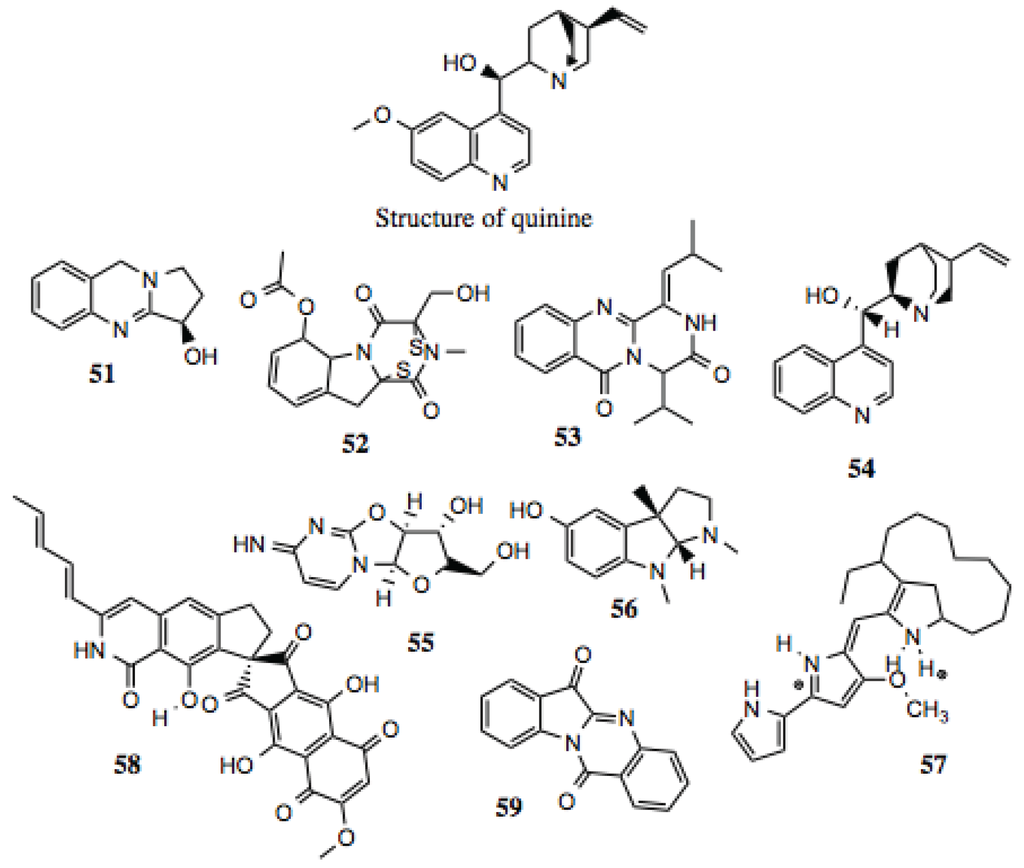
Figure 7.
Alkaloid compounds.
Aporphines also demonstrate in vitro anti-viral activity. A number of compounds inhibit polio-virus infection of cultured mammalian cells by 50% at low micromolar concentrations [121,122]. Additionally, the aporphines dicentrine, glaucine, corydine, and apomorphine, which are analogs of the aporphines presented in this study, demonstrate anti-proliferative activity against mouse leukemia, melanoma, bladder cancer, and colon cancer cells [123].
As discussed earlier, Hsp90 contains a distinct ATP-binding domain, the specificity of which has been exploited for the development of inhibitors. This domain contains a Bergerat fold, and is shared by only a few protein families, which include DNA gyrase, a type II topoisomerase. Some aporphines are inhibitors of topoisomerase II, increasing the likelihood that highly similar compounds from this screen are also Hsp90 inhibitors. One such example is liriodenine (Figure 7), which manifests activity against human cancer cells [124], Gram-positive bacteria, yeast, and fungi [125,126,127].
Table 7.
Benzylisoquinoline Alkaloids.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 60 | Sanguinarine | ~70 | Induces cell cycle arrest and apoptosis in human cervical cancer cells [128]; anti-inflammatory [129]; anti-fungal [130]. |
| 61 | 3-hydroxymethyl-glaucine | ~80 | |
| 62 | Thaliporphine; thalicmidine | ~9 | Inhibits the activity of LPS-induced NOS; inhibits LPS-induced nuclear translocation of NF-κB [131]. |
| 63 | Isoboldine | ~30–80 | Anti-viral [121]. |
| 64 | Bracteoline | ~80 | |
| 65 | 7-oxoglaucine | ~80 | Anti-plasmodial [132]; anti-inflammatory [133]. |
| 66 | 7-Formyl-dehydroglaucine | ~1–3 | |
| 67 | Dehydroglaucine | 0.9 | Anti-microbial, some anti-fungal activity [126]. |
| 68 | Dehydroglaucine dimer | 0.1–0.4 | |
| 69 | Glaucine derivative | >30 | |
| 70 | Dehydroglaucinylphenone | ~7–20 | |
| 71 | Berberine | ~40 | Anti-tumor, anti-metastatic, inhibits HIF1α [129,134,135,136]. |
| 72 | 3-Formyl-glaucine | ~8–20 | |
| 73 | Dihydrosanguinarine derivative | ~40 | Anti-fungal [130]. |
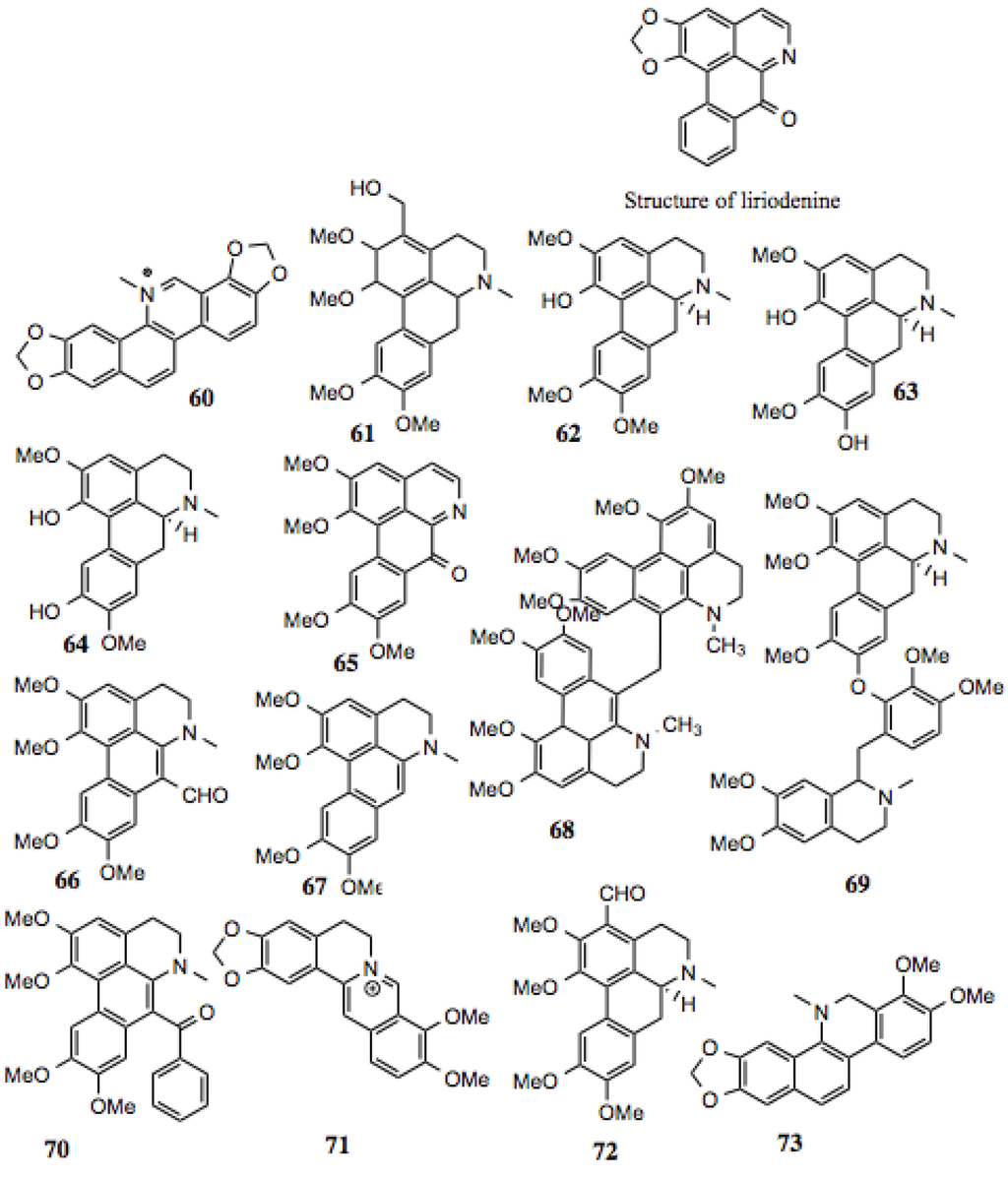
Figure 8.
Benzylisoquinoline Alkaloids.
3.1.8. Cyclic Peptides
The McAlpine Laboratory has investigated the anti-cancer and Hsp90-inhibiting activities of naturally occurring macrocyclic peptides. These molecules and their derivatives have demonstrated modest activity against a number of cancers [137]. The rationale behind the pursuit of these particular scaffolds is that similar compounds have been identified as antibiotics and anti-fungals, while maintaining anti-cancer activity. The polypeptide nature of these compounds may confer the ability to mimic hydrophobic portions of client proteins. Four of the compounds identified from this screen (Table 8, Figure 9) contain moieties that could mimic hydrophobic amino acids. Of particular interest is antibiotic A83586 C (compound 75), which manifest potent anti-proliferative activity against cancer cells [138].
Table 8.
Cyclic peptides.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 74 | Tyrothricin | ~5 | Antibiotic [139]. |
| 75 | Antibiotic A83586C | ~10 | Anti-proliferative activity against cancer cells [138]. |
| 76 | P12 | ~10 | |
| 77 | Cyclopeptide L-156373 | ~10 |
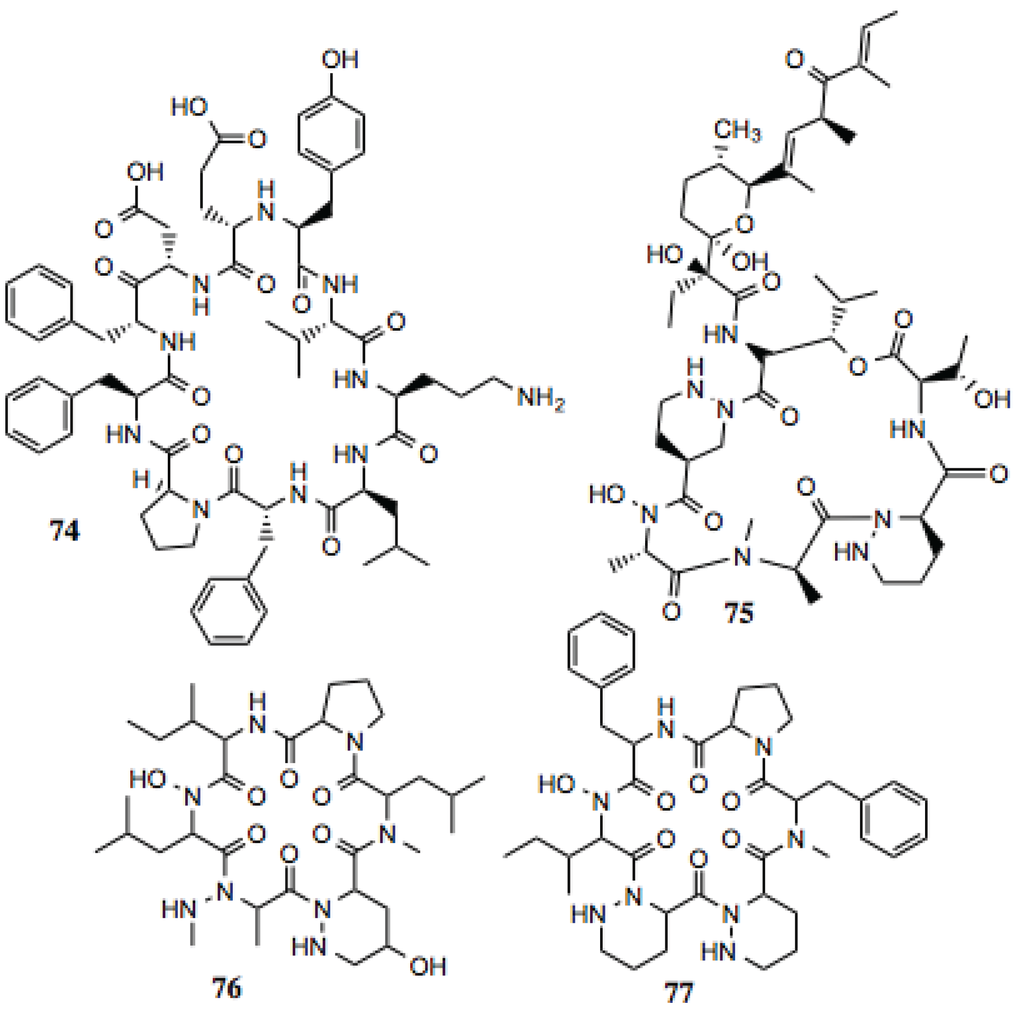
Figure 9.
Cyclic peptides.
3.1.9. Quinones
Quinones represent an extremely large and diverse family of compounds. Essentially, the only thing that differentiates a quinone from other classes of compounds is the presence of two keto groups on an unsaturated six-membered ring. As such, an enormous array of functional and structural groups that decorate this motif is possible. Generally, quinones are redox-active, making them promising compounds with which to treat cancer, but also potential liabilities that may arise as a consequence of this activity. The production of reactive oxygen species resulting from exposure to quinone-containing compounds is a process that is potentially destructive to any cell. It has not been established whether the redox potential of these compounds contributes to the anti-cancer activity by increasing their ability to inhibit Hsp90, or by increasing cellular stress alongside Hsp90 inhibition. It should be noted, that reduction of 17-DMAG and 17-AAG to the corresponding hydroquinones resulted in increased Hsp90 inhibitory activity. [140]
Some well-established Hsp90 inhibitors, such as geldanamycin (Figure 10) and its derivatives, contain quinone moieties. Consequently, we have observed reticulocyte lysate treated with these inhibitors to possess a distinctive dark red color, attributable to met-hemoglobin formation resulting from the oxidative activity of such compounds. Similarly, geldanamycin has been shown to generate reactive oxygen species in vitro and in cell culture [141]. Structural studies demonstrate direct binding of these compounds to the ATP-binding site at the N-terminus of Hsp90. Not surprisingly, the antibiotic, rifamycin, contains a reduced quinone moiety within a similar anthroquinone ansamycin structure, and has similar activity as geldanamycin. Seventeen quinones were identified in this screen (Table 9, Figure 10). As indicated in Table 9, a number of these compounds display anti-cancer, anti-trypanosomal, anti-viral, and anti-inflammatory activities, as well as having the capacity to inhibit the activities of several well-known Hsp90-dependent proteins.
3.1.10. Other Compounds
Compounds identified in this section (Figure 11, Table 10) do not fit well into any of the previously described families. Some of these are known biochemicals, such as Vitamin D2, 9-cis-retionioc acid, prostaglandin J2 and L-adrenaline, and have not been implicated as Hsp90 inhibitors, although Vitamin D2 [142,143] and retinoic acid [144,145] have demonstrated anti-cancer activity. Others are more exotic and little, if anything is known about their mechanism of action. Recently, the anti-cancer activity of hypericin has been tied to its ability to inhibit Hsp90 stabilization of HIF-1α [146].
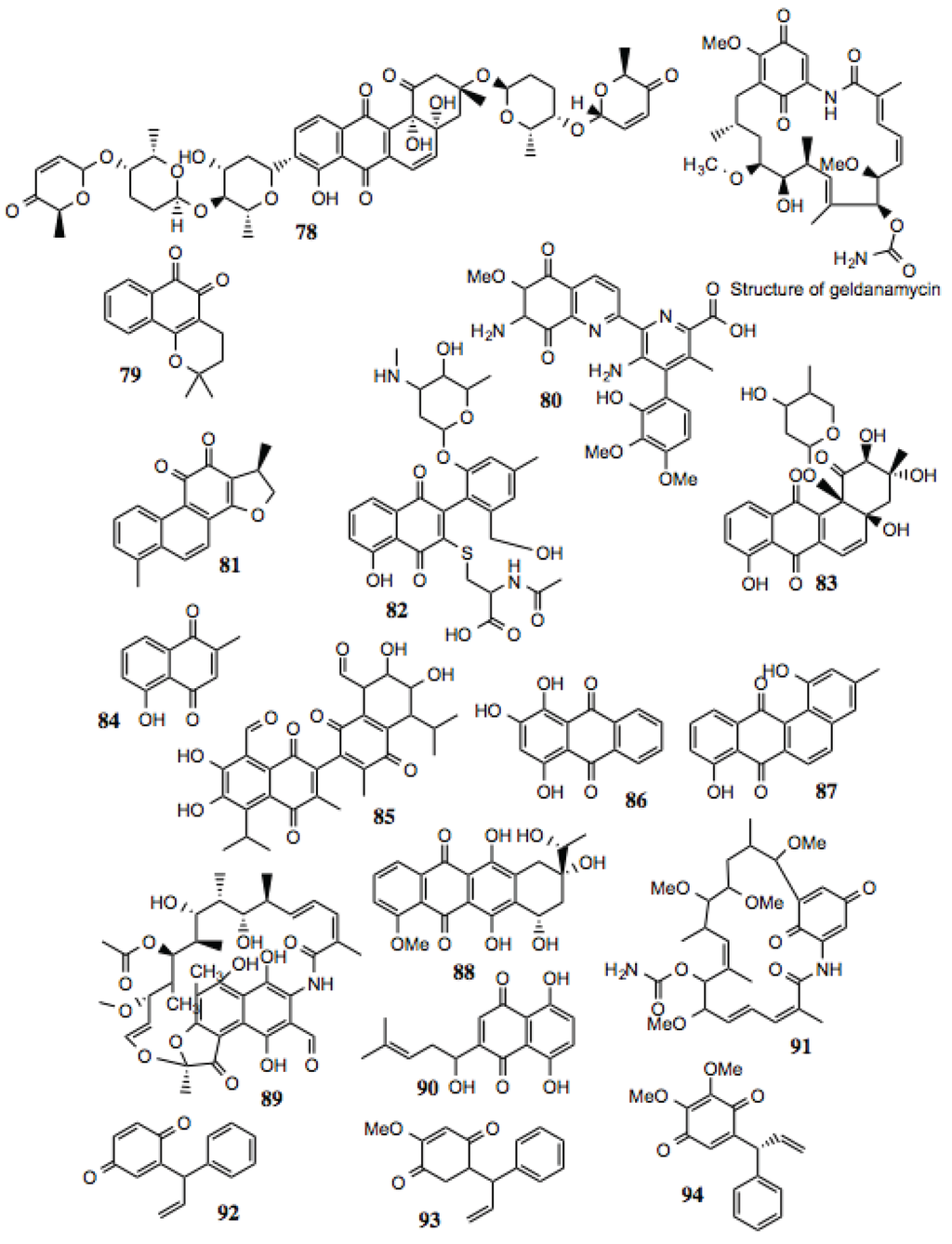
Figure 10.
Quinones.
Table 9.
Quinones.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 78 | Vineomycin A1 | ~100 | |
| 79 | β-Lapachone (CID 3885) | ~20 | Anti-cancer; anti-trypanosomal activity; anti-viral [147,148,149,150,151,152,153,154,155]. |
| 80 | Streptonigrin (CID 5351165) | ~15 | Disrupts NF-κB activation; antibacterial, antifungal, antiviral, anti-glioma [156,157,158,159,160,161]. |
| 81 | 15.16-Dihydrotanshinone | ~80 | Anti-cancer; inhibition of HIF-1α; depletion of Bcl-2 [162,163,164]. |
| 82 | 2-A5: 1,4-Napthoquinone derivative | ~10 | |
| 83 | 30-D10: 9,10-Anthraquinone derivative | ~2 | |
| 84 | Plumbagin | <0.05 | Widely studied anti-cancer activity; reported to target EGFR, STAT-3, Akt, and NF-κB pathways [165,166]. |
| 85 | 7-[8-formyl-6,7-dihydroxy-3-methyl-5-(methylethyl)-1,4-dioxo(2-naphthyl)]-2,3- dihydroxy-6-methyl-4-(methylethyl)-5,8-dioxonaphthalenecarbaldehyde | ~6 | |
| 86 | 1,2,4-trihydroxyanthracene-9,10-dione | ~40–100 | |
| 87 | Tetrangulol G2 | ~20 | |
| 88 | Dihydodaunomycinone; Leukaemomycin-D | >60 | |
| 89 | 3-Formyl Rifamycin SV | ~15–35 | Inhibition of Vaccinia virus assembly [167]. |
| 90 | Shikonin | ~75 | Anti-inflammatory, anti-tumor, and wide-ranging activities reported [166,168]. |
| 91 | Herbimycin | ~40 | Hsp90 inhibitor [1,169,170,171]. |
| 92 | Dalbergione | ~2 | |
| 93 | 4'-Methoxydalbergione | ~5 | Anti-trypanosomal [172]. |
| 94 | 3'4'-Dimethoxydalbergione | ~5 | Anti-trypanosomal [172]. |
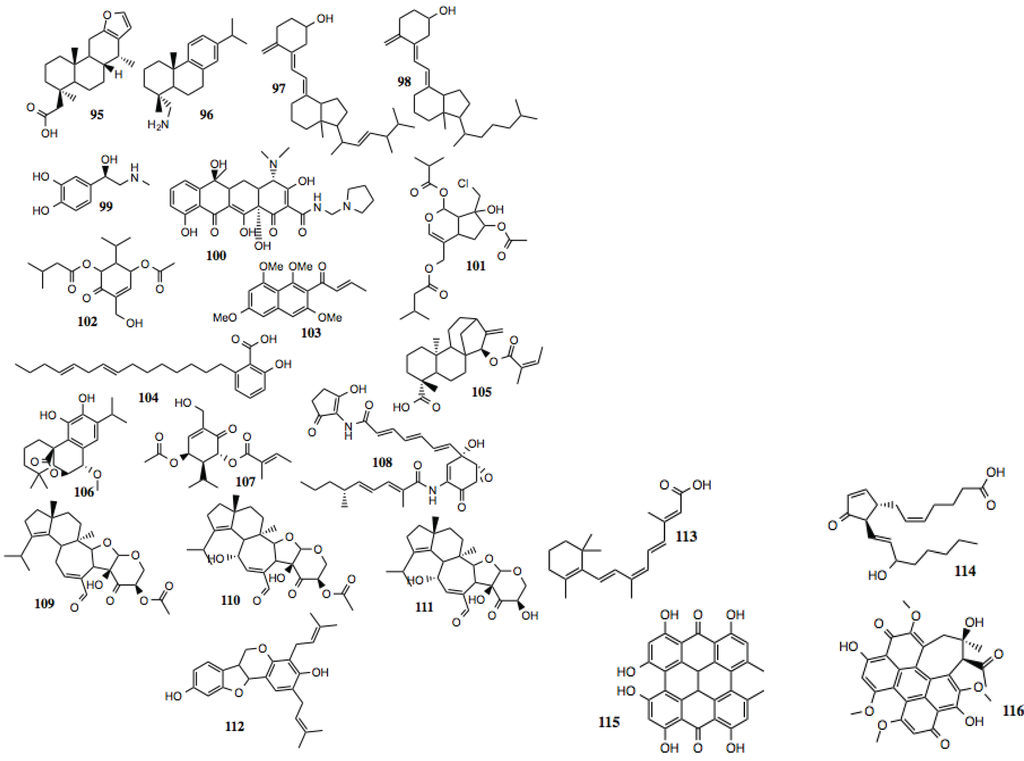
Figure 11.
Other miscellaneous compounds.
Table 10.
Other compounds.
| # | Location and/or name | IC50 (µM) | Properties |
|---|---|---|---|
| 95 | 2-((4R,6aS,7R,11bR)-4,7,11b-trimethyl-1,2,3,4,4a,5,6,6a,7,11,11a,11b-dodecahydro-phenanthro[3,2-b]furan-4-yl) acetic acid | >75 | |
| 96 | (+)-Dehydroabietylamine; Leelamine | ~30–75 | |
| 97 | Vitamin D2 | ~60 | |
| 98 | Vitamin D2 metabolite | ~60 | |
| 99 | L-Adrenaline | ~55 | |
| 100 | Rolitetracycline | ~20 | |
| 101 | MolPort-005-945-572 | ~50 | |
| 102 | CID 45359640 | ~50 | |
| 103 | (E)-1-(1,3,6,8-tetramethoxy- naphthalen-2-yl)but-2-en-1-one | ~5 | |
| 104 | CID 53984538: 8,11- anacardic acid | ~10 | Anti-cancer; induction of the UPR [173,174]. |
| 105 | (4R,7R,11bS)-4,11b-dimethyl-7-(((Z)-2-methylbut-2-enoyl)oxy)-8-methylenetetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylic acid | ~40 | |
| 106 | (4bR,9S,10S)-3,4-dihydroxy-2-isopropyl-10-methoxy-8,8-dimethyl-6,7,8,8a,9,10-hexahydro-5H-9,4b-(epoxymethano) phenanthren-12-one | ~30 | |
| 107 | CID 13818684 | ~15 | |
| 108 | Manumycin A | ~30–50 | Inhibition of STAT-3 and telomerase; down-regulation of Akt and MEK; anti-cancer [175]; induces autophagy [176]. |
| 109 | Striatal A | ~2 | |
| 110 | Striatal B | ~2 | Inhibition of growth of multiple cancer cell lines- NCI cell line growth inhibition assay [PubChem CID 329431]. |
| 111 | Striatal C | ~2 | |
| 112 | CID 45360154 | ~50 | |
| 113 | 9-Cis-retinoic acid | ~30–50 | |
| 114 | Prostaglandin J2 | ~75–100 | |
| 115 | Hypericin | ~50–75 | Inhibits HIF-1α [146]. |
| 116 | Hypocrellin A | ~5–10 |
3.2. Further Characterization of Select Putative Inhibitors
The HTS of commercially available natural product libraries identified over a 100 compounds as potential Hsp90 Inhibitors. This raises the question inherent to hits identified in HTS: which of the compounds should one select for further characterization using secondary screens? Of these compounds, derrubone [77], compounds containing the 1,4-naphthoquinone scaffold [177], and gambogic acid [33] have been characterized, in addition to silybin [178], which was identified as an Hsp90 inhibitor by mining the literature. Based on the results from our high-throughput screen and data mining, anthothecol and rottlerin were selected for further characterization. In addition, during the search through the literature for natural products that displayed broad biological activities, garcinol, a gambogic acid-like compound, and piplartine/piperlongumine, a polyphenol chalcone-like compound were identified as potential Hsp90 inhibitors. An additional consideration in the selection of rottlerin, garcinol, and piplartine/piperlongumine was that the literature indicated that these compounds were being discussed as possible candidates for clinical trials.
Of the four compounds chosen for additional investigation, rottlerin (Figure 5 and Figure 12) is the best studied, and, as noted above has been discussed as a possible candidate for clinical trials [101]. It has been used in traditional medicine and demonstrates many physiologically significant biological activities. Rottlerin is isolated from the tropical tree Mallotus philippinensis and displays cytotoxicity against a number of cancer types, including lung, breast, lymphocytic leukemia, and multiple myelomas. While the activity of rottlerin was initially attributed to inhibition of PKCδ [101], this mechanism of action has largely been called into question [101,103]. Rottlerin appears to inhibit a combination of signal transduction pathways at multiple levels [101], making it a good Hsp90 inhibitor candidate. For example, an array of human malignant tumor cells was treated with rottlerin, and all lines were found to undergo apoptosis mediated by Death Receptor 5 (DR5) [179]. Rottlerin has also been reported to inhibit the kinases PRAK, MAPKAP-K2, Akt, and CaMK [180].
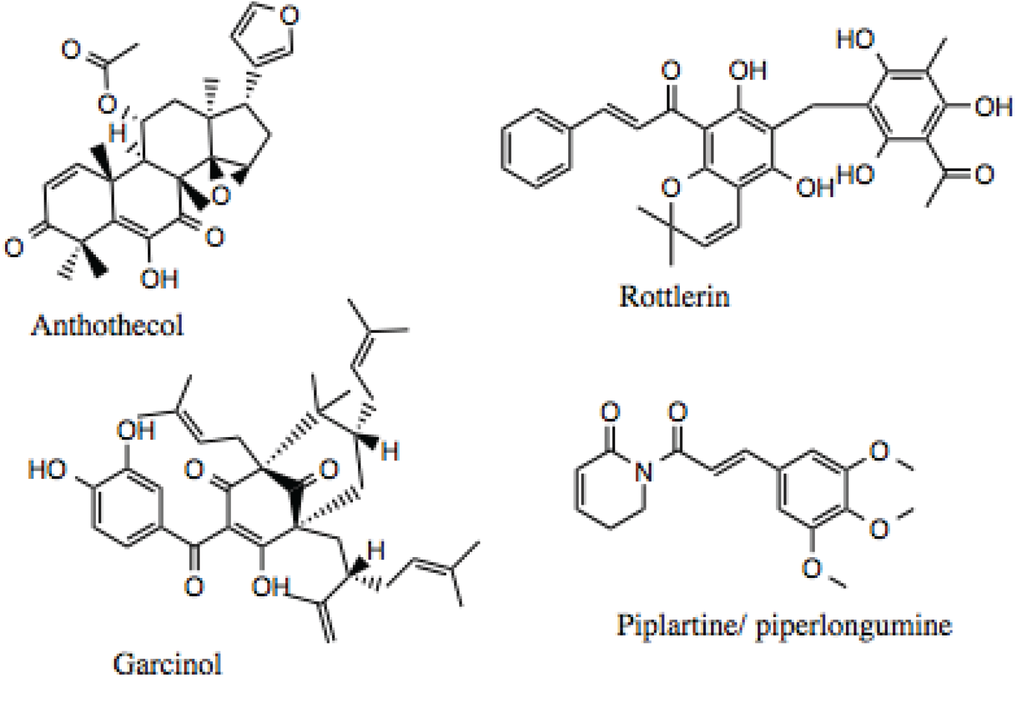
Figure 12.
Structure of anthothecol, rottlerin garcinol and piplartine/piperlongumine.
Anthothecol (Figure 6 and Figure 12) is a limonoid natural product isolated from the Khaya anthotheca tree, and possesses low micromolar activity against the growth of Plasmodium falciparum in erythrocytes [105], which is another property manifested by Hsp90 inhibitors. The compound’s structure is similar to that of the Hsp90 inhibitor, gedunin [88], but it has approximately 10-fold higher activity, as will be demonstrated below.
Garcinol (Figure 12) was chosen for further study, because it demonstrated the ability to induce apoptosis in a number of cancers, including breast, colon, kidney, prostate, leukemia [181], pancreatic [182], and others. The anti-cancer activity of garcinol has been attributed, in part, to its inhibition of STAT and NF-κB signaling [183,184]. Additionally, garcinol can inhibit angiogenesis through down-regulation of Prostaglandin E2, VEGF, and IL-8 [182], which are bonafide Hsp90-dependent clients. Garcinol also exhibits anti-oxidant and anti-inflammatory properties, as it inhibits the production of ROS and nitric oxide [185].
Piplartine (a.k.a., piperlongumine, Figure 12) has been shown to suppress platelet-derived growth factor (PDGF) signaling [186], and inhibit the proliferation of prostate cancer cells through depletion of the androgen receptor, a well-known Hsp90 client protein [187]. A recent review noted that the pharmacological activities for piplartine reported in the literature include cytotoxic, anti-tumor, anti-angiogenic, anti-metastatic, anti-bacterial, anti-fungal, anti-leishmanial, anti-trypanosomal, and anti-schistosomal activities among others [188].
In our study, we provide evidence that further implicates these compounds as inhibitors of the Hsp90 complex. We show that, in addition to inhibiting the proliferation of cancer cells, these compounds also inhibit the Hsp90-dependent folding of thermally denatured luciferase in a dose-dependent manner, block the Hsp90-dependent maturation of the heme-regulated eIF2α kinase (HRI), and deplete cells of Hsp90-dependent clients.
3.2.1. Inhibition of Hsp90-Mediated Refolding of Denatured Luciferase
Anthothecol and rottlerin were identified as potential Hsp90 inhibitors by their ability to inhibit refolding of thermally denatured luciferase in screens of natural compound libraries. To more accurately define their inhibitory activity, the compounds were titrated into the assay in a three-fold dilution series. The compounds inhibited refolding of luciferase in a concentration-dependent manner with low µM IC50s (Table 11). Similarly, garcinol, and piplartine display the ability to inhibit the refolding of luciferase at micromolar concentrations in reticulocyte lysate (Table 11), implicating them as Hsp90 inhibitors.
Table 11.
IC50 values for inhibition of luciferase refolding and proliferation of MCF7 cells.
| Compound | IC50 luciferase refolding (µM) 1 | IC50 MCF7 proliferation (µM) 2 |
|---|---|---|
| Anthothecol | 12 ± 2 | 0.5 ± 0.06 |
| Rottlerin | 63 ± 5 | 7 ± 3 |
| Garcinol | 12 ± 5 | 4 ± 0.6 |
| Piplartine | 80 ± 7 | 10 ± 3 |
1 Anthothecol, rottlerin, garcinol and piplartine were titrated into wells containing denatured luciferase and reticulocyte lysate. After a two-hour incubation period, assay buffer containing luciferin was added, and relative luminescence was measured. IC50 is the concentration of the compound that inhibited luciferase refolding by 50% compared to the DMSO control; 2 MCF-7 cells were treated in culture with anthothecol, rottlerin, garcinol and piplartine and DMSO as a control. Proliferation was assessed at 48 h using an MTS assay. Proliferation is defined as the colorimetric intensity difference between wells treated with DMSO and wells treated with the compounds.
3.2.2. Compounds Inhibit Proliferation of Human Cancer Cells
While piplartine, rottlerin and garcinol have well characterized anti-proliferative properties, there were no reports in the literature with regards to the anti-proliferative activity anthothecol. Therefore, the anti-proliferative activity of these four compounds on the growth of MCF7 cells was determined using the MTS assay (Table 11). In MCF-7 cells, all the compounds cause a 50% reduction in growth in the 0.5–10 µM range (Table 11). These results confirm the anti-proliferative properties of piplartine, rottlerin and garcinol, and establish anthothecol as anti-proliferative drug.
3.2.3. Inhibition of HRI Maturation
The heme-regulated eIF2α kinase (HRI) is an Hsp90 client kinase which, upon folding by Hsp90, will activate, or mature, by autophosphorylation when heme is deficient [189]. This activation is dependent on functional Hsp90, and can be detected as an electophoretic mobility shift [189] when separated by polyacrylamide gel electrophoresis (PAGE) (Figure 13). Similar to the known Hsp90 inhibitors, geldanamycin (GA), molybdate and novobiocin, anthothecol, garcinol, rottlerin, and piplartine inhibited the maturation of HRI, as observed by the absence of the slower mobility form of HRI upon PAGE. This result further supports the hypothesis that physiological effects of these four compounds on cells are mediated, at least in part, through their ability to inhibit Hsp90.
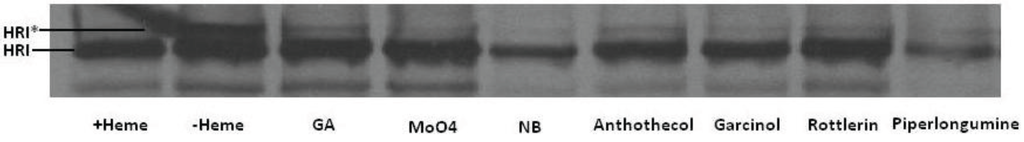
Figure 13.
Effect of compounds on HRI’s Hsp90-dependent maturation. [35S]Labeled-HRI was synthesized by TnT in RRL and transferred to heme-deficient lysate for maturation. Translated protein was separated by SDS PAGE, transferred to PVDF membrane, and visualized by X-ray film exposure. The phosphorylated active form of the kinase is indicated with an asterisk. Lanes were treated as follows: heme, no heme, 20 µM geldanamycin (GA), 20 mM sodium molybdate, 20 mM novobiocin, and 100 µM each of anthothecol, garcinol, rottlerin, and piplartine/piperlongumine
3.2.4. Compounds Induce Depletion of Hsp90-Dependent Clients
Inhibition of Hsp90 is well known to cause the depletion of Hsp90-depedent client proteins from inhibitor-treated cells. To further test the hypothesis that anthothecol, garcinol, rottlerin and piplartine are inhibitors of Hsp90, the effect of varying concentrations of the compounds on the expression of Hsp90-dependent client proteins Cdk6, pAkt, and Her2 in MCF7 cells was examined (Figure 14). Consistent with our hypothesis, all four compounds were observed to cause a concentration-dependent depletion of Hsp90 clients from MCF7 cells after 24 h of treatment. None of the compounds increased the expression of Hsp90. However, similar to geldanamycin, anthothecol and rottlerin caused a concentration-dependent increase in the expression of Hsp70, indicating that the compounds likely interact with the ATP binding site in Hsp90’s N-terminal domain. On the other hand, garcinol and piplartine had no effect on the expression of Hsp70, which is a property of compounds that inhibit Hsp90 by binding to its C-terminal domain [190].
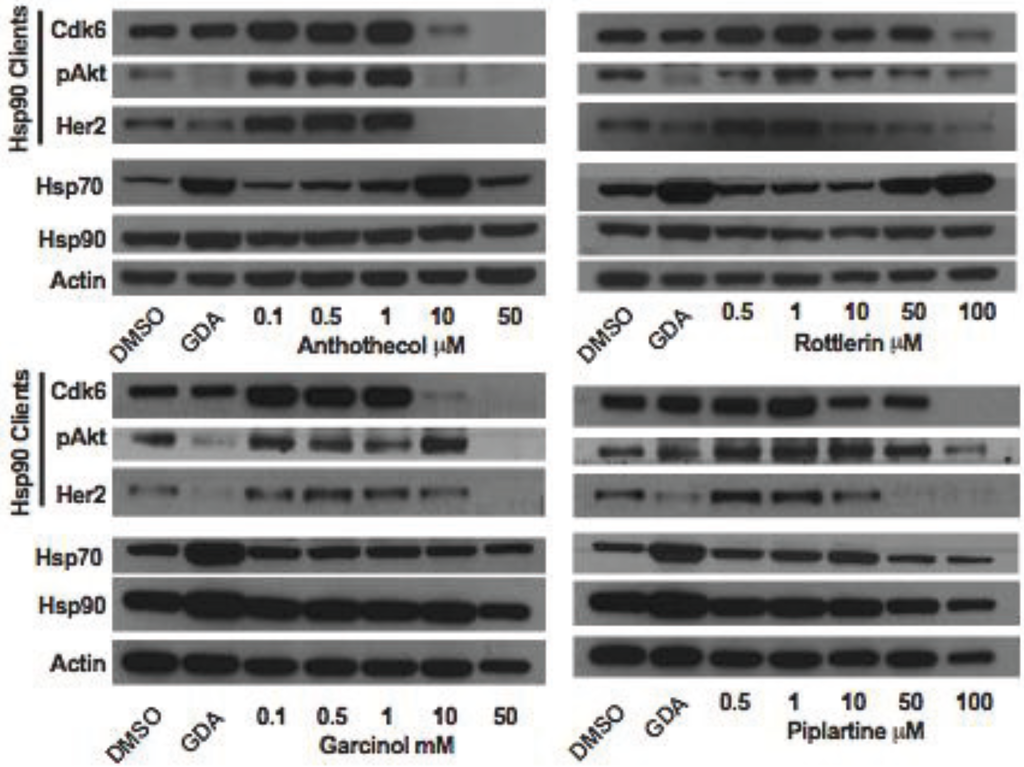
Figure 14.
Western blot for Hsp90-dependent client proteins (Cdk2, pAkt and Her2), and Hsp90 and Hsp70 present in extracts prepared from MCF7 cells treated for 24 h with DMSO (vehicle control), 0.5 µM geldanamycin, or the indicated concentrations of anthothecol, rottlerin, garcinol, or piplartine. Actin was used as the loading control.
4. Conclusions
The compounds identified from this screen represent diverse structures. Compounds with the greatest potential to be Hsp90 inhibitors are those that have already been identified as having specific activities, but against multiple targets. Often, reports by separate laboratories will demonstrate the activity of a compound against a certain kinase or pathway, while showing that related proteins or pathways are unaffected. When multiple targets with such effects are demonstrated, it is a strong indicator that the compound in question may be an Hsp90 inhibitor. Hsp90 is essential for the function of kinases, receptors, and other proteins from varied and wide-ranging pathways in the cell, but often plays no part in the function of closely related proteins. This scattered, yet specific, involvement of Hsp90 often precludes the detection of Hsp90’s importance by groups studying a narrow portion of the proteomic landscape. Accompanying the effects of these compounds is that they usually manifest anti-proliferative cytotoxic activities against cancer cells. Compounds that demonstrate these somewhat pleiotropic effects are amongst the first that should be considered for subsequent evaluations.
Another tell-tale sign of an Hsp90 inhibitor among screen hits is its anti-microbial activity. The earliest Hsp90 inhibitors, such as radicicol, geldanamycin, and novobiocin, were identified as antibiotics long before their activity against Hsp90 was elucidated.
It should be noted here that a portion of the compounds identified in the screening have structural properties that likely eliminate them from consideration for further characterization. Some compounds closely resemble compounds that are known intercalating agents. While these compounds could theoretically have some value as anti-cancer and anti-microbial drugs, we generally chose to bypass them, given their potential for off-target toxicity. Other compounds contain aldehyde moieties that can nonspecifically react with amino groups found in both proteins and nucleic acids. Another characteristic of compounds that potentially limit their usefulness is that some possess redox active quinones. While this may or may not affect their activity as Hsp90 inhibitors, the compounds can potentially induce oxidative stress, as well as chelate metal ions, and thus contribute to nonspecific effects in cells. An additional problem with a number of the compounds is that they contain α,β-unsaturated ketones, which can readily undergo Michael addition.
Recent reviews have noted that aldehydes, quinones and α,β-unsaturated ketones are undesirable moieties and are generally avoided during drug development [191,192,193,194]. The presence of such moieties in small synthetic molecules often present in HTS libraries, has led to identification of a class of compounds that are pan-assay interference compounds (PAINS, [193,194]): compounds identified as hits in multiple screening assays due to their nonspecific biological activity. Indeed, it has been emphasized that development of compounds containing undesirable moieties as potential drug leads should proceed with caution, if they are to be pursued at all. In the field of development of Hsp90 inhibitors as potential chemotherapeutics this issue raises an interesting question in regards to the “poly”-pharmacological property expected from Hsp90 inhibitors: does the observed “poly”-pharmacology of a compound indicate that it is an Hsp90 inhibitor, or does it reflect the fact that it is a PAIN and is nonspecifically inhibiting multiple biological pathways in the cell?
With regards to HTS, it should be noted that natural products are not indicative of the types of small synthetic molecules usually present in libraries utilized for HTS [191,195,196]. Natural products represent complex structures and, generally, a higher molecular weight. Natural products also occupy under-represented regions of biologically relevant chemical space: space that is often non-compliant with the rules of five [191,195,196]. Indeed, some drugs on the market based on natural products contain undesirable moieties [191]. In the context of the natural products presented herein, quinones are redox-active species that in some cases have been shown to exhibit preferential selectivity toward cancer cells [197]. In addition, we note that electrophilic moieties such as those found in α,β-unsaturated ketones are reactive species and further modification or retardation of their activity could be beneficial. However, it should be noted that a small set of compounds exhibiting such properties have advanced through clinical trials and have gained FDA approval [198,199].
The results presented here implicate the compounds anthothecol, garcinol, rottlerin, and piplartine/piperlongumine as inhibitors of the Hsp90 chaperone complex. Further study of the compounds will be necessary to confirm their status as inhibitors and Hsp90 as their target [200]. Similar studies will also be required to confirm or disprove whether the other compounds identified in the screening are inhibitors of the Hsp90 chaperone machine. The evidence presented for the +100 compounds being inhibitors of the Hsp90 chaperone machine obviously requires further support, and in no manner do we wish to imply that the compounds represent good lead compounds for therapeutic development. Instead, we offer this screening and literature mining as a resource for investigators interested in the possible mechanism of action of these natural compounds.
Acknowledgments
The authors gratefully acknowledge support of this project by NIH (R01 CA125392), the Sarkeys Foundation, and the Oklahoma Agricultural Experiment Station (Project No. 1975).
Author Contributions
Conceived and designed the experiments: JD, BSJB, RLM. Performed the experiments: JD, MB, LG, JH. Analyzed the data: JD, MB, LG, JH, BSJB, RLM. Contributed reagents/materials: RLM, BSJB. Wrote the paper: JD, AG, BSJS, RLM.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Whitesell, L.; Mimnaugh, E.G.; de Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein hsp90-pp60v-Src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar]
- Grenert, J.P.; Sullivan, W.P.; Fadden, P.; Haystead, T.A.; Clark, J.; Mimnaugh, E.; Krutzsch, H.; Ochel, H.J.; Schulte, T.W.; Sausville, E.; et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J. Biol. Chem. 1997, 272, 23843–23850. [Google Scholar]
- Sharma, S.V.; Agatsuma, T.; Nakano, H. Targeting of the protein chaperone, Hsp90, by the transformation suppressing agent, radicicol. Oncogene 1998, 16, 2639–2645. [Google Scholar]
- Schulte, T.W.; Akinaga, S.; Soga, S.; Sullivan, W.; Stensgard, B.; Toft, D.; Neckers, L.M. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3, 100–108. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O'Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Theodoraki, M.A.; Caplan, A.J. Quality control and fate determination of Hsp90 client proteins. Biochim. Biophys. Acta 2012, 1823, 683–688. [Google Scholar]
- Richter, K.; Buchner, J. Hsp90: Chaperoning signal transduction. J. Cell. Physiol. 2001, 188, 281–290. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Bishop, S.C.; Burlison, J.A.; Blagg, B.S. Hsp90: A novel target for the disruption of multiple signaling cascades. Curr. Cancer Drug Targets 2007, 7, 369–388. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Whitesell, L.; Shifrin, S.D.; Schwab, G.; Neckers, L.M. Benzoquinonoid ansamycins possess selective tumoricidal activity unrelated to Src kinase inhibition. Cancer Res. 1992, 52, 1721–1728. [Google Scholar]
- Calcul, L.; Zhang, B.; Jinwal, U.K.; Dickey, C.A.; Baker, B.J. Natural products as a rich source of tau-targeting drugs for Alzheimer's disease. Future Med. Chem. 2012, 4, 1751–1761. [Google Scholar]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Tanaka, F.; Doyu, M.; Sobue, G. Modulation of Hsp90 function in neurodegenerative disorders: A molecular-targeted therapy against disease-causing protein. J. Mol. Med. 2006, 84, 635–646. [Google Scholar] [CrossRef]
- Ansar, S.; Burlison, J.A.; Hadden, M.K.; Yu, X.M.; Desino, K.E.; Bean, J.; Neckers, L.; Audus, K.L.; Michaelis, M.L.; Blagg, B.S. A non-toxic Hsp90 inhibitor protects neurons from Abeta-induced toxicity. Bioorg. Med. Chem. Lett. 2007, 17, 1984–1990. [Google Scholar] [CrossRef]
- Chen, G.; Cao, P.; Goeddel, D.V. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell 2002, 9, 401–410. [Google Scholar] [CrossRef]
- Salminen, A.; Paimela, T.; Suuronen, T.; Kaarniranta, K. Innate immunity meets with cellular stress at the IKK complex: Regulation of the IKK complex by Hsp70 and Hsp90. Immunol. Lett. 2008, 117, 9–15. [Google Scholar] [CrossRef]
- Garcia-Cardena, G.; Fan, R.; Shah, V.; Sorrentino, R.; Cirino, G.; Papapetropoulos, A.; Sessa, W.C. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 1998, 392, 821–824. [Google Scholar] [CrossRef]
- Chatterjee, A.; Black, S.M.; Catravas, J.D. Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vascul. Pharmacol. 2008, 49, 134–140. [Google Scholar] [CrossRef]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef]
- Hong, D.S.; Banerji, U.; Tavana, B.; George, G.C.; Aaron, J.; Kurzrock, R. Targeting the molecular chaperone heat shock protein 90 (Hsp90): Lessons learned and future directions. Cancer Treat. Rev. 2013, 39, 375–387. [Google Scholar] [CrossRef]
- Geller, R.; Taguwa, S.; Frydman, J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 2012, 1823, 698–706. [Google Scholar] [CrossRef]
- Wirk, B. Heat shock protein inhibitors for the treatment of fungal infections. Recent Pat. Antiinfect. Drug Discov. 2011, 6, 38–44. [Google Scholar] [CrossRef]
- Shonhai, A.; Maier, A.G.; Przyborski, J.M.; Blatch, G.L. Intracellular protozoan parasites of humans: The role of molecular chaperones in development and pathogenesis. Protein Pept. Lett. 2011, 18, 143–157. [Google Scholar] [CrossRef]
- Shonhai, A. Plasmodial heat shock proteins: Targets for chemotherapy. FEMS Immunol. Med. Microbiol. 2010, 58, 61–74. [Google Scholar] [CrossRef]
- Matts, R.; Manjarrez, J.R. Assays for identification of Hsp90 inhibitors and biochemical methods for discriminating their mechanism of action. Curr. Top. Med. Chem. 2009, 9, 1462–1478. [Google Scholar] [CrossRef]
- Thulasiraman, V.; Matts, R.L. Luciferase renaturation assays of chaperones and chaperone antagonists. In Methods in Molecular biology: Bioluminescent Protocols; LaRossa, R., Ed.; Humana Press, Inc.: Totowa, NJ, USA, 1997; Chapter 11. [Google Scholar]
- Thulasiraman, V.; Matts, R.L. Effect of geldanamycin on the kinetics of chaperone-mediated renaturation of firefly luciferase in rabbit reticulocyte lysate. Biochemistry 1996, 35, 13443–13450. [Google Scholar]
- Galam, L.; Hadden, M.K.; Ma, Z.; Ye, Q.Z.; Yun, B.G.; Blagg, B.S.; Matts, R.L. High-throughput assay for the identification of Hsp90 inhibitors based on Hsp90-dependent refolding of firefly luciferase. Bioorg. Med. Chem. 2007, 15, 1939–1946. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Huber, P.J. Robust Statistics; Wiley: New York, NY, USA, 1981; p. 308. [Google Scholar]
- Davenport, J.; Manjarrez, J.R.; Peterson, L.; Krumm, B.; Blagg, B.S.; Matts, R.L. Gambogic acid, a natural product inhibitor of Hsp90. J. Nat. Prod. 2011, 74, 1085–1092. [Google Scholar] [CrossRef]
- Schumacher, R.J.; Hurst, R.; Sullivan, W.P.; McMahon, N.J.; Toft, D.O.; Matts, R.L. ATP-dependent chaperoning activity of reticulocyte lysate. J. Biol. Chem. 1994, 269, 9493–9499. [Google Scholar]
- Koga, F.; Kihara, K.; Neckers, L. Inhibition of cancer invasion and metastasis by targeting the molecular chaperone heat-shock protein 90. Anticancer Res. 2009, 29, 797–807. [Google Scholar]
- Burch, A.D.; Weller, S.K. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone Hsp90 for proper localization to the nucleus. J. Virol. 2005, 79, 10740–10749. [Google Scholar]
- Kampmueller, K.M.; Miller, D.J. The cellular chaperone heat shock protein 90 facilitates flock house virus rna replication in drosophila cells. J. Virol. 2005, 79, 6827–6837. [Google Scholar]
- Bruns, A.F.; Yuldasheva, N.; Latham, A.M.; Bao, L.; Pellet-Many, C.; Frankel, P.; Stephen, S.L.; Howell, G.J.; Wheatcroft, S.B.; Kearney, M.T.; et al. A heat-shock protein axis regulates VEGFR2 proteolysis, blood vessel development and repair. PLoS One 2012, 7, e48539. [Google Scholar]
- Staufer, K.; Stoeltzing, O. Implication of heat shock protein 90 (Hsp90) in tumor angiogenesis: A molecular target for anti-angiogenic therapy? Curr. Cancer Drug Targets 2010, 10, 890–897. [Google Scholar] [CrossRef]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678. [Google Scholar] [CrossRef]
- Geetha, B.S.; Nair, M.S.; Latha, P.G.; Remani, P. Sesquiterpene lactones isolated from Elephantopus scaber l. Inhibits human lymphocyte proliferation and the growth of tumour cell lines and induces apoptosis in vitro. J. Biomed. Biotechnol. 2012, 2012, 721285. [Google Scholar]
- Duraipandiyan, V.; Al-Harbi, N.A.; Ignacimuthu, S.; Muthukumar, C. Antimicrobial activity of sesquiterpene lactones isolated from traditional medicinal plant, Costus speciosus (koen ex.Retz.) sm. BMC Complement. Altern. Med. 2012, 12, 13. [Google Scholar]
- Kim, Y.H.; Choo, S.J.; Ryoo, I.J.; Ahn, J.S.; Yoo, I.D. Eudesmanolides from Taraxacum mongolicum and their inhibitory effects on the production of nitric oxide. Arch. Pharm. Res. 2011, 34, 37–41. [Google Scholar]
- Yamashita, Y.; Ikeda, T.; Matsuda, M.; Maji, D.; Hoshino, T.; Mizushima, T. Purification and characterization of Hsp-inducers from Eupatorium lindleyanum. Biochem. Pharmacol. 2012, 83, 909–922. [Google Scholar] [CrossRef]
- Kwok, B.H.; Koh, B.; Ndubuisi, M.I.; Elofsson, M.; Crews, C.M. The anti-inflammatory natural product parthenolide from the medicinal herb feverfew directly binds to and inhibits IkappaB kinase. Chem. Biol. 2001, 8, 759–766. [Google Scholar] [CrossRef]
- Carlisi, D.; D'Anneo, A.; Angileri, L.; Lauricella, M.; Emanuele, S.; Santulli, A.; Vento, R.; Tesoriere, G. Parthenolide sensitizes hepatocellular carcinoma cells to TRIAL by inducing the expression of death receptors through inhibition of STAT3 activation. J. Cell. Physiol. 2011, 226, 1632–1641. [Google Scholar] [CrossRef]
- Hoffmann, R.; von Schwarzenberg, K.; Lopez-Anton, N.; Rudy, A.; Wanner, G.; Dirsch, V.M.; Vollmar, A.M. Helenalin bypasses Bcl-2-mediated cell death resistance by inhibiting NF-kappaB and promoting reactive oxygen species generation. Biochem. Pharmacol. 2011, 82, 453–463. [Google Scholar] [CrossRef]
- Berges, C.; Fuchs, D.; Opelz, G.; Daniel, V.; Naujokat, C. Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms. Mol. Immunol. 2009, 46, 2892–2901. [Google Scholar]
- Huang, P.R.; Yeh, Y.M.; Wang, T.C. Potent inhibition of human telomerase by helenalin. Cancer Lett. 2005, 227, 169–174. [Google Scholar] [CrossRef]
- Li, J.; Du, L.; Kelly, M.; Zhou, Y.D.; Nagle, D.G. Structures and potential antitumor activity of sesterterpenes from the marine sponge Hyrtios communis. J. Nat. Prod. 2013, 76, 1492–1497. [Google Scholar]
- Xie, Y.; Liu, L.; Huang, X.; Guo, Y.; Lou, L. Scalaradial inhibition of epidermal growth factor receptor-mediated Akt phosphorylation is independent of secretory phospholipase A2. J. Pharmacol. Exp. Ther. 2005, 314, 1210–1217. [Google Scholar]
- Liu, H.; Edrada-Ebel, R.; Ebel, R.; Wang, Y.; Schulz, B.; Draeger, S.; Muller, W.E.; Wray, V.; Lin, W.; Proksch, P. Drimane sesquiterpenoids from the fungus Aspergillus ustus isolated from the marine sponge Suberites domuncula. J. Nat. Prod. 2009, 72, 1585–1588. [Google Scholar] [CrossRef]
- Kaur, K.; Jain, M.; Kaur, T.; Jain, R. Antimalarials from nature. Bioorg. Med. Chem. 2009, 17, 3229–3256. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cragg, G.M. Antineoplastic agents 32. The pseudoguaianolide helenalin. Experientia 1973, 29, 781. [Google Scholar] [CrossRef]
- Lim, C.B.; Fu, P.Y.; Ky, N.; Zhu, H.S.; Feng, X.; Li, J.; Srinivasan, K.G.; Hamza, M.S.; Zhao, Y. NF-kappaB p65 repression by the sesquiterpene lactone, helenalin, contributes to the induction of autophagy cell death. BMC Complement. Altern. Med. 2012, 12, 93. [Google Scholar] [CrossRef]
- Halder, B.; Das Gupta, S.; Gomes, A. Black tea polyphenols induce human leukemic cell cycle arrest by inhibiting Akt signaling: Possible involvement of hsp90, Wnt/beta-catenin signaling and FOXO1. FEBS J. 2012, 279, 2876–2891. [Google Scholar] [CrossRef]
- Miyamoto, K.; Nomura, M.; Murayama, T.; Furukawa, T.; Hatano, T.; Yoshida, T.; Koshiura, R.; Okuda, T. Antitumor activities of ellagitannins against sarcoma-180 in mice. Biol. Pharm. Bull. 1993, 16, 379–387. [Google Scholar]
- Nakashima, H.; Murakami, T.; Yamamoto, N.; Sakagami, H.; Tanuma, S.; Hatano, T.; Yoshida, T.; Okuda, T. Inhibition of human immunodeficiency viral replication by tannins and related compounds. Antiviral Res. 1992, 18, 91–103. [Google Scholar] [CrossRef]
- Zu, M.; Yang, F.; Zhou, W.; Liu, A.; Du, G.; Zheng, L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antiviral Res. 2012, 94, 217–224. [Google Scholar] [CrossRef]
- Kim, S.; Joo, Y.E. Theaflavin inhibits LPS-induced IL-6, MCP-1, and ICAM-1 expression in bone marrow-derived macrophages through the blockade of NF-kappaB and MAPK signaling pathways. Chonnam. Med. J. 2011, 47, 104–110. [Google Scholar]
- Wang, X.; Howell, C.P.; Chen, F.; Yin, J.; Jiang, Y. Gossypol—A polyphenolic compound from cotton plant. Adv. Food Nutr. Res. 2009, 58, 215–263. [Google Scholar] [CrossRef]
- Krenn, L.; Presser, A.; Pradhan, R.; Bahr, B.; Paper, D.H.; Mayer, K.K.; Kopp, B. Sulfemodin 8-O-beta-D-glucoside, a new sulfated anthraquinone glycoside, and antioxidant phenolic compounds from Rheum emodi. J. Nat. Prod. 2003, 66, 1107–1109. [Google Scholar] [CrossRef]
- Kageura, T.; Matsuda, H.; Morikawa, T.; Toguchida, I.; Harima, S.; Oda, M.; Yoshikawa, M. Inhibitors from rhubarb on lipopolysaccharide-induced nitric oxide production in macrophages: Structural requirements of stilbenes for the activity. Bioorg. Med. Chem. 2001, 9, 1887–1893. [Google Scholar] [CrossRef]
- Tereschuk, M.L.; Riera, M.V.; Castro, G.R.; Abdala, L.R. Antimicrobial activity of flavonoids from leaves of Tagetes minuta. J. Ethnopharmacol. 1997, 56, 227–232. [Google Scholar] [CrossRef]
- Ikigai, H.; Nakae, T.; Hara, Y.; Shimamura, T. Bactericidal catechins damage the lipid bilayer. Biochim. Biophys. Acta 1993, 1147, 132–136. [Google Scholar] [CrossRef]
- Yin, Z.; Henry, E.C.; Gasiewicz, T.A. (−)-Epigallocatechin-3-gallate is a novel Hsp90 inhibitor. Biochemistry 2009, 48, 336–345. [Google Scholar] [CrossRef]
- Sanchez, I.; Gomez-Garibay, F.; Taboada, J.; Ruiz, B.H. Antiviral effect of flavonoids on the dengue virus. Phytother. Res. 2000, 14, 89–92. [Google Scholar] [CrossRef]
- Kole, L.; Giri, B.; Manna, S.K.; Pal, B.; Ghosh, S. Biochanin-A, an isoflavon, showed anti-proliferative and anti-inflammatory activities through the inhibition of iNOS expression, p38-MAPK and ATF-2 phosphorylation and blocking NFkappaB nuclear translocation. Eur. J. Pharmacol. 2011, 653, 8–15. [Google Scholar] [CrossRef]
- Choi, A.Y.; Choi, J.H.; Yoon, H.; Hwang, K.Y.; Noh, M.H.; Choe, W.; Yoon, K.S.; Ha, J.; Yeo, E.J.; Kang, I. Luteolin induces apoptosis through endoplasmic reticulum stress and mitochondrial dysfunction in neuro-2A mouse neuroblastoma cells. Eur. J. Pharmacol. 2011, 668, 115–126. [Google Scholar]
- Xagorari, A.; Papapetropoulos, A.; Mauromatis, A.; Economou, M.; Fotsis, T.; Roussos, C. Luteolin inhibits an endotoxin-stimulated phosphorylation cascade and proinflammatory cytokine production in macrophages. J. Pharmacol. Exp. Ther. 2001, 296, 181–187. [Google Scholar]
- Lee, E.J.; Oh, S.Y.; Sung, M.K. Luteolin exerts anti-tumor activity through the suppression of epidermal growth factor receptor-mediated pathway in MDA-MB-231 ER-negative breast cancer cells. Food Chem. Toxicol. 2012, 50, 4136–4143. [Google Scholar]
- Fu, J.; Chen, D.; Zhao, B.; Zhao, Z.; Zhou, J.; Xu, Y.; Xin, Y.; Liu, C.; Luo, L.; Yin, Z. Luteolin induces carcinoma cell apoptosis through binding Hsp90 to suppress constitutive activation of STAT3. PLoS One 2012, 7, e49194. [Google Scholar]
- Watanapokasin, R.; Jarinthanan, F.; Nakamura, Y.; Sawasjirakij, N.; Jaratrungtawee, A.; Suksamrarn, S. Effects of alpha-mangostin on apoptosis induction of human colon cancer. World J. Gastroenterol. 2011, 17, 2086–2095. [Google Scholar] [CrossRef]
- Johnson, J.J.; Petiwala, S.M.; Syed, D.N.; Rasmussen, J.T.; Adhami, V.M.; Siddiqui, I.A.; Kohl, A.M.; Mukhtar, H. Alpha-mangostin, a xanthone from mangosteen fruit, promotes cell cycle arrest in prostate cancer and decreases xenograft tumor growth. Carcinogenesis 2012, 33, 413–419. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Iinuma, M.; Nozawa, Y. Anti-cancer effects of xanthones from pericarps of mangosteen. Int. J. Mol. Sci. 2008, 9, 355–370. [Google Scholar]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef]
- Hadden, M.K.; Galam, L.; Gestwicki, J.E.; Matts, R.L.; Blagg, B.S. Derrubone, an inhibitor of the Hsp90 protein folding machinery. J. Nat. Prod. 2007, 70, 2014–2018. [Google Scholar] [CrossRef]
- Cushnie, T.P.; Lamb, A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef]
- Liu, L.Z.; Fang, J.; Zhou, Q.; Hu, X.; Shi, X.; Jiang, B.H. Apigenin inhibits expression of vascular endothelial growth factor and angiogenesis in human lung cancer cells: Implication of chemoprevention of lung cancer. Mol. Pharmacol. 2005, 68, 635–643. [Google Scholar]
- Fang, J.; Zhou, Q.; Liu, L.Z.; Xia, C.; Hu, X.; Shi, X.; Jiang, B.H. Apigenin inhibits tumor angiogenesis through decreasing HIF-1alpha and VEGF expression. Carcinogenesis 2007, 28, 858–864. [Google Scholar]
- Opletalova, V.; Sedivy, D. [Chalcones and their heterocyclic analogs as potential antifungal chemotherapeutic agents]. Ceska. Slov. Farm. 1999, 48, 252–255. [Google Scholar]
- Herencia, F.; Ferrandiz, M.L.; Ubeda, A.; Guillen, I.; Dominguez, J.N.; Charris, J.E.; Lobo, G.M.; Alcaraz, M.J. Novel anti-inflammatory chalcone derivatives inhibit the induction of nitric oxide synthase and cyclooxygenase-2 in mouse peritoneal macrophages. FEBS Lett. 1999, 453, 129–134. [Google Scholar] [CrossRef]
- Makita, H.; Tanaka, T.; Fujitsuka, H.; Tatematsu, N.; Satoh, K.; Hara, A.; Mori, H. Chemoprevention of 4-nitroquinoline 1-oxide-induced rat oral carcinogenesis by the dietary flavonoids chalcone, 2-hydroxychalcone, and quercetin. Cancer Res. 1996, 56, 4904–4909. [Google Scholar]
- Casano, G.; Dumetre, A.; Pannecouque, C.; Hutter, S.; Azas, N.; Robin, M. Anti-HIV and antiplasmodial activity of original flavonoid derivatives. Bioorg. Med. Chem. 2010, 18, 6012–6023. [Google Scholar] [CrossRef]
- Yadav, V.R.; Prasad, S.; Sung, B.; Aggarwal, B.B. The role of chalcones in suppression of NF-kappaB-mediated inflammation and cancer. Int. Immunopharmacol. 2011, 11, 295–309. [Google Scholar] [CrossRef]
- Salminen, A.; Lehtonen, M.; Paimela, T.; Kaarniranta, K. Celastrol: Molecular targets of thunder god vine. Biochem. Biophys. Res. Commun. 2010, 394, 439–442. [Google Scholar] [CrossRef]
- Patwardhan, C.A.; Fauq, A.; Peterson, L.B.; Miller, C.; Blagg, B.S.; Chadli, A. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J. Biol. Chem. 2013, 288, 7313–7325. [Google Scholar] [CrossRef]
- Brandt, G.E.; Schmidt, M.D.; Prisinzano, T.E.; Blagg, B.S. Gedunin, a novel hsp90 inhibitor: Semisynthesis of derivatives and preliminary structure-activity relationships. J. Med. Chem. 2008, 51, 6495–6502. [Google Scholar] [CrossRef]
- Kim, M.S.; Kwon, J.Y.; Kang, N.J.; Lee, K.W.; Lee, H.J. Phloretin induces apoptosis in H-Ras MCF10A human breast tumor cells through the activation of p53 via Jnk and p38 mitogen-activated protein kinase signaling. Ann. N. Y. Acad. Sci. 2009, 1171, 479–483. [Google Scholar]
- Esatbeyoglu, T.; Huebbe, P.; Ernst, I.M.; Chin, D.; Wagner, A.E.; Rimbach, G. Curcumin—From molecule to biological function. Angew. Chem. Int. Ed. Engl. 2012, 51, 5308–5332. [Google Scholar]
- Giommarelli, C.; Zuco, V.; Favini, E.; Pisano, C.; Dal Piaz, F.; de Tommasi, N.; Zunino, F. The enhancement of antiproliferative and proapoptotic activity of HDAC inhibitors by curcumin is mediated by Hsp90 inhibition. Cell. Mol. Life Sci. 2010, 67, 995–1004. [Google Scholar] [CrossRef]
- Han, A.R.; Kang, Y.J.; Windono, T.; Lee, S.K.; Seo, E.K. Prenylated flavonoids from the heartwood of Artocarpus communis with inhibitory activity on lipopolysaccharide-induced nitric oxide production. J. Nat. Prod. 2006, 69, 719–721. [Google Scholar]
- Gafner, S.; Wolfender, J.L.; Mavi, S.; Hostettmann, K. Antifungal and antibacterial chalcones from Myrica serrata. Planta Med. 1996, 62, 67–69. [Google Scholar] [CrossRef]
- Belofsky, G.; Percivill, D.; Lewis, K.; Tegos, G.P.; Ekart, J. Phenolic metabolites of Dalea versicolor that enhance antibiotic activity against model pathogenic bacteria. J. Nat. Prod. 2004, 67, 481–484. [Google Scholar] [CrossRef]
- Ko, H.; Kim, Y.J.; Amor, E.C.; Lee, J.W.; Kim, H.C.; Kim, H.J.; Yang, H.O. Induction of autophagy by dimethyl cardamonin is associated with proliferative arrest in human colorectal carcinoma HCT116 and LOVO cells. J. Cell. Biochem. 2011, 112, 2471–2479. [Google Scholar] [CrossRef]
- Kim, Y.J.; Ko, H.; Park, J.S.; Han, I.H.; Amor, E.C.; Lee, J.W.; Yang, H.O. Dimethyl cardamonin inhibits lipopolysaccharide-induced inflammatory factors through blocking NF-kappaB p65 activation. Int. Immunopharmacol. 2010, 10, 1127–1134. [Google Scholar]
- Ye, C.L.; Qian, F.; Wei, D.Z.; Lu, Y.H.; Liu, J.W. Induction of apoptosis in K562 human leukemia cells by 2',4'-dihydroxy-6'-methoxy-3',5'-dimethylchalcone. Leuk. Res. 2005, 29, 887–892. [Google Scholar] [CrossRef]
- Ye, C.L.; Liu, J.W.; Wei, D.Z.; Lu, Y.H.; Qian, F. In vivo antitumor activity by 2',4'-dihydroxy-6'-methoxy-3',5'-dimethylchalcone in a solid human carcinoma xenograft model. Cancer Chemother. Pharmacol. 2005, 56, 70–74. [Google Scholar]
- Friedlander, T.W.; Weinberg, V.K.; Huang, Y.; Mi, J.T.; Formaker, C.G.; Small, E.J.; Harzstark, A.L.; Lin, A.M.; Fong, L.; Ryan, C.J. A phase II study of insulin-like growth factor receptor inhibition with nordihydroguaiaretic acid in men with non-metastatic hormone-sensitive prostate cancer. Oncol. Rep. 2012, 27, 3–9. [Google Scholar]
- Zhang, Y.; Xu, S.; Lin, J.; Yao, G.; Han, Z.; Liang, B.; Zou, Z.; Chen, Z.; Song, Q.; Dai, Y.; et al. mTORC1 is a target of nordihydroguaiaretic acid to prevent breast tumor growth in vitro and in vivo. Breast Cancer Res. Treat. 2012, 136, 379–388. [Google Scholar] [CrossRef]
- Maioli, E.; Torricelli, C.; Valacchi, G. Rottlerin and cancer: Novel evidence and mechanisms. Sci. World J. 2012, 2012, 350826. [Google Scholar]
- Torricelli, C.; Salvadori, S.; Valacchi, G.; Soucek, K.; Slabakova, E.; Muscettola, M.; Volpi, N.; Maioli, E. Alternative pathways of cancer cell death by rottlerin: Apoptosis versus autophagy. Evid. Based Complement. Alternat. Med. 2012, 2012, 980658. [Google Scholar]
- Singh, B.N.; Kumar, D.; Shankar, S.; Srivastava, R.K. Rottlerin induces autophagy which leads to apoptotic cell death through inhibition of PI3K/Akt/mTOR pathway in human pancreatic cancer stem cells. Biochem. Pharmacol. 2012, 84, 1154–1163. [Google Scholar] [CrossRef]
- Maioli, E.; Torricelli, C.; Valacchi, G. Rottlerin and curcumin: A comparative analysis. Ann. N. Y. Acad. Sci. 2012, 1259, 65–76. [Google Scholar]
- Lee, S.E.; Kim, M.R.; Kim, J.H.; Takeoka, G.R.; Kim, T.W.; Park, B.S. Antimalarial activity of anthothecol derived from Khaya anthotheca (meliaceae). Phytomedicine 2008, 15, 533–535. [Google Scholar] [CrossRef]
- Souto, A.L.; Tavares, J.F.; da Silva, M.S.; Diniz Mde, F.; de Athayde-Filho, P.F.; Barbosa Filho, J.M. Anti-inflammatory activity of alkaloids: An update from 2000 to 2010. Molecules 2011, 16, 8515–8534. [Google Scholar] [CrossRef]
- Cortijo, J.; Villagrasa, V.; Pons, R.; Berto, L.; Marti-Cabrera, M.; Martinez-Losa, M.; Domenech, T.; Beleta, J.; Morcillo, E.J. Bronchodilator and anti-inflammatory activities of glaucine: In vitro studies in human airway smooth muscle and polymorphonuclear leukocytes. Br. J. Pharmacol. 1999, 127, 1641–1651. [Google Scholar] [CrossRef]
- Lamchouri, F.; Zemzami, M.; Jossang, A.; Abdellatif, A.; Israili, Z.H.; Lyoussi, B. Cytotoxicity of alkaloids isolated from Peganum harmala seeds. Pak. J. Pharm. Sci. 2013, 26, 699–706. [Google Scholar]
- Ignacimuthu, S.; Shanmugam, N. Antimycobacterial activity of two natural alkaloids, vasicine acetate and 2-acetyl benzylamine, isolated from indian shrub Adhatoda vasica ness. Leaves. J. Biosci. 2010, 35, 565–570. [Google Scholar]
- Misra, P.; Khaliq, T.; Dixit, A.; SenGupta, S.; Samant, M.; Kumari, S.; Kumar, A.; Kushawaha, P.K.; Majumder, H.K.; Saxena, A.K.; et al. Antileishmanial activity mediated by apoptosis and structure-based target study of peganine hydrochloride dihydrate: An approach for rational drug design. J. Antimicrob. Chemother. 2008, 62, 998–1002. [Google Scholar] [CrossRef]
- Khaliq, T.; Misra, P.; Gupta, S.; Reddy, K.P.; Kant, R.; Maulik, P.R.; Dube, A.; Narender, T. Peganine hydrochloride dihydrate an orally active antileishmanial agent. Bioorg. Med. Chem. Lett. 2009, 19, 2585–2586. [Google Scholar]
- Trebec-Reynolds, D.P.; Voronov, I.; Heersche, J.N.; Manolson, M.F. VEGF-A expression in osteoclasts is regulated by NF-kappaB induction of HIF-1alpha. J. Cell. Biochem. 2010, 110, 343–351. [Google Scholar]
- Rightsel, W.A.; Schneider, H.G.; Sloan, B.J.; Graf, P.R.; Miller, F.A.; Bartz, O.R.; Ehrlich, J.; Dixon, G.J. Antiviral activity of gliotoxin and gliotoxin acetate. Nature 1964, 204, 1333–1334. [Google Scholar]
- Frenzel, A.; Zirath, H.; Vita, M.; Albihn, A.; Henriksson, M.A. Identification of cytotoxic drugs that selectively target tumor cells with Myc overexpression. PLoS One 2011, 6, e27988. [Google Scholar]
- Genne, P.; Duchamp, O.; Solary, E.; Magnette, J.; Belon, J.P.; Chauffert, B. Cinchonine per os: Efficient circumvention of P-glycoprotein-mediated multidrug resistance. Anticancer Drug Des. 1995, 10, 103–118. [Google Scholar]
- Furstner, A. Chemistry and biology of roseophilin and the prodigiosin alkaloids: A survey of the last 2500 years. Angew. Chem. Int. Ed. Engl. 2003, 42, 3582–3603. [Google Scholar]
- Liu, R.; Cui, C.B.; Duan, L.; Gu, Q.Q.; Zhu, W.M. Potent in vitro anticancer activity of metacycloprodigiosin and undecylprodigiosin from a sponge-derived Actinomycete saccharopolyspora sp. Nov. Arch. Pharm. Res. 2005, 28, 1341–1344. [Google Scholar] [CrossRef]
- Warnick-Pickle, D.J.; Byrne, K.M.; Pandey, R.C.; White, R.J. Fredericamycin a, a new antitumor antibiotic. II. Biological properties. J. Antibiot. 1981, 34, 1402–1407. [Google Scholar] [CrossRef]
- Latham, M.D.; King, C.K.; Gorycki, P.; Macdonald, T.L.; Ross, W.E. Inhibition of topoisomerases by fredericamycin A. Cancer Chemother. Pharmacol. 1989, 24, 167–171. [Google Scholar] [CrossRef]
- Miao, S.; Shi, X.; Zhang, H.; Wang, S.; Sun, J.; Hua, W.; Miao, Q.; Zhao, Y.; Zhang, C. Proliferation-attenuating and apoptosis-inducing effects of tryptanthrin on human chronic myeloid leukemia K562 cell line in vitro. Int. J. Mol. Sci. 2011, 12, 3831–3845. [Google Scholar] [CrossRef]
- Boustie, J.; Stigliani, J.L.; Montanha, J.; Amoros, M.; Payard, M.; Girre, L. Antipoliovirus structure-activity relationships of some aporphine alkaloids. J. Nat. Prod. 1998, 61, 480–484. [Google Scholar] [CrossRef]
- Spasova, M.; Philipov, S.; Nikolaeva-Glomb, L.; Galabov, A.S.; Milkova, T. Cinnamoyl- and hydroxycinnamoyl amides of glaucine and their antioxidative and antiviral activities. Bioorg. Med. Chem. 2008, 16, 7457–7461. [Google Scholar]
- Kondo, Y.; Imai, Y.; Hojo, H.; Endo, T.; Nozoe, S. Suppression of tumor cell growth and mitogen response by aporphine alkaloids, dicentrine, glaucine, corydine, and apomorphine. J. Pharmacobiodyn. 1990, 13, 426–431. [Google Scholar] [CrossRef]
- Dong, X.P.; Mondranondra, I.O.; Che, C.T.; Fong, H.S.; Farnsworth, N.R. Kmeriol and other aromatic constituents of Kmeria duperreana. Pharm. Res. 1989, 6, 637–640. [Google Scholar] [CrossRef]
- Clark, A.M.; Watson, E.S.; Ashfaq, M.K.; Hufford, C.D. In vivo efficacy of antifungal oxoaporphine alkaloids in experimental disseminated candidiasis. Pharm. Res. 1987, 4, 495–498. [Google Scholar] [CrossRef]
- Hufford, C.D.; Funderburk, M.J.; Morgan, J.M.; Robertson, L.W. Two antimicrobial alkaloids from heartwood of Liriodendron tulipifera l. J. Pharm. Sci. 1975, 64, 789–792. [Google Scholar] [CrossRef]
- Woo, S.H.; Reynolds, M.C.; Sun, N.J.; Cassady, J.M.; Snapka, R.M. Inhibition of topoisomerase II by liriodenine. Biochem. Pharmacol. 1997, 54, 467–473. [Google Scholar] [CrossRef]
- Xu, J.Y.; Meng, Q.H.; Chong, Y.; Jiao, Y.; Zhao, L.; Rosen, E.M.; Fan, S. Sanguinarine inhibits growth of human cervical cancer cells through the induction of apoptosis. Oncol. Rep. 2012, 28, 2264–2270. [Google Scholar]
- Gupta, S.C.; Kim, J.H.; Prasad, S.; Aggarwal, B.B. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010, 29, 405–434. [Google Scholar] [CrossRef]
- Yang, X.J.; Miao, F.; Yao, Y.; Cao, F.J.; Yang, R.; Ma, Y.N.; Qin, B.F.; Zhou, L. In vitro antifungal activity of sanguinarine and chelerythrine derivatives against phytopathogenic fungi. Molecules 2012, 17, 13026–13035. [Google Scholar] [CrossRef]
- Yu, S.M. Thaliporphine selectively inhibits expression of the inducible, but not the constitutive, nitric oxide synthase. Biochem. J. 1994, 303, 289–294. [Google Scholar]
- Graziose, R.; Rathinasabapathy, T.; Lategan, C.; Poulev, A.; Smith, P.J.; Grace, M.; Lila, M.A.; Raskin, I. Antiplasmodial activity of aporphine alkaloids and sesquiterpene lactones from Liriodendron tulipifera l. J. Ethnopharmacol. 2011, 133, 26–30. [Google Scholar] [CrossRef]
- Remichkova, M.; Dimitrova, P.; Philipov, S.; Ivanovska, N. Toll-like receptor-mediated anti-inflammatory action of glaucine and oxoglaucine. Fitoterapia 2009, 80, 411–414. [Google Scholar]
- Yang, X.; Yang, B.; Cai, J.; Zhang, C.; Zhang, Q.; Xu, L.; Qin, Q.; Zhu, H.; Ma, J.; Tao, G.; et al. Berberine enhances radiosensitivity of esophageal squamous cancer by targeting HIF-1alpha in vitro and in vivo. Cancer Biol. Ther. 2013, 14, 1066–1073. [Google Scholar]
- Pierpaoli, E.; Arcamone, A.G.; Buzzetti, F.; Lombardi, P.; Salvatore, C.; Provinciali, M. Antitumor effect of novel berberine derivatives in breast cancer cells. Biofactors 2013, 39, 672–679. [Google Scholar] [CrossRef]
- Wu, C.M.; Li, T.M.; Tan, T.W.; Fong, Y.C.; Tang, C.H. Berberine reduces the metastasis of chondrosarcoma by modulating the alpha V beta 3 integrin and the PKC delta , c-Src, and AP-1 signaling pathways. Evid. Based Complement. Alternat. Med. 2013, 2013, 423164. [Google Scholar]
- Sellers, R.P.; Alexander, L.D.; Johnson, V.A.; Lin, C.C.; Savage, J.; Corral, R.; Moss, J.; Slugocki, T.S.; Singh, E.K.; Davis, M.R.; et al. Design and synthesis of hsp90 inhibitors: Exploring the SAR of sansalvamide A derivatives. Bioorg. Med. Chem. 2010, 18, 6822–6856. [Google Scholar] [CrossRef]
- Hale, K.J.; Manaviazar, S.; Lazarides, L.; George, J.; Walters, M.A.; Cai, J.; Delisser, V.M.; Bhatia, G.S.; Peak, S.A.; Dalby, S.M.; et al. Synthesis of A83586C analogs with potent anticancer and beta-catenin/ TCF4/osteopontin inhibitory effects and insights into how A83586C modulates E2Fs and PRB. Org. Lett. 2009, 11, 737–740. [Google Scholar]
- Korting, H.C.; Schollmann, C.; Stauss-Grabo, M.; Schafer-Korting, M. Antimicrobial peptides and skin: A paradigm of translational medicine. Skin Pharmacol. Physiol. 2012, 25, 323–334. [Google Scholar]
- Guo, W.; Reigan, P.; Siegel, D.; Zirrolli, J.; Gustafson, D.; Ross, D. The bioreduction of a series of benzoquinone ansamycins by NAD(P)H:Quinone oxidoreductase 1 to more potent heat shock protein 90 inhibitors, the hydroquinone ansamycins. Mol. Pharmacol. 2006, 70, 1194–1203. [Google Scholar]
- Dikalov, S.; Landmesser, U.; Harrison, D.G. Geldanamycin leads to superoxide formation by enzymatic and non-enzymatic redox cycling. Implications for studies of Hsp90 and endothelial cell nitric-oxide synthase. J. Biol. Chem. 2002, 277, 25480–25485. [Google Scholar] [CrossRef]
- Wimalawansa, S.J. Vitamin D in the new millennium. Curr. Osteoporos. Rep. 2012, 10, 4–15. [Google Scholar] [CrossRef]
- Wigington, D.P.; Strugnell, S.A.; Knutson, J.C. Pamidronate and 1,24(S)-dihydroxyvitamin D2 synergistically inhibit the growth of myeloma, breast and prostate cancer cells. Anticancer Res. 2005, 25, 1909–1917. [Google Scholar]
- Simeone, A.M.; Tari, A.M. How retinoids regulate breast cancer cell proliferation and apoptosis. Cell. Mol. Life Sci. 2004, 61, 1475–1484. [Google Scholar]
- Abu, J.; Batuwangala, M.; Herbert, K.; Symonds, P. Retinoic acid and retinoid receptors: Potential chemopreventive and therapeutic role in cervical cancer. Lancet Oncol. 2005, 6, 712–720. [Google Scholar] [CrossRef]
- Barliya, T.; Mandel, M.; Livnat, T.; Weinberger, D.; Lavie, G. Degradation of HIF-1alpha under hypoxia combined with induction of Hsp90 polyubiquitination in cancer cells by hypericin: A unique cancer therapy. PLoS One 2011, 6, e22849. [Google Scholar]
- Sacau, E.P.; Estevez-Braun, A.; Ravelo, A.G.; Ferro, E.A.; Tokuda, H.; Mukainaka, T.; Nishino, H. Inhibitory effects of lapachol derivatives on epstein-barr virus activation. Bioorg. Med. Chem. 2003, 11, 483–488. [Google Scholar]
- Park, E.J.; Choi, K.S.; Kwon, T.K. Beta-lapachone-induced reactive oxygen species (ROS) generation mediates autophagic cell death in glioma U87 MG cells. Chem. Biol. Interact. 2011, 189, 37–44. [Google Scholar] [CrossRef]
- Pardee, A.B.; Li, Y.Z.; Li, C.J. Cancer therapy with beta-lapachone. Curr Cancer Drug Targets 2002, 2, 227–242. [Google Scholar] [CrossRef]
- Li, C.J.; Zhang, L.J.; Dezube, B.J.; Crumpacker, C.S.; Pardee, A.B. Three inhibitors of type 1 human immunodeficiency virus long terminal repeat-directed gene expression and virus replication. Proc. Natl. Acad. Sci. USA 1993, 90, 1839–1842. [Google Scholar]
- Guiraud, P.; Steiman, R.; Campos-Takaki, G.M.; Seigle-Murandi, F.; Simeon de Buochberg, M. Comparison of antibacterial and antifungal activities of lapachol and beta-lapachone. Planta Med. 1994, 60, 373–374. [Google Scholar] [CrossRef]
- Duvoix, A.; Delhalle, S.; Blasius, R.; Schnekenburger, M.; Morceau, F.; Fougere, M.; Henry, E.; Galteau, M.M.; Dicato, M.; Diederich, M. Effect of chemopreventive agents on glutathione S-transferase P1-1 gene expression mechanisms via activating protein 1 and nuclear factor kappaB inhibition. Biochem. Pharmacol. 2004, 68, 1101–1111. [Google Scholar] [CrossRef]
- Cavalcanti, B.C.; Barros, F.W.; Cabral, I.O.; Ferreira, J.R.; Magalhaes, H.I.; Junior, H.V.; da Silva Junior, E.N.; de Abreu, F.C.; Costa, C.O.; Goulart, M.O.; et al. Preclinical genotoxicology of nor-beta-lapachone in human cultured lymphocytes and chinese hamster lung fibroblasts. Chem. Res. Toxicol. 2011, 24, 1560–1574. [Google Scholar]
- Boothman, D.A.; Greer, S.; Pardee, A.B. Potentiation of halogenated pyrimidine radiosensitizers in human carcinoma cells by beta-lapachone (3,4-dihydro-2,2-dimethyl-2H-naphtho[1,2-b]pyran- 5,6-dione), a novel DNA repair inhibitor. Cancer Res. 1987, 47, 5361–5366. [Google Scholar]
- Salas, C.O.; Faundez, M.; Morello, A.; Maya, J.D.; Tapia, R.A. Natural and synthetic naphthoquinones active against Trypanosoma cruzi: An initial step towards new drugs for Chagas disease. Curr. Med. Chem. 2011, 18, 144–161. [Google Scholar]
- Wilson, J.B.; Johnson, M.A.; Stuckert, A.P.; Trueman, K.L.; May, S.; Bryant, P.E.; Meyn, R.E.; D'Andrea, A.D.; Jones, N.J. The chinese hamster FANCG/XRCC9 mutant NM3 fails to express the monoubiquitinated form of the FANCD2 protein, is hypersensitive to a range of DNA damaging agents and exhibits a normal level of spontaneous sister chromatid exchange. Carcinogenesis 2001, 22, 1939–1946. [Google Scholar] [CrossRef]
- Take, Y.; Oogose, K.; Kubo, T.; Inouye, Y.; Nakamura, S.; Kitahara, Y.; Kubo, A. Comparative study on biological activities of heterocyclic quinones and streptonigrin. J. Antibiot. 1987, 40, 679–684. [Google Scholar]
- Ross, D.; Beall, H.; Traver, R.D.; Siegel, D.; Phillips, R.M.; Gibson, N.W. Bioactivation of quinones by DT-diaphorase, molecular, biochemical, and chemical studies. Oncol. Res. 1994, 6, 493–500. [Google Scholar]
- Okada, H.; Mukai, H.; Inouye, Y.; Nakamura, S. Biological properties of streptonigrin derivatives. II. Inhibition of reverse transcriptase activity. J. Antibiot. 1986, 39, 306–308. [Google Scholar]
- Gu, B.; DeAngelis, L.M. Enhanced cytotoxicity of bioreductive antitumor agents with dimethyl fumarate in human glioblastoma cells. Anticancer Drugs 2005, 16, 167–174. [Google Scholar] [CrossRef]
- Beall, H.D.; Liu, Y.; Siegel, D.; Bolton, E.M.; Gibson, N.W.; Ross, D. Role of NAD(P)H:Quinone oxidoreductase (DT-diaphorase) in cytotoxicity and induction of DNA damage by streptonigrin. Biochem. Pharmacol. 1996, 51, 645–652. [Google Scholar] [CrossRef]
- Lee, W.Y.; Cheung, C.C.; Liu, K.W.; Fung, K.P.; Wong, J.; Lai, P.B.; Yeung, J.H. Cytotoxic effects of tanshinones from Salvia miltiorrhiza on doxorubicin-resistant human liver cancer cells. J. Nat. Prod. 2010, 73, 854–859. [Google Scholar] [CrossRef]
- Dat, N.T.; Jin, X.; Lee, J.H.; Lee, D.; Hong, Y.S.; Lee, K.; Kim, Y.H.; Lee, J.J. Abietane diterpenes from Salvia miltiorrhiza inhibit the activation of hypoxia-inducible factor-1. J. Nat. Prod. 2007, 70, 1093–1097. [Google Scholar]
- Suk, F.M.; Jou, W.J.; Lin, R.J.; Lin, S.Y.; Tzeng, F.Y.; Liang, Y.C. 15,16-Dihydrotanshinone I-induced apoptosis in human colorectal cancer cells: Involvement of ATF3. Anticancer Res. 2013, 33, 3225–3231. [Google Scholar]
- Padhye, S.; Dandawate, P.; Yusufi, M.; Ahmad, A.; Sarkar, F.H. Perspectives on medicinal properties of plumbagin and its analogs. Med. Res. Rev. 2012, 32, 1131–1138. [Google Scholar] [CrossRef]
- Lu, J.J.; Bao, J.L.; Wu, G.S.; Xu, W.S.; Huang, M.Q.; Chen, X.P.; Wang, Y.T. Quinones derived from plant secondary metabolites as anti-cancer agents. Anticancer Agents Med. Chem. 2013, 13, 456–463. [Google Scholar] [CrossRef]
- Grimley, P.M.; Moss, B. Similar effect of rifampin and other rifamycin derivatives on vaccinia virus morphogenesis. J. Virol. 1971, 8, 225–231. [Google Scholar]
- Chen, X.; Yang, L.; Oppenheim, J.J.; Howard, M.Z. Cellular pharmacology studies of shikonin derivatives. Phytother. Res. 2002, 16, 199–209. [Google Scholar]
- Uehara, Y.; Hori, M.; Takeuchi, T.; Umezawa, H. Screening of agents which convert “transformed morphology” of rous sarcoma virus-infected rat kidney cells to “normal morphology”: Identification of an active agent as herbimycin and its inhibition of intracellular Src kinase. Jpn. J. Cancer Res. 1985, 76, 672–675. [Google Scholar]
- Sakagami, M.; Morrison, P.; Welch, W.J. Benzoquinoid ansamycins (herbimycin A and geldanamycin) interfere with the maturation of growth factor receptor tyrosine kinases. Cell Stress Chaperones 1999, 4, 19–28. [Google Scholar] [CrossRef]
- Omura, S.; Iwai, Y.; Takahashi, Y.; Sadakane, N.; Nakagawa, A.; Oiwa, H.; Hasegawa, Y.; Ikai, T. Herbimycin, a new antibiotic produced by a strain of streptomyces. J. Antibiot. 1979, 32, 255–261. [Google Scholar]
- Chiari, E.; de Oliveira, A.B.; Raslan, D.S.; Mesquita, A.A.; Tavares, K.G. Screening in vitro of natural products against blood forms of Trypanosoma cruzi. Trans. R. Soc. Trop. Med. Hyg. 1991, 85, 372–374. [Google Scholar] [CrossRef]
- Tan, J.; Chen, B.; He, L.; Tang, Y.; Jiang, Z.; Yin, G.; Wang, J.; Jiang, X. Anacardic acid (6-pentadecylsalicylic acid) induces apoptosis of prostate cancer cells through inhibition of androgen receptor and activation of p53 signaling. Chin. J. Cancer Res. 2012, 24, 275–283. [Google Scholar] [CrossRef]
- Seong, Y.A.; Shin, P.G.; Yoon, J.S.; Yadunandam, A.K.; Kim, G.D. Induction of the endoplasmic reticulum stress and autophagy in human lung carcinoma A549 cells by anacardic acid. Cell Biochem. Biophys. 2013. [Google Scholar] [CrossRef]
- Dixit, D.; Sharma, V.; Ghosh, S.; Koul, N.; Mishra, P.K.; Sen, E. Manumycin inhibits STAT3, telomerase activity, and growth of glioma cells by elevating intracellular reactive oxygen species generation. Free Radic. Biol. Med. 2009, 47, 364–374. [Google Scholar] [CrossRef]
- Pan, J.; Chen, B.; Su, C.H.; Zhao, R.; Xu, Z.X.; Sun, L.; Lee, M.H.; Yeung, S.C. Autophagy induced by farnesyltransferase inhibitors in cancer cells. Cancer Biol. Ther. 2008, 7, 1679–1684. [Google Scholar]
- Hadden, M.K.; Hill, S.A.; Davenport, J.; Matts, R.L.; Blagg, B.S. Synthesis and evaluation of Hsp90 inhibitors that contain the 1,4-naphthoquinone scaffold. Bioorg. Med. Chem. 2009, 17, 634–640. [Google Scholar] [CrossRef]
- Zhao, H.; Brandt, G.E.; Galam, L.; Matts, R.L.; Blagg, B.S. Identification and initial SAR of silybin: An Hsp90 inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 2659–2664. [Google Scholar] [CrossRef]
- Lim, J.H.; Park, J.W.; Choi, K.S.; Park, Y.B.; Kwon, T.K. Rottlerin induces apoptosis via death receptor 5 (DR5) upregulation through CHOP-dependent and PKC delta-independent mechanism in human malignant tumor cells. Carcinogenesis 2009, 30, 729–736. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Prasad, S.; Ravindran, J.; Sung, B.; Pandey, M.K.; Aggarwal, B.B. Garcinol potentiates TRIAL-induced apoptosis through modulation of death receptors and antiapoptotic proteins. Mol. Cancer Ther. 2010, 9, 856–868. [Google Scholar] [CrossRef]
- Parasramka, M.A.; Gupta, S.V. Garcinol inhibits cell proliferation and promotes apoptosis in pancreatic adenocarcinoma cells. Nutr. Cancer 2011, 63, 456–465. [Google Scholar]
- Ahmad, A.; Sarkar, S.H.; Aboukameel, A.; Ali, S.; Biersack, B.; Seibt, S.; Li, Y.; Bao, B.; Kong, D.; Banerjee, S.; et al. Anticancer action of garcinol in vitro and in vivo is in part mediated through inhibition of STAT-3 signaling. Carcinogenesis 2012, 33, 2450–2456. [Google Scholar] [CrossRef]
- Hong, J.; Sang, S.; Park, H.J.; Kwon, S.J.; Suh, N.; Huang, M.T.; Ho, C.T.; Yang, C.S. Modulation of arachidonic acid metabolism and nitric oxide synthesis by garcinol and its derivatives. Carcinogenesis 2006, 27, 278–286. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Shimada, R.; Kagami, S.; Yamaguchi, F.; Kataoka, S.; Ariga, T.; Murakami, A.; Koshimizu, K.; Ohigashi, H. Prevention of colonic aberrant crypt foci by dietary feeding of garcinol in male F344 rats. Carcinogenesis 2000, 21, 1183–1189. [Google Scholar]
- Son, D.J.; Kim, S.Y.; Han, S.S.; Kim, C.W.; Kumar, S.; Park, B.S.; Lee, S.E.; Yun, Y.P.; Jo, H.; Park, Y.H. Piperlongumine inhibits atherosclerotic plaque formation and vascular smooth muscle cell proliferation by suppressing PDGF receptor signaling. Biochem. Biophys. Res. Commun. 2012, 427, 349–354. [Google Scholar] [CrossRef]
- Golovine, K.V.; Makhov, P.B.; Teper, E.; Kutikov, A.; Canter, D.; Uzzo, R.G.; Kolenko, V.M. Piperlongumine induces rapid depletion of the androgen receptor in human prostate cancer cells. Prostate 2013, 73, 23–30. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2012, 48, 453–463. [Google Scholar]
- Uma, S.; Hartson, S.D.; Chen, J.J.; Matts, R.L. Hsp90 is obligatory for the heme-regulated eIF-2alpha kinase to acquire and maintain an activable conformation. J. Biol. Chem. 1997, 272, 11648–11656. [Google Scholar] [CrossRef]
- Eskew, J.D.; Sadikot, T.; Morales, P.; Duren, A.; Dunwiddie, I.; Swink, M.; Zhang, X.; Hembruff, S.; Donnelly, A.; Rajewski, R.A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. [Google Scholar] [CrossRef]
- Axerio-Cilies, P.; Castaneda, I.P.; Mirza, A.; Reynisson, J. Investigation of the incidence of "Undesirable" molecular moieties for high-throughput screening compound libraries in marketed drug compounds. Eur. J. Med. Chem. 2009, 44, 1128–1134. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C. Mechanisms of chemical carcinogenicity and mutagenicity: A review with implications for predictive toxicology. Chem. Rev. 2011, 111, 2507–2536. [Google Scholar] [CrossRef]
- Baell, J.B. Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2010, 2, 1529–1546. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Bauer, R.A.; Wurst, J.M.; Tan, D.S. Expanding the range of 'druggable' targets with natural product-based libraries: An academic perspective. Curr. Opin. Chem. Biol. 2010, 14, 308–314. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Ross, D. Quinone reductases multitasking in the metabolic world. Drug Metab. Rev. 2004, 36, 639–654. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/Her2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef]
- Hong, Y.; Yu, B.; Sherman, M.; Yuan, Y.C.; Zhou, D.; Chen, S. Molecular basis for the aromatization reaction and exemestane-mediated irreversible inhibition of human aromatase. Mol. Endocrinol. 2007, 21, 401–414. [Google Scholar] [CrossRef]
- Matts, R.L.; Brandt, G.E.; Lu, Y.; Dixit, A.; Mollapour, M.; Wang, S.; Donnelly, A.C.; Neckers, L.; Verkhivker, G.; Blagg, B.S. A systematic protocol for the characterization of Hsp90 modulators. Bioorg. Med. Chem. 2011, 19, 684–692. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).