Understanding the Dynamics of Gene Regulatory Systems; Characterisation and Clinical Relevance of cis-Regulatory Polymorphisms

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Importance of Non-Coding DNA
2.1. cis-Regulatory Sequences Have Shaped Human Evolution and Development
2.2. cis-Regulatory Sequences are Implicated in Human Pathologies
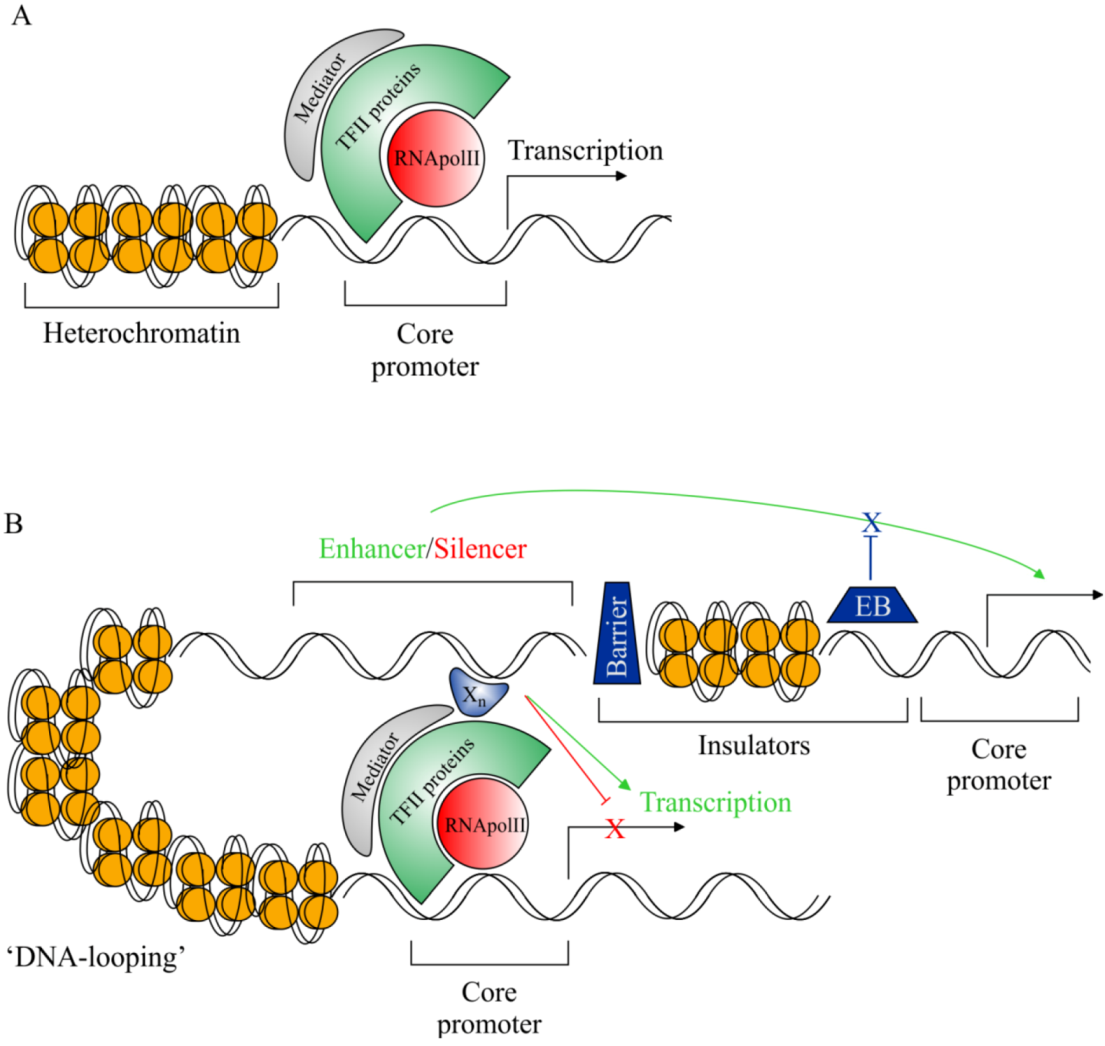
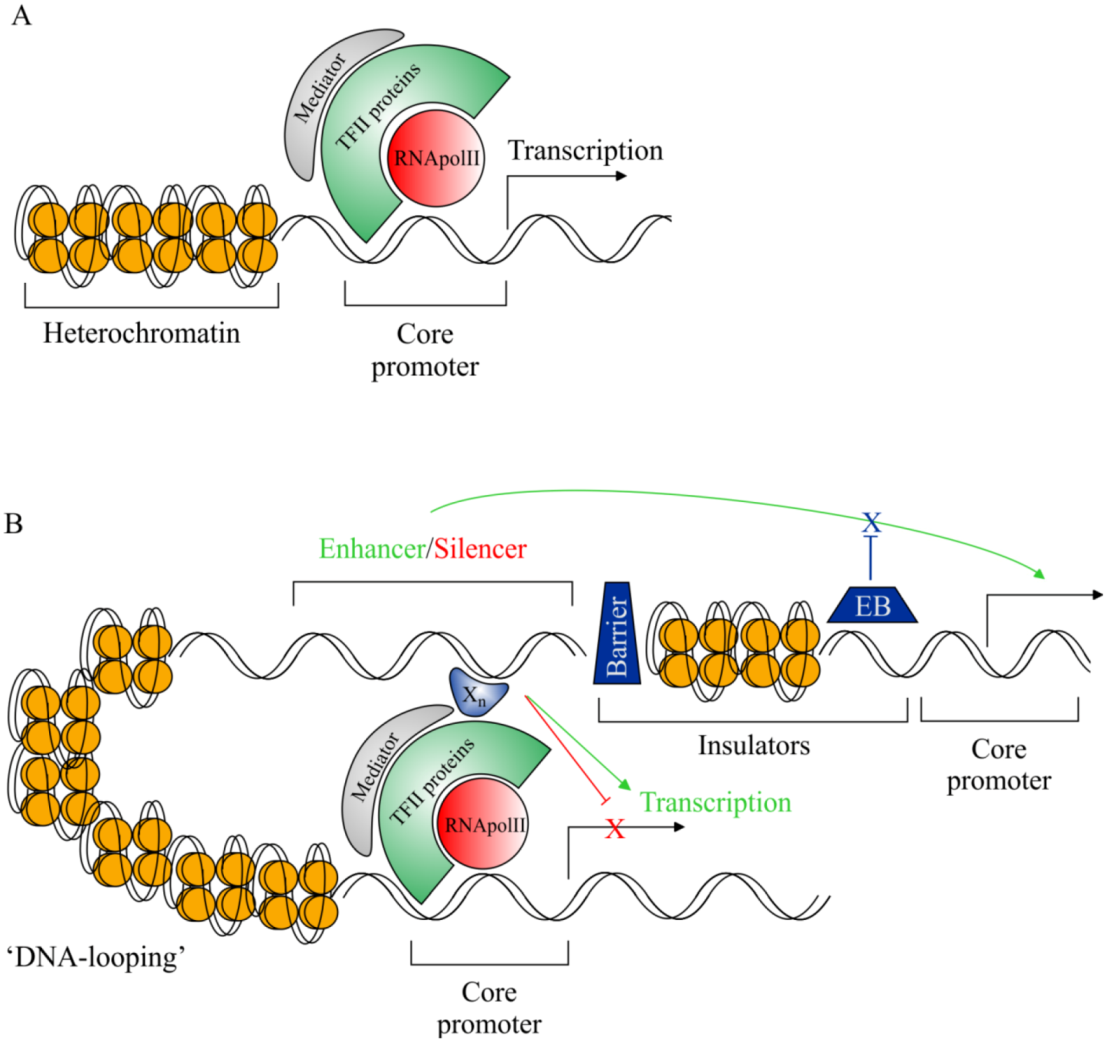
2.3. Rationale for cis-Regulatory Sequence Research
3. cis--Regulatory Sequence Identification—Comparative Genomics
3.1. Evolutionary Distant Species Comparisons
3.2. Evolutionary Related Species Comparisons
4. cis--Regulatory Sequence Identification—Experimental Approaches
4.1. Transcriptional Associations: Chromatin Immunoprecipitation Techniques
4.2. Active Chromatin Signatures: DNaseI Hypersensitivity and Formaldehyde-Assisted Identification of Regulatory Elements
4.3. Chromosome Interactions: Chromosome Conformation Capture Strategies
4.4. Towards a Map of the Genome’s Regulatory Landscape: The ENCODE Consortium
5. Analysis of cis-Regulatory Sequences
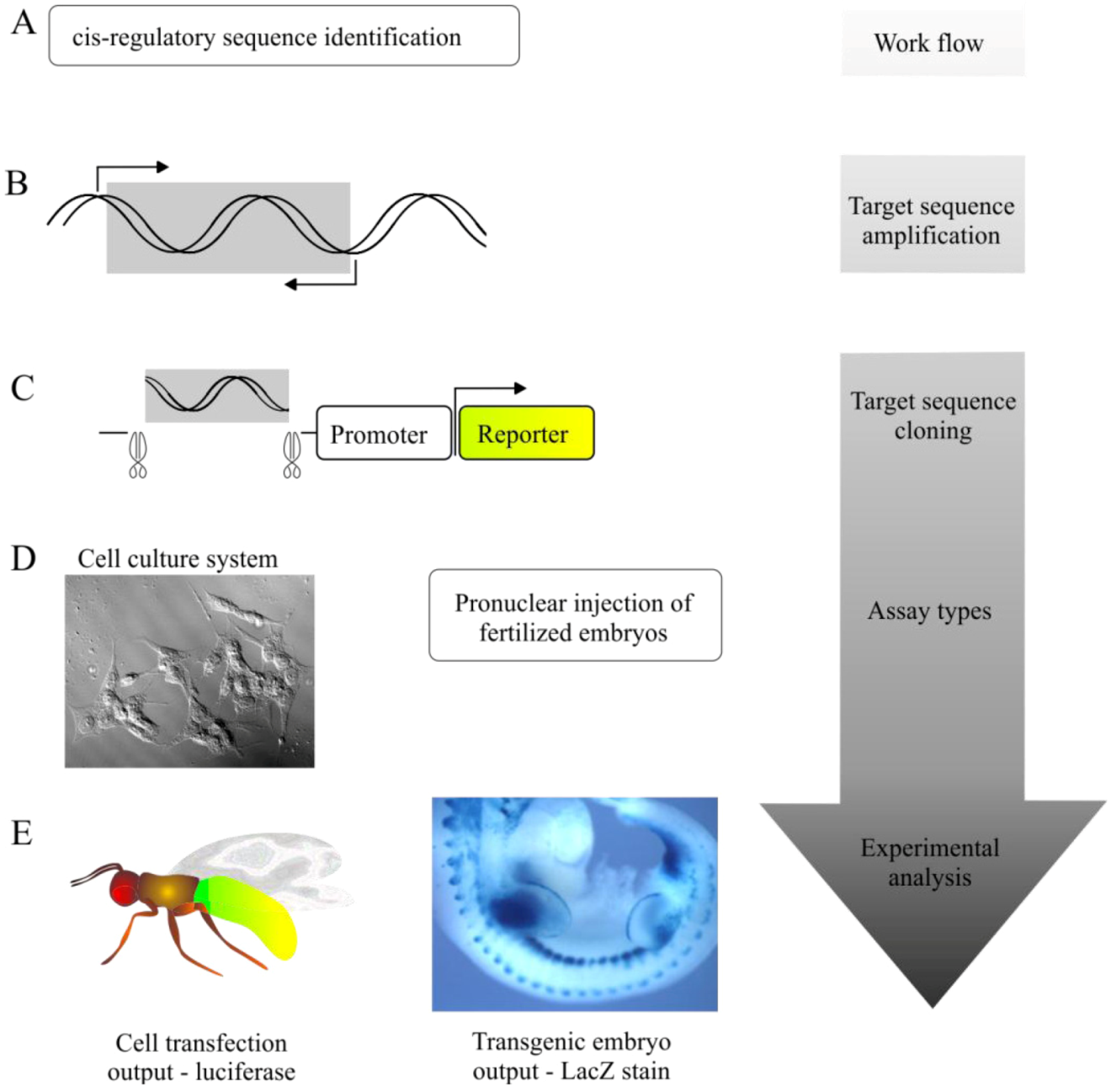
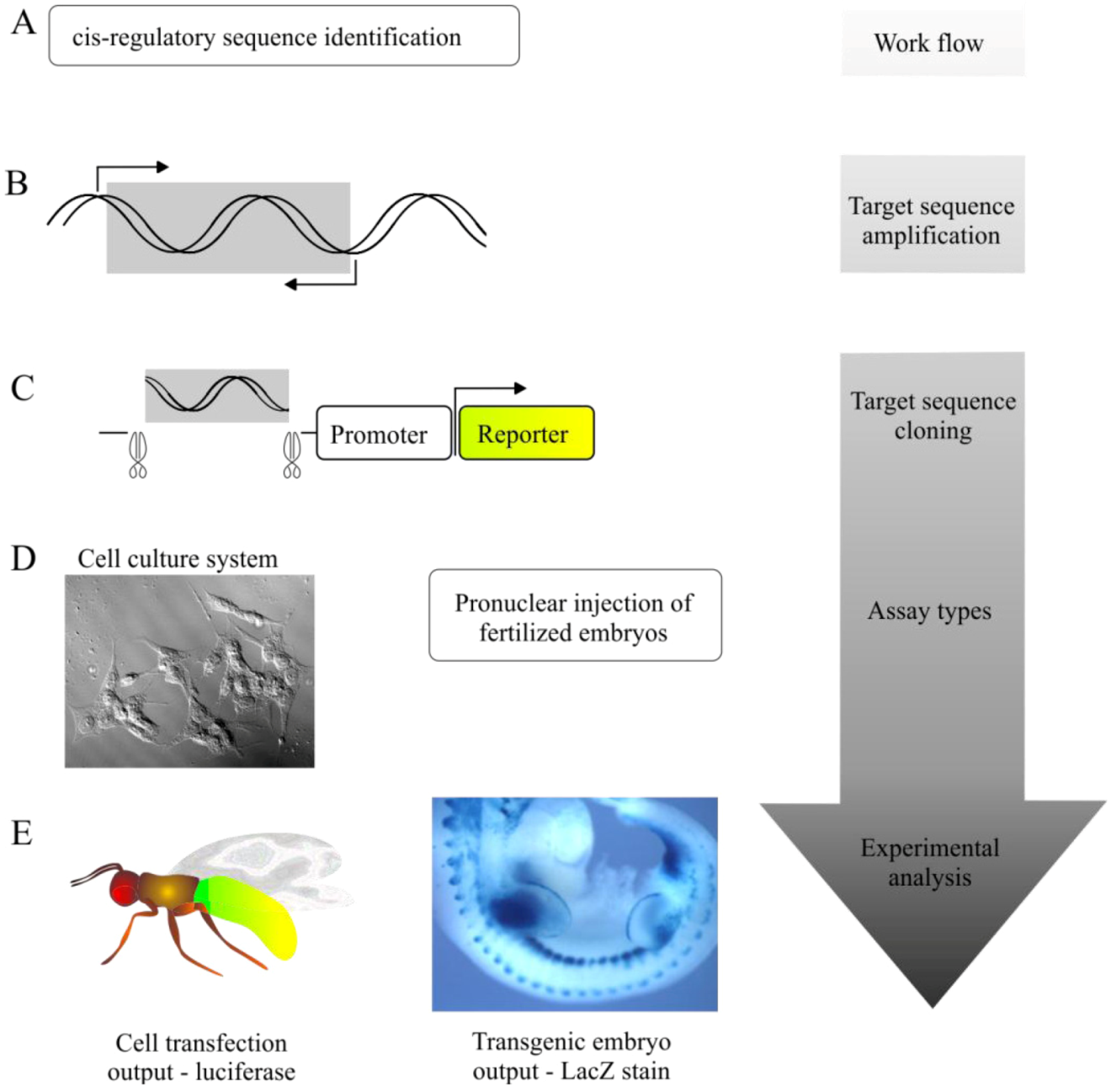
5.1. Transgenic Animal Reporter Assays
5.2. Cell-Based Reporter Gene Assays
6. Beyond Identification: cis-Regulatory Sequence Characterisation

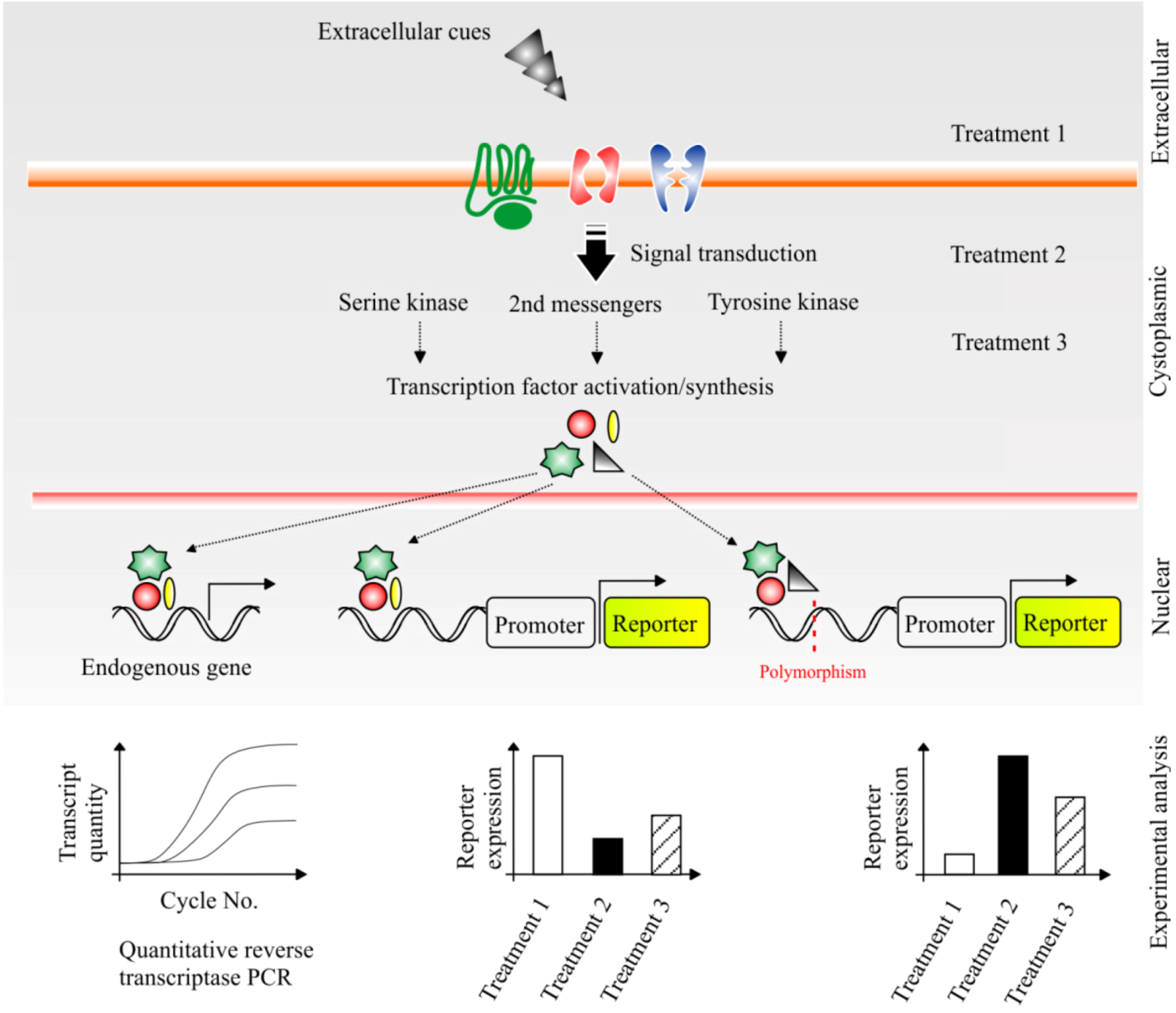
6.1. Dissecting the Impact of Cellular Signalling
6.2. Embryonic Stem Cell Targeting
6.3. A Question of Specificity?
7. Novel Considerations of cis-Regulatory Sequence Polymorphic Variation
7.1. cis-Regulatory Sequence Variation and Drug Response Stratification
7.2. Genetic and Epigenetic Interaction within CRSs and Disease Susceptibility
Acknowledgements
References
- Levine, M.; Tjian, R. Transcription regulation and animal diversity. Nature 2003, 424, 147–151. [Google Scholar] [CrossRef]
- Moore, M.J. From birth to death: The complex lives of eukaryotic mrnas. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef]
- Davidson, E. The Regulatory Genome: Gene Regulatory Networks in Development and Evolution; Academic Press: Burlington, San Diego, USA, London, UK, 2006. [Google Scholar]
- Ong, C.-T.; Corces, V.G. Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Collins, F.S.; Lander, E.S.; Rogers, J.; Waterson, R.H. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- O’Brien, S.J.; Menotti-Raymond, M.; Murphy, W.J.; Nash, W.G.; Wienberg, J.; Stanyon, R.; Copeland, N.G.; Jenkins, N.A.; Womack, J.E.; Marshall Graves, J.A. The promise of comparative genomics in mammals. Science 1999, 286, 458–481. [Google Scholar]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [Green Version]
- Davidson, S.; Miller, K.A.; Dowell, A.; Gildea, A.; MacKenzie, A. A remote and highly conserved enhancer supports amygdala specific expression of the gene encoding the anxiogenic neuropeptide substance-p. Mol. Psychiatry 2006, 11, 410–421. [Google Scholar] [CrossRef]
- Visel, A.; Bristow, J.; Pennacchio, L.A. Enhancer identification through comparative genomics. Semi. Cell Dev. Biol. 2007, 18, 140–152. [Google Scholar]
- Boffelli, D.; Nobrega, M.A.; Rubin, E.M. Comparative genomics at the vertebrate extremes. Nat. Rev. Genet. 2004, 5, 456–465. [Google Scholar] [CrossRef]
- Pennacchio, L.A.; Ahituv, N.; Moses, A.M.; Prabhakar, S.; Nobrega, M.A.; Shoukry, M.; Minovitsky, S.; Dubchak, I.; Holt, A.; Lewis, K.D.; et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 2006, 444, 499–502. [Google Scholar] [CrossRef]
- Maston, G.A.; Evans, S.K.; Green, M.R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Human Genet. 2006, 7, 29–59. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74.
- Singleton, A.B.; Hardy, J.; Traynor, B.J.; Houlden, H. Towards a complete resolution of the genetic architecture of disease. Trends Genet. 2010, 26, 438–442. [Google Scholar] [CrossRef]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef]
- Wang, W.Y.S.; Barratt, B.J.; Clayton, D.G.; Todd, J.A. Genome-wide association studies: Theoretical and practical concerns. Nat. Rev. Genet. 2005, 6, 109–118. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar]
- Stern, D.L. Perspective: Evolutionary developmental biology and the problem of variation. Evolution 2000, 54, 1079–1091. [Google Scholar]
- Simonet, W.S.; Bucay, N.; Lauer, S.J.; Taylor, J.M. A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein e and c-i genes in transgenic mice. J. Biol. Chem. 1993, 268, 8221–8229. [Google Scholar]
- Allan, C.M.; Walker, D.; Taylor, J.M. Evolutionary duplication of a hepatic control region in the human apolipoprotein e gene locus. J. Biol. Chem. 1995, 270, 26278–26281. [Google Scholar]
- Simonet, W.S.; Bucay, N.; Pitas, R.E.; Lauer, S.J.; Taylor, J.M. Multiple tissue-specific elements control the apolipoprotein e/c-i gene locus in transgenic mice. J. Biol. Chem. 1991, 266, 8651–8654. [Google Scholar]
- Grehan, S.; Tse, E.; Taylor, J.M. Two distal downstream enhancers direct expression of the human apolipoprotein e gene to astrocytes in the brain. J. Neurosci. 2001, 21, 812–822. [Google Scholar]
- Shih, S.-J.; Allan, C.; Grehan, S.; Tse, E.; Moran, C.; Taylor, J.M. Duplicated downstream enhancers control expression of the human apolipoprotein e gene in macrophages and adipose tissue. J. Biol. Chem. 2000, 275, 31567–31572. [Google Scholar]
- Chaudhuri, A.; Zbrzezna, V.; Polyakova, J.; Pogo, A.O.; Hesselgesser, J.; Horuk, R. Expression of the duffy antigen in k562 cells. Evidence that it is the human erythrocyte chemokine receptor. J. Biol. Chem. 1994, 269, 7835–7838. [Google Scholar]
- Horuk, R.; Chitnis, C.; Darbonne, W.; Colby, T.; Rybicki, A.; Hadley, T.; Miller, L. A receptor for the malarial parasite plasmodium vivax: The erythrocyte chemokine receptor. Science 1993, 261, 1182–1184. [Google Scholar]
- Tournamille, C.; Blancher, A.; Le Van Kim, C.; Gane, P.; Apoil, P.; Nakamoto, W.; Cartron, J.; Colin, Y. Sequence, evolution and ligand binding properties of mammalian duffy antigen/receptor for chemokines. Immunogenetics 2004, 55, 682–694. [Google Scholar] [CrossRef]
- Iwamoto, S.; Li, J.; Sugimoto, N.; Okuda, H.; Kajii, E. Characterization of the duffy gene promotor: Evidence for tissue-specific abolishment of expression in fy(a−b−) of black individuals. Biochem. Biophys. Res. Commun. 1996, 222, 852–859. [Google Scholar] [CrossRef]
- Tournamille, C.; Colin, Y.; Cartron, J.P.; Le Van Kim, C. Disruption of a gata motif in the duffy gene promoter abolishes erythroid gene expression in duffy-negative individuals. Nat. Genet. 1995, 10, 224–228. [Google Scholar] [CrossRef]
- Hadley, T.J.; Peiper, S.C. From malaria to chemokine receptor: The emerging physiologic role of the duffy blood group antigen. Blood 1997, 89, 3077–3091. [Google Scholar]
- Miller, L.H.; Mason, S.J.; Clyde, D.F.; McGinniss, M.H. The resistance factor to plasmodium vivax in blacks. N. Engl. J. Med. 1976, 295, 302–304. [Google Scholar] [CrossRef]
- Oscar Pogo, A.; Chaudhuri, A. The duffy protein: A malarial and chemokine receptor. Semi. Hematol. 2000, 37, 122–129. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Polyakova, J.; Zbrzezna, V.; Pogo, A. The coding sequence of duffy blood group gene in humans and simians: Restriction fragment length polymorphism, antibody and malarial parasite specificities, and expression in nonerythroid tissues in duffy-negative individuals. Blood 1995, 85, 615–621. [Google Scholar]
- Noonan, J.P.; McCallion, A.S. Genomics of long-range regulatory elements. Annu. Rev. Genomics Hum. Genet. 2010, 11, 1–23. [Google Scholar] [CrossRef]
- De Gobbi, M.; Viprakasit, V.; Hughes, J.R.; Fisher, C.; Buckle, V.J.; Ayyub, H.; Gibbons, R.J.; Vernimmen, D.; Yoshinaga, Y.; de Jong, P.; et al. A regulatory snp causes a human genetic disease by creating a new transcriptional promoter. Science 2006, 312, 1215–1217. [Google Scholar]
- Savic, D.; Park, S.; Bailey, K.; Bell, G.; Nobrega, M. In vitro scan for enhancers at the TCF7L2 locus. Diabetologia 2012, 56, 121–125. [Google Scholar]
- Emison, E.S.; McCallion, A.S.; Kashuk, C.S.; Bush, R.T.; Grice, E.; Lin, S.; Portnoy, M.E.; Cutler, D.J.; Green, E.D.; Chakravarti, A. A common sex-dependent mutation in a ret enhancer underlies hirschsprung disease risk. Nature 2005, 434, 857–863. [Google Scholar]
- Grice, E.A.; Rochelle, E.S.; Green, E.D.; Chakravarti, A.; McCallion, A.S. Evaluation of the ret regulatory landscape reveals the biological relevance of a hscr-implicated enhancer. Hum. Mol. Genet. 2005, 14, 3837–3845. [Google Scholar] [CrossRef]
- Monod, J.; Jacob, F. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb. Symp. Quant. Biol. 1961, 26, 389–401. [Google Scholar] [CrossRef]
- Britten, R.J.; Davidson, E.H. Gene regulation for higher cells: A theory. Science 1969, 165, 349–357. [Google Scholar]
- Britten, R.J.; Davidson, E.H. Repetitive and non-repetitive DNA sequences and a speculation on the origins of evolutionary novelty. Q. Rev. Biol. 1971, 46, 111–138. [Google Scholar]
- King, M.; Wilson, A. Evolution at two levels in humans and chimpanzees. Science 1975, 188, 107–116. [Google Scholar]
- Davidson, S.; Starkey, A.; MacKenzie, A. Evidence of uneven selective pressure on different subsets of the conserved human genome; implications for the significance of intronic and intergenic DNA. BMC Genomics 2009, 10, 614. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, S.; Morrison, A.; Gould, A.; Gilthorpe, J.; Chaudhuri, C.; Rigby, P.; Krumlauf, R.; Brenner, S. Detecting conserved regulatory elements with the model genome of the japanese puffer fish, fugu rubripes. Proc. Natl. Acad. Sci. USA 1995, 92, 1684–1688. [Google Scholar]
- Miller, K.A.; Davidson, S.; Liaros, A.; Barrow, J.; Lear, M.; Heine, D.; Hoppler, S.; MacKenzie, A. Prediction and characterisation of a highly conserved, remote and camp responsive enhancer that regulates msx1 gene expression in cardiac neural crest and outflow tract. Dev. Biol. 2008, 317, 686–694. [Google Scholar] [CrossRef]
- Miller, K.A.; Barrow, J.; Collinson, J.M.; Davidson, S.; Lear, M.; Hill, R.E.; MacKenzie, A. A highly conserved wnt-dependent tcf4 binding site within the proximal enhancer of the anti-myogenic msx1 gene supports expression within pax3-expressing limb bud muscle precursor cells. Dev. Biol. 2007, 311, 665–678. [Google Scholar] [CrossRef]
- Nobrega, M.A.; Ovcharenko, I.; Afzal, V.; Rubin, E.M. Scanning human gene deserts for long-range enhancers. Science 2003, 302, 413. [Google Scholar] [CrossRef]
- Woolfe, A.; Goodson, M.; Goode, D.K.; Snell, P.; McEwen, G.K.; Vavouri, T.; Smith, S.F.; North, P.; Callaway, H.; Kelly, K.; et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2004, 3, e7. [Google Scholar]
- Ovcharenko, I.; Loots, G.G.; Nobrega, M.A.; Hardison, R.C.; Miller, W.; Stubbs, L. Evolution and functional classification of vertebrate gene deserts. Genome Res. 2005, 15, 137–145. [Google Scholar] [CrossRef]
- Prabhakar, S.; Poulin, F.; Shoukry, M.; Afzal, V.; Rubin, E.M.; Couronne, O.; Pennacchio, L.A. Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res. 2006, 16, 855–863. [Google Scholar] [CrossRef]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved elements in the human genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef]
- Poulin, F.; Nobrega, M.A.; Plajzer-Frick, I.; Holt, A.; Afzal, V.; Rubin, E.M.; Pennacchio, L.A. In vivo characterization of a vertebrate ultraconserved enhancer. Genomics 2005, 85, 774–781. [Google Scholar] [CrossRef]
- Sandelin, A.; Bailey, P.; Bruce, S.; Engstrom, P.; Klos, J.; Wasserman, W.; Ericson, J.; Lenhard, B. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics 2004, 5, 99. [Google Scholar] [CrossRef] [Green Version]
- Visel, A.; Prabhakar, S.; Akiyama, J.A.; Shoukry, M.; Lewis, K.D.; Holt, A.; Plajzer-Frick, I.; Afzal, V.; Rubin, E.M.; Pennacchio, L.A. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat. Genet. 2008, 40, 158–160. [Google Scholar] [CrossRef]
- Maston, G.A.; Landt, S.G.; Snyder, M.; Green, M.R. Characterization of enhancer function from genome-wide analyses. Annu. Rev. Genomics Hum. Genet. 2012, 13, 29–57. [Google Scholar] [CrossRef]
- Birney, E.; Stamatoyannopoulos, J.; Dutta, A.; Guigó, R.; Gingeras, T.; Margulies, E.; Weng, Z.; Snyder, M.; Dermitzakis, E.; Thurman, R.; et al. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Blow, M.J.; McCulley, D.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. Chip-seq identification of weakly conserved heart enhancers. Nat. Genet. 2010, 42, 806–810. [Google Scholar] [CrossRef]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus e1a-associated 300-kd protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef]
- Iyer, V.R.; Horak, C.E.; Scafe, C.S.; Botstein, D.; Snyder, M.; Brown, P.O. Genomic binding sites of the yeast cell-cycle transcription factors sbf and mbf. Nature 2001, 409, 533–538. [Google Scholar] [CrossRef]
- Ren, B.; Robert, F.O.; Wyrick, J.J.; Aparicio, O.; Jennings, E.G.; Simon, I.; Zeitlinger, J.; Schreiber, J.R.; Hannett, N.; Kanin, E.; et al. Genome-wide location and function of DNA binding proteins. Science 2000, 290, 2306–2309. [Google Scholar]
- Impey, S.; McCorkle, S.R.; Cha-Molstad, H.; Dwyer, J.M.; Yochum, G.S.; Boss, J.M.; McWeeney, S.; Dunn, J.J.; Mandel, G.; Goodman, R.H. Defining the creb regulon: A genome-wide analysis of transcription factor regulatory regions. Cell 2004, 119, 1041–1054. [Google Scholar]
- Wei, C.-L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef]
- Visel, A.; Blow, M.J.; Li, Z.; Zhang, T.; Akiyama, J.A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C.; Chen, F.; et al. Chip-seq accurately predicts tissue-specific activity of enhancers. Nature 2009, 457, 854–858. [Google Scholar]
- Wu, C. The 5' ends of drosophila heat shock genes in chromatin are hypersensitive to dnase i. Nature 1980, 286, 854–860. [Google Scholar] [CrossRef]
- Boyle, A.P.; Song, L.; Lee, B.-K.; London, D.; Keefe, D.; Birney, E.; Iyer, V.R.; Crawford, G.E.; Furey, T.S. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2011, 21, 456–464. [Google Scholar] [CrossRef]
- Crawford, G.E.; Holt, I.E.; Whittle, J.; Webb, B.D.; Tai, D.; Davis, S.; Margulies, E.H.; Chen, Y.; Bernat, J.A.; Ginsburg, D.; et al. Genome-wide mapping of dnase hypersensitive sites using massively parallel signature sequencing (mpss). Genome Res. 2006, 16, 123–131. [Google Scholar]
- McDaniell, R.; Lee, B.-K.; Song, L.; Liu, Z.; Boyle, A.P.; Erdos, M.R.; Scott, L.J.; Morken, M.A.; Kucera, K.S.; Battenhouse, A.; et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science 2010, 328, 235–239. [Google Scholar]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Schadt, E.; Chang, R. A gps for navigating DNA. Science 2012, 337, 1179–1180. [Google Scholar] [CrossRef]
- Giresi, P.G.; Lieb, J.D. Isolation of active regulatory elements from eukaryotic chromatin using faire (formaldehyde assisted isolation of regulatory elements). Methods 2009, 48, 233–239. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar]
- De Wit, E.; de Laat, W. A decade of 3c technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef]
- Simonis, M.; Kooren, J.; de Laat, W. An evaluation of 3c-based methods to capture DNA interactions. Nat. Meth. 2007, 4, 895–901. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Wei, C.-L.; Liu, E.T.; Ruan, Y. Next-generation DNA sequencing of paired-end tags (pet) for transcriptome and genome analyses. Genome Res. 2009, 19, 521–532. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Ruan, Y. Chip-based methods for the identification of long-range chromatin interactions. J. Cell. Biochem. 2009, 107, 30–39. [Google Scholar] [CrossRef]
- Zhang, J.; Poh, H.M.; Peh, S.Q.; Sia, Y.Y.; Li, G.; Mulawadi, F.H.; Goh, Y.; Fullwood, M.J.; Sung, W.-K.; Ruan, X.; et al. Chia-pet analysis of transcriptional chromatin interactions. Methods 2012, 58, 289–299. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at ucsc. Genome Res. 2002, 12, 996–1006. [Google Scholar]
- Rosenbloom, K.R.; Dreszer, T.R.; Long, J.C.; Malladi, V.S.; Sloan, C.A.; Raney, B.J.; Cline, M.S.; Karolchik, D.; Barber, G.P.; Clawson, H. Encode whole-genome data in the ucsc genome browser. Nucleic Acids Res. 2010, 38, 620–625. [Google Scholar] [CrossRef]
- Kothary, R.; Clapoff, S.; Darling, S.; Perry, M.D.; Moran, L.A.; Rossant, J. Inducible expression of an hsp68-lacz hybrid gene in transgenic mice. Development 1989, 105, 707–714. [Google Scholar]
- Davidson, S.; Lear, M.; Shanley, L.; Hing, B.; Baizan-Edge, A.; Herwig, A.; Quinn, J.P.; Breen, G.; McGuffin, P.; Starkey, A.; et al. Differential activity by polymorphic variants of a remote enhancer that supports galanin expression in the hypothalamus and amygdala: Implications for obesity, depression and alcoholism. Neuropsychopharmacology 2011, 36, 2211–2221. [Google Scholar] [CrossRef]
- Hing, B.; Davidson, S.; Lear, M.; Breen, G.; Quinn, J.; McGuffin, P.; MacKenzie, A. A polymorphism associated with depressive disorders differentially regulates brain derived neurotrophic factor promoter iv activity. Biol. Psychiatry 2012, 71, 618–626. [Google Scholar] [CrossRef]
- Brivanlou, A.H.; Darnell, J.E. Signal transduction and the control of gene expression. Science 2002, 295, 813–818. [Google Scholar] [CrossRef]
- Nicoll, G.; Davidson, S.; Shanley, L.; Hing, B.; Lear, M.; McGuffin, P.; Ross, R.; MacKenzie, A. Allele-specific differences in activity of a novel cannabinoid receptor 1 (cnr1) gene intronic enhancer in hypothalamus, dorsal root ganglia, and hippocampus. J. Biol. Chem. 2012, 287, 12828–12834. [Google Scholar]
- Swanson, C.I.; Evans, N.C.; Barolo, S. Structural rules and complex regulatory circuitry constrain expression of a notch- and egfr-regulated eye enhancer. Dev. Cell 2010, 18, 359–370. [Google Scholar] [CrossRef]
- Shanley, L.; Davidson, S.; Lear, M.; Thotakura, A.K.; McEwan, I.J.; Ross, R.A.; MacKenzie, A. Long-range regulatory synergy is required to allow control of the tac1 locus by mek/erk signalling in sensory neurones. Neurosignals 2010, 18, 173–185. [Google Scholar] [CrossRef]
- Shanley, L.; Lear, M.; Davidson, S.; Ross, R.; MacKenzie, A. Evidence for regulatory diversity and auto-regulation at the tac1 locus in sensory neurones. J. Neuroinflammation 2011, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Sauer, B. Functional expression of the cre-lox site-specific recombination system in the yeast saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7, 2087–2096. [Google Scholar]
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the cre recombinase of bacteriophage p1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef]
- Orban, P.C.; Chui, D.; Marth, J.D. Tissue- and site-specific DNA recombination in transgenic mice. Proc. Natl. Acad. Sci. USA 1992, 89, 6861–6865. [Google Scholar] [CrossRef]
- Gu, H.; Zou, Y.-R.; Rajewsky, K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through cre-loxp-mediated gene targeting. Cell 1993, 73, 1155–1164. [Google Scholar] [CrossRef]
- Gu, H.; Marth, J.; Orban, P.; Mossmann, H.; Rajewsky, K. Deletion of a DNA polymerase beta gene segment in t cells using cell type-specific gene targeting. Science 1994, 265, 103–106. [Google Scholar]
- Lettice, L.A.; Horikoshi, T.; Heaney, S.J.H.; van Baren, M.J.; van der Linde, H.C.; Breedveld, G.J.; Joosse, M.; Akarsu, N.; Oostra, B.A.; Endo, N.; et al. Disruption of a long-range cis-acting regulator for shh causes preaxial polydactyly. Proc. Natl. Acad. Sci. USA 2002, 99, 7548–7553. [Google Scholar]
- Lomvardas, S.; Barnea, G.; Pisapia, D.J.; Mendelsohn, M.; Kirkland, J.; Axel, R. Interchromosomal interactions and olfactory receptor choice. Cell 2006, 126, 403–413. [Google Scholar] [CrossRef]
- Li, Q.; Barkess, G.I.; Qian, H. Chromatin looping and the probability of transcription. Trends Genet. 2006, 22, 197–202. [Google Scholar] [CrossRef]
- Jackson, D.A.; Hassan, A.B.; Errington, R.J.; Cook, P.R. Visualization of focal sites of transcription within human nuclei. EMBO J. 1993, 12, 1059. [Google Scholar]
- Fraser, P.; Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 2007, 447, 413–417. [Google Scholar] [CrossRef]
- Hu, Q.; Kwon, Y.-S.; Nunez, E.; Cardamone, M.D.; Hutt, K.R.; Ohgi, K.A.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G.; et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and lsd1-dependent gene networking in interchromatin granules. Proc. Natl. Acad. Sci. USA 2008, 105, 19199–19204. [Google Scholar]
- Gondor, A.; Ohlsson, R. Chromosome crosstalk in three dimensions. Nature 2009, 461, 212–217. [Google Scholar] [CrossRef]
- Maranville, J.C.; Luca, F.; Richards, A.L.; Wen, X.; Witonsky, D.B.; Baxter, S.; Stephens, M.; di Rienzo, A. Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genet. 2011, 7, e1002162. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmuhl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.X.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cowie, P.; Ross, R.; MacKenzie, A. Understanding the Dynamics of Gene Regulatory Systems; Characterisation and Clinical Relevance of cis-Regulatory Polymorphisms. Biology 2013, 2, 64-84. https://doi.org/10.3390/biology2010064
Cowie P, Ross R, MacKenzie A. Understanding the Dynamics of Gene Regulatory Systems; Characterisation and Clinical Relevance of cis-Regulatory Polymorphisms. Biology. 2013; 2(1):64-84. https://doi.org/10.3390/biology2010064
Chicago/Turabian StyleCowie, Philip, Ruth Ross, and Alasdair MacKenzie. 2013. "Understanding the Dynamics of Gene Regulatory Systems; Characterisation and Clinical Relevance of cis-Regulatory Polymorphisms" Biology 2, no. 1: 64-84. https://doi.org/10.3390/biology2010064