Genetic and Epigenetic Regulation of CCR5 Transcription

1
Department of Immunohematology and Blood Transfusion, Leiden University Medical Center, Leiden, Albinusdreef 2, 2333 ZA, Leiden, The Netherlands
2
Department of Pathology, VU University Medical Center, De Boelelaan 1117, 1081 HV Amsterdam, The Netherlands
*
Author to whom correspondence should be addressed.
Biology 2012, 1(3), 869-879; https://doi.org/10.3390/biology1030869
Submission received: 7 October 2012
/
Revised: 27 November 2012
/
Accepted: 3 December 2012
/
Published: 13 December 2012
(This article belongs to the Special Issue Gene Expression and Regulation)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The chemokine receptor CCR5 regulates trafficking of immune cells of the lymphoid and the myeloid lineage (such as monocytes, macrophages and immature dendritic cells) and microglia. Because of this, there is an increasing recognition of the important role of CCR5 in the pathology of (neuro-) inflammatory diseases such as atherosclerosis and multiple sclerosis. Expression of CCR5 is under the control of a complexly organized promoter region upstream of the gene. The transcription factor cAMP-responsive element binding protein 1 (CREB-1) transactivates the CCR5 P1 promoter. The cell-specific expression of CCR5 however is realized by using various epigenetic marks providing a multivalent chromatin state particularly in monocytes. Here we discuss the transcriptional regulation of CCR5 with a focus on the epigenetic peculiarities of CCR5 transcription.
1. Introductions
CC chemokine receptor 5 (CCR5), a receptor for the CC chemokines macrophage inflammatory protein-1α (MIP-1α), macrophage inflammatory protein-1β (MIP-1β), and regulated and normal T cell expressed and secreted (RANTES), regulates trafficking of lymphoid cells such as memory/effector Th1 lymphocytes, or cells of the myeloid lineage (e.g., monocytes, macrophages, immature dendritic cells) and microglia [1,2,3]. As such, CCR5 is implicated in the pathogenesis of various inflammatory diseases such as atherosclerosis and multiple sclerosis [4,5,6,7]. This is illustrated in atherosclerotic mouse models, in which Ccr5 knock-out mice show less neointima formation and an increase in production of the anti-inflammatory IL-10 molecule by vascular smooth muscle cells (vSMCs) [8,9]. One of the ligands for the CCR5 receptor, CCL5 or RANTES, has also been shown to be involved in unstable angina pectoris, which usually results from atherosclerosis [10]. Furthermore, CCR5 also functions as a co-receptor for HIV-1 [11,12,13]. Importantly, individuals with a mutated CCR5 receptor appear to be near-completely protected for HIV-1 infection [10,13]. Notably, CCR5 expression is markedly upregulated upon T cell activation, which allows the activated T cells to migrate towards site(s) of inflammation [14,15,16,17,18].
Upon encountering a pathogen, antigen-presenting cells will present the antigenic peptide to resting naive T cells, which results in the generation and activation of antigen-specific T cells [19,20]. After activation, the T cells migrate to the site of inflammation, guided by chemokine receptors [21]. Similarly, circulating monocytes are also attracted to inflammatory sites by chemokine receptors, where they then can differentiate into e.g., macrophages or microglia [22,23,24]. Atherosclerosis and multiple sclerosis are greatly characterized by inflammatory lesions, consisting of T cells and macrophages or microglia, respectively [25,26,27]. The chemokine receptor CCR5 is implicated in the pathogenesis of both of these diseases [8,9,28,29].
With CCR5 being implicated in various (inflammatory) diseases and as co-receptor for HIV-1 infection, much work has been put into the transcriptional regulation of CCR5. CCR5 transcription is controlled by a complex promoter structure. Although a number of transcription factors, including CREB-1, have been shown to play a role in CCR5 transcriptional regulation, its cell type specific expression is also controlled by epigenetic mechanisms. In some cell types the chromatin status of CCR5 is rather peculiar in that it is hallmarked by various multivalent states. In this review we will discuss the regulation of CCR5 expression, with the focus on epigenetic regulation and the possible pharmacological intervention in these epigenetic regulatory processes.
2. Genomic Organization
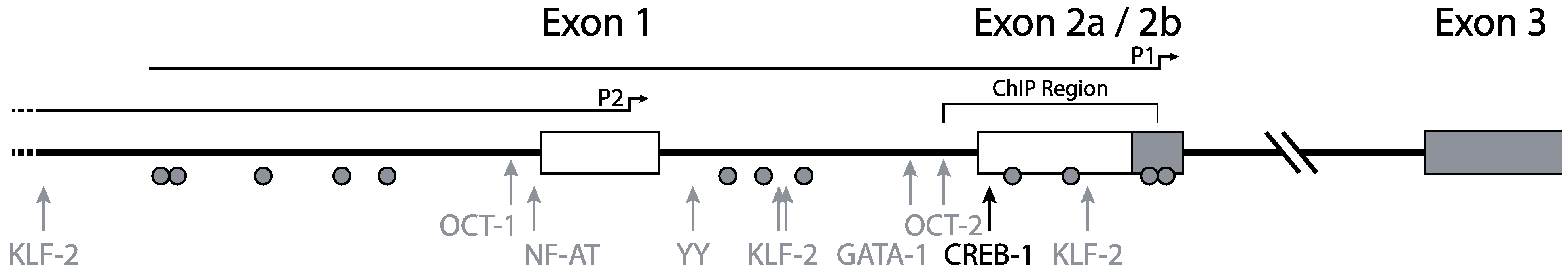
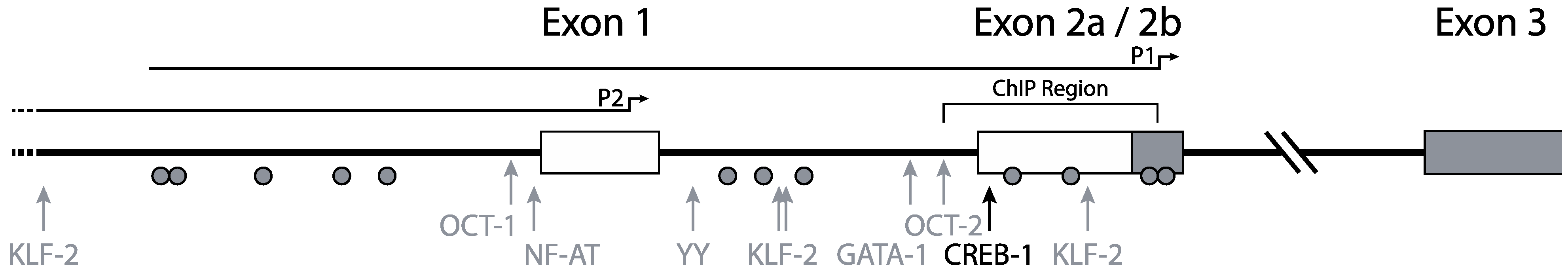
Mummidi et al. previously elucidated the organization and promoter usage of the CCR5 gene (Figure 1) [16,30]. Two distinct functional promoter regions for the CCR5 gene were identified using luciferase-reporter constructs containing CCR5 regulatory regions: a downstream promoter region, nowadays designated as P1, and an upstream promoter region, designated P2. The P2 promoter in front of exon 1 was identified as a weaker promoter than the P1 promoter, which is located near exon 2b (Figure 1) [16]. Using 5' RACE and RT-PCR the authors found four exons and two introns in the genomic organization of CCR5. The authors suggested that two full-length transcripts arose from promoter P1 and numerous truncated transcripts (i.e., missing exon 1) arose from promoter P2. Between exon 2a and exon 2b there is no intron. Both the truncated and the full-length transcripts give rise to the full length CCR5 protein. The two different full-length transcripts designated CCR5A and CCR5B differ by 235 nucleotides, corresponding to a lack of exon 2 in the CCR5B transcript.
Both P1 and P2 lack the canonical TATA and CCAAT motifs, although in P1 a non-classical TATA-box can be found. Unlike other TATA-less promoters, the CCR5 promoters have a relatively low CpG content.
Figure 1.
Promoter organization of the CCR5 gene. CCR5 transcription is initiated from two distinct promoters. Full-length transcripts arise from promoter P1 and truncated transcripts from promoter P2. The black arrow indicates the most responsive CREB-1 site. Other transcription factors attributed to CCR5 transcription regulation are depicted with gray arrows. Grey circles indicate CpG residues. The region investigated for epigenetic regulation by chromatin immune precipitation is annotated as “ChIP region”. Adapted from [31].
Figure 1.
Promoter organization of the CCR5 gene. CCR5 transcription is initiated from two distinct promoters. Full-length transcripts arise from promoter P1 and truncated transcripts from promoter P2. The black arrow indicates the most responsive CREB-1 site. Other transcription factors attributed to CCR5 transcription regulation are depicted with gray arrows. Grey circles indicate CpG residues. The region investigated for epigenetic regulation by chromatin immune precipitation is annotated as “ChIP region”. Adapted from [31].
3. CCR5 Regulation by Transcription Factors
In their initial characterization of the CCR5 promoter, Liu and coworkers suggested that CCR5 transcription could be up-regulated by NF-κB [32]. Indeed, others and we found several potential binding sites for NF-κB in the CCR5 P1-promoter [33]. However, the results of the study by Kuipers et al. indicate that CCR5 expression is neither induced nor modulated by NF-κB [33]. In addition, these authors also found binding sites for interferon regulatory factors (IRFs) and CREB-1 in the CCR5 P1- and P2-promoters. Like for NF-κB and in contrast to CREB-1, Kuipers et al. could not establish a role for the IFN-γ induced regulatory pathway in CCR5 transcription. By using various reporter assays, as well as by competition for CREB-1 binding-sites by inducible cAMP early repressor (ICER), which is induced by forskolin treatment, the authors concluded that CCR5 transcription is regulated by CREB-1 [33]. More recently, Banerjee et al. also showed that in the TF-1 human bone marrow progenitor cell line, CCR5 is regulated at the transcriptional level by the cAMP/PKA/CREB pathway [34].
In line with an important role for CREB-1 in CCR5 expression are the transcription levels of CREB-1 isoforms and ICER in activated versus naïve CD4+ T cells. Upon activation of CD4+ T cells, the relative transcription levels of CREB-1 isoforms increase whereas transcription levels of ICER decrease [31].
The site most responsive to the CREB-1-mediated transactivation is located in the downstream P1-promoter (Figure 1). Promoter constructs using the upstream promoter however were unresponsive to CREB-1. This suggests a repressive function of the upstream promoter region. This hypothesis is underscored by the fact that the longest downstream promoter construct, including part of the upstream region, is less responsive to CREB-1 than truncated downstream promoter constructs. These findings corroborate those of others that mapped a repressive element, corresponding to a region upstream, affecting the CCR5 promoter [32,35,36]. Besides an important role for CREB-1 in the transcriptional regulation of CCR5, several other studies have revealed also a contribution for Oct1 and Oct2 [15,37], NF-AT [32], YY1 [38], GATA-1 [39], and KLF2 [40] in the modulation of CCR5 transcription.
4. Epigenetic Regulation
In addition to the transcription factors discussed above, most if not all genes are also regulated at the transcriptional level by epigenetic processes. Since CCR5 is only expressed in a subset of T lymphocytes, monocytes, macrophages, or dendritic cells the contribution of epigenetic mechanisms in the cell-type specific transcriptional regulation of CCR5 was proposed and investigated [31,33].
In the cell nucleus, DNA is present as a protein-DNA complex called chromatin. The basic repeating unit of chromatin is the nucleosome, which consists of 146bp of DNA wrapped around an octamer of histones, comprising pairs of the core-histones H2A, H2B, H3 and H4. The structure of the chromatin can be altered by epigenetic regulation through modification of DNA and of histones. In this way, epigenetic regulation controls gene transcription by modifying the chromatin architecture in such a way that it influences the accessibility of DNA for transcription factors. Note, these epigenetic modifications are reversible allowing the chromatin structure to switch between open and closed states. This form of transcriptional control is thought to form also the basis for cell-to-cell inheritance of gene expression profiles [41]. Epigenetic modifications include methylation of DNA at CpG residues in gene promoters and post-translational modifications of histone tails such as acetylation and methylation [42].
DNA methylation is perhaps one of the best-studied epigenetic modifications. CpG residues are underrepresented in the human genome but are highly enriched in so-called CpG islands in most gene promoters [43,44]. As a general rule of thumb, gene expression is associated with unmethylated CpGs in gene promoters, while CpG methylation in gene promoters is associated with transcriptional repression [45]. More recently, the involvement of additional CpG containing regions, the so-called CpG island shores, at more distal locations from gene promoters in gene transcription has become apparent [46]. Mechanisms that underlie gene repression by histone methylation involve, amongst others, tri-methylation of histone H3 at lysine 9 (3MeK9H3) and at lysine 27 (3MeK27H3), and of histone H4 at lysine 20 (3MeK20H4) in proximal promoter chromatin [47,48,49,50]. Counteracting these repressive modifications are the transcriptionally permissive modifications tri-methylation of histone H3 at lysine 4 (3MeK4H3) and histone acetylation [50,51,52].
Together these modifications form a “histone code”, like the genetic code, that controls transcription levels of genes [53]. As mentioned above, one important trait of epigenetic regulation is that modifications to DNA and to histone tails are reversible. Furthermore, these activities are functionally linked [54].
4.1. “Classical” Epigenetic Regulation
In the case of the CCR5 locus, non-expressing cells such as Jurkat (a T leukemia cell line) cells have a densely methylated promoter. Conversely, cells on which CCR5 is highly expressed display a virtually unmethylated CCR5 promoter region [31].
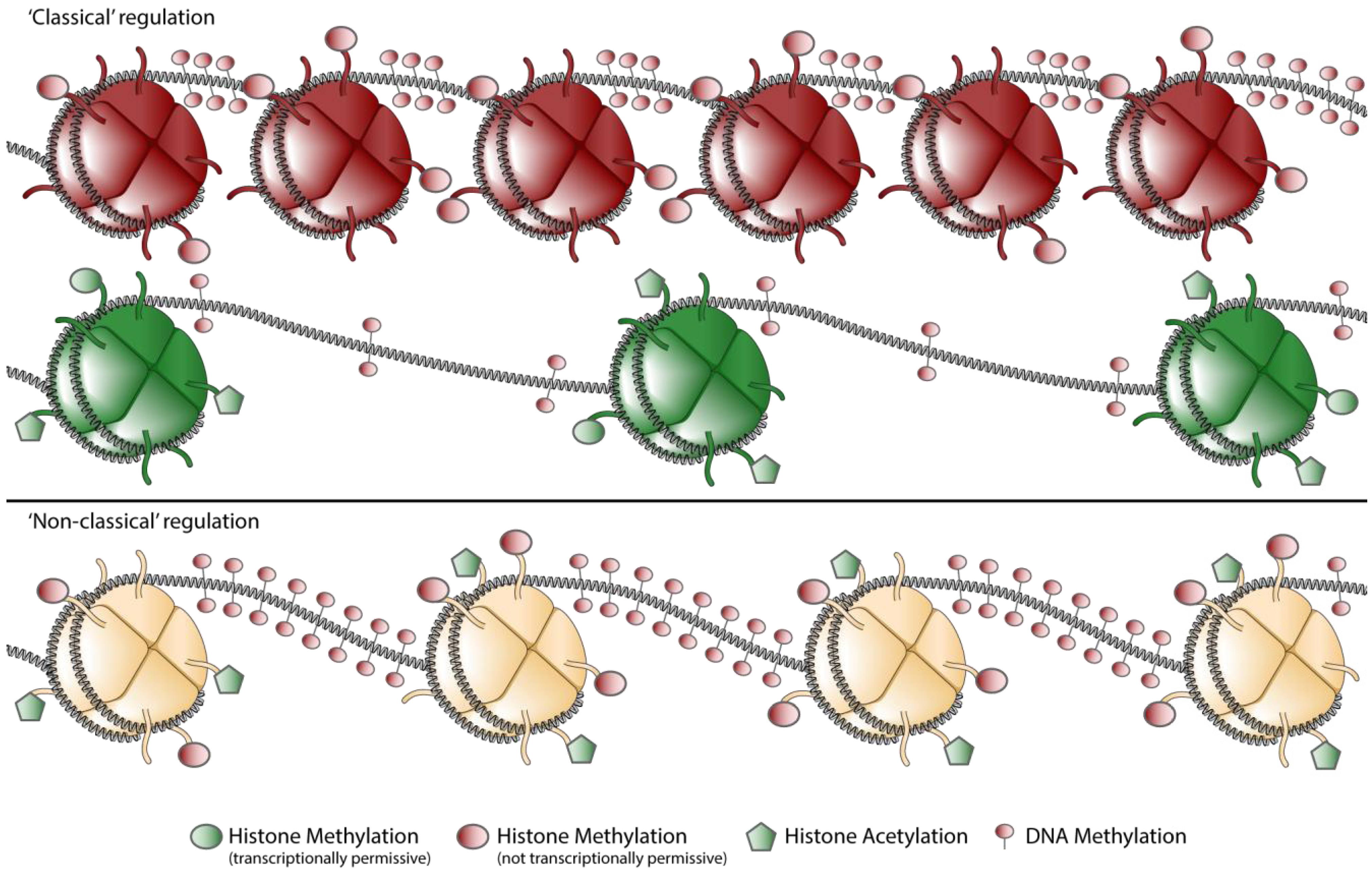
In a traditional view of epigenetic regulation, chromatin could either be in open (euchromatin) or closed (heterochromatin) conformation (Figure 2) [55,56]. Intermediate chromatin states were thought to be a transition between either hetero- or euchromatin and were thought to be only short lived [57]. When transcription was occurring, a gene promoter was enriched in acetylated lysine 9 in histone H3 (AcK9H3) and 3MeK4H3, whereas transcriptionally silenced genes where enriched in 3MeH3K27 and 3MeH3K9 [52,58].
Figure 2.
Schematic representation of chromatin states encountered in the CCR5 locus. Chromatin can be marked by mainly repressive or mainly permissive marks, regarded as the classical euchromatin (green) and heterochromatin (red) states (“classical” regulation). Nowadays it is widely appreciated that more complex forms of chromatin exist, hallmarked by both repressive and permissive marks in the same locus (“non classical” regulation). Note: For clarity DNA-methylation is drawn on the internucleosomal-DNA, whereas it has been shown that methylated DNA co-localizes also with nucleosomes [59].
Figure 2.
Schematic representation of chromatin states encountered in the CCR5 locus. Chromatin can be marked by mainly repressive or mainly permissive marks, regarded as the classical euchromatin (green) and heterochromatin (red) states (“classical” regulation). Nowadays it is widely appreciated that more complex forms of chromatin exist, hallmarked by both repressive and permissive marks in the same locus (“non classical” regulation). Note: For clarity DNA-methylation is drawn on the internucleosomal-DNA, whereas it has been shown that methylated DNA co-localizes also with nucleosomes [59].

When comparing activated and naïve CD4+ T cells, the CCR5 locus displays such a classical pattern [31,33]. Naïve CD4+ T cells are characterized be relatively low levels of AcH3 and relatively high levels of the repressive mark 3MeK20H4. When compared to activated T cells, naïve T cells also contain high levels of 3MeK27H3. Upon activation the repressive marks in the chromatin of the CCR5 promoter are replaced by activating marks. Chromatin of the CCR5 promoter in activated CD4+ T cells is almost exclusively covered with the activating marks AcH3 and 3MeK4H3. In other cell types however, the chromatin makeup of the CCR5 locus is more complex.
4.2. “Non-classical” Epigenetic Regulation
Bivalent chromatin structures, composed of both 3MeK4H3 and 3MeK27H3, were first found in embryonic stem cells. In these cells, bivalent states marked genes encoding transcription factors that play an essential role in development. It was proposed that these bivalent states would silence developmental genes while keeping them poised for activation. Upon differentiation, this bivalent state would be lost [60]. Later work not only identified bivalent, but also tri- and tetravalent chromatin states [61]. Furthermore, multivalent chromatin states where found to also occur in differentiated cells [31,61]. Lastly it turned out that bivalent, “poised” chromatin, is not completely silenced as proposed initially. Some transcripts arise from multivalent chromatin, thus multivalent chromatin might be a mechanism for transcriptional fine-tuning [62]. It is thought therefore that these multivalent states may be of importance for the control of gene expression in the activation of T cells and the differentiation of monocytes [62]. Chromatin Immunoprecipitation (ChIP) experiments performed on the CCR5 promoter region revealed that this promoter is extensively covered by multivalent marks [31].
In CCR5-expressing monocytes, the CCR5 promoter is covered with high levels of AcH3, but also with relative high levels of 3MeK9H3 and 3MeK27H3 and an intermediate level of 3MeK20H4. The histone modifications in monocytes are accompanied with relatively high levels of DNA methylation (Figure 2). Yet despite all these repressive modifications monocytes show CCR5 transcription levels that are markedly higher then naïve CD4+ T cells, albeit at a lower level than in activated CD4+ T cells [31].
Monocytes have the ability to differentiate into macrophages and dendritic cells. Upon differentiation CCR5 expression is lost. The fact that monocytes display high amounts of repressive marks in conjunction with histone acetylation may reflect the potential to rapidly shut down CCR5 transcription upon differentiation. The observed multivalent chromatin state of CCR5 as such might reflect the central role of CCR5 in the regulation of lymphoid cell trafficking.
4.3. Epigenetic Intervention
Epigenetic modifications are reversible and can be modulated by small molecule inhibitors. With CCR5 cell-type specific transcription being achieved by epigenetic regulation, small molecule inhibitors could modulate CCR5 transcription. Jurkat cells do not express CCR5, however it was demonstrated by using promoter constructs that Jurkat cells contain all the necessary transcription factors to achieve CCR5 transcription [32].
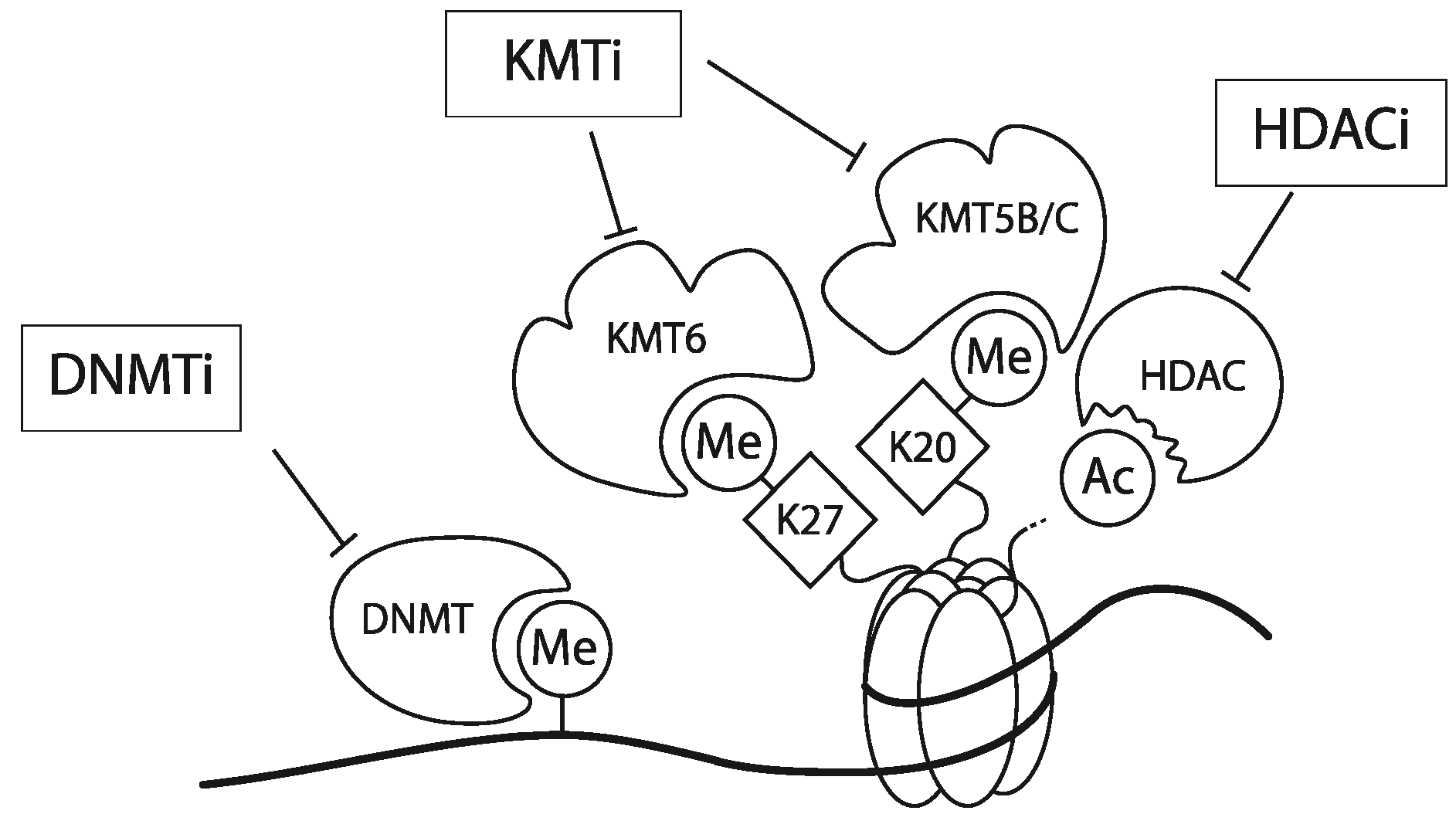
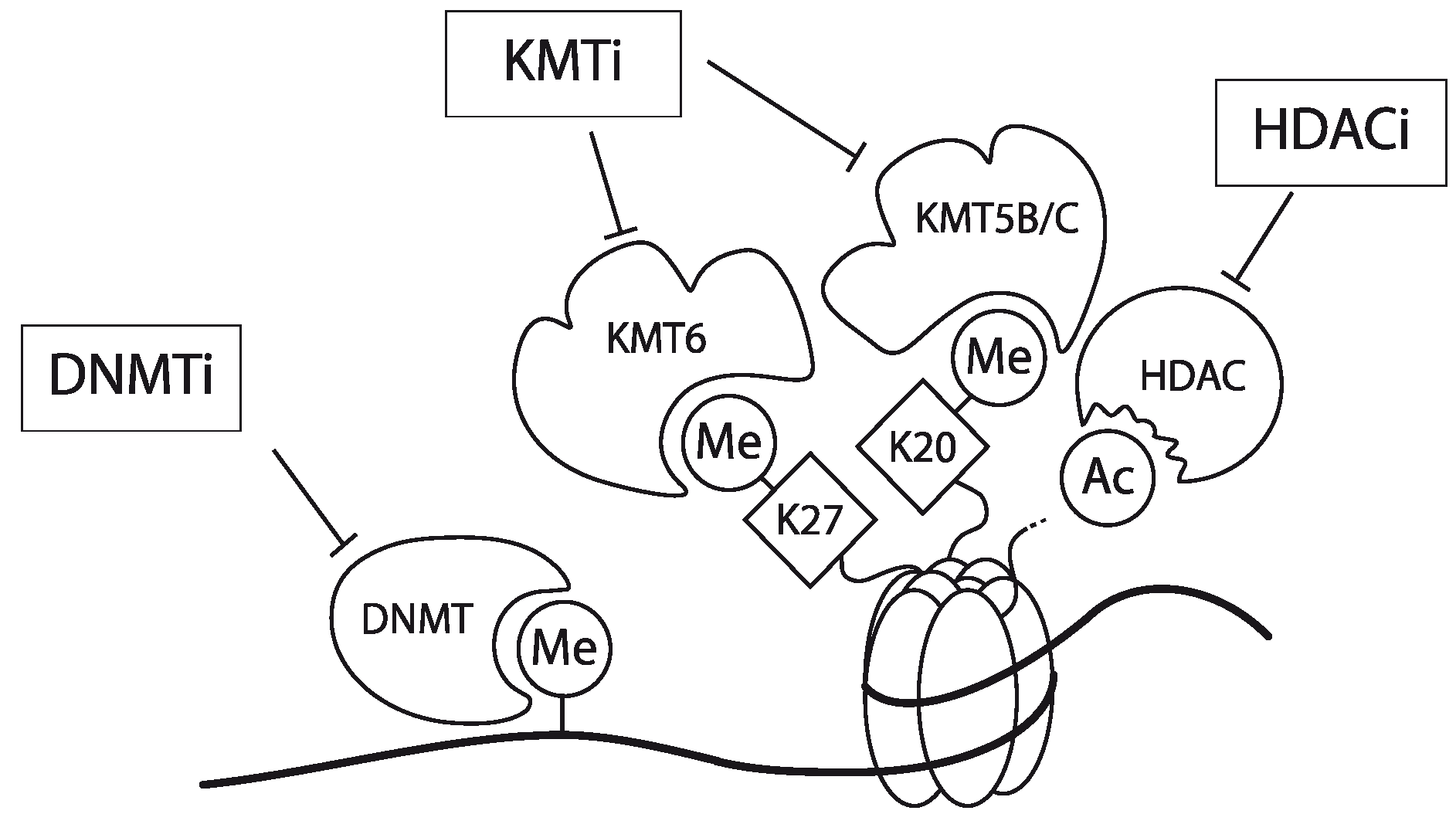
In Jurkat cells, DNA of the CCR5 promoter is densely methylated. Furthermore, chromatin encompassing the promoter region hardly contains AcH3 and is devoid of 3MeK4H3. Finally, the CCR5 promoter is enriched for the repressive chromatin marks 3MeK9H3, 3MeK27H3 and 3MeK20H4. Using a combination of a DNMT inhibitor (Zebularine), a lysine methyltransferase inhibitor (DZNep) and a histone deacetylase (HDAC) inhibitor (MS-275; specific for the class 1 HDACs 1 and 3 [63,64,65]) the expression of the CCR5 gene could be activated in Jurkat cells but also in other CCR5-deficient leukemic T cell-lines (Figure 3). Interestingly mono-treatment with these inhibitors showed only a very small effect [31]. Matalon et al. also have shown that treatment of CD4+ T cells and monocytes with HDAC inhibitors modulates the expression of CCR5 in these cell types [66]. Together, these observations underscore that CCR5 transcription is actively controlled by epigenetic mechanisms. It should be noted however, that these inhibitors act genome-wide and as such targeting these inhibitors to specific genes may prove to be challenging. Meanwile, some of these epigenetic inhibitors have been in clinical use already for decades (e.g., for the management of epilepsy with Depakene, or valproic acid), despite the fact that they act genome-wide and they seem to be well tolerated [67,68].
Figure 3.
Schematic representation of the working mechanism for the pharmacological intervention in CCR5 transcription. Re-expression was achieved by combinatorial treatment with a DNMT inhibitor (DNMTi, Zebularine), a lysine methyltransferase inhibitor (KMTi, DZNep) and a HDAC inhibitor (HDACi, MS-275). Adapted from [31].
Figure 3.
Schematic representation of the working mechanism for the pharmacological intervention in CCR5 transcription. Re-expression was achieved by combinatorial treatment with a DNMT inhibitor (DNMTi, Zebularine), a lysine methyltransferase inhibitor (KMTi, DZNep) and a HDAC inhibitor (HDACi, MS-275). Adapted from [31].
5. Conclusions
Transcription of CCR5 is achieved by a complex interplay of transcription factors and various forms of epigenetic regulation. Cell type-specific transcription of CCR5 is achieved by epigenetic regulation. Epigenetic regulation also allows for rapid transcriptional shutdown or initiation of the CCR5 locus upon differentiation of various cell types. This epigenetic regulation is a perfect entry point for pharmaceutical intervention. Given the importance of CCR5 in numerous inflammatory diseases and as co-receptor for HIV-1 gaining entrance into cells, pharmaceutical intervention in the epigenetic regulation of CCR5 transcription may prove to be valuable in future disease management.
Acknowledgments
This research was supported by grants from the Dutch MS Research Foundation and the Translation of Excellence in Regenerative Medicine (TeRM) Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science.
References and Notes
- Samson, M.; Labbe, O.; Mollereau, C.; Vassart, G.; Parmentier, M. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry 1996, 35, 3362–3367. [Google Scholar] [CrossRef]
- Combadiere, C.; Ahuja, S.K.; Tiffany, H.L.; Murphy, P.M. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1(alpha), MIP-1(beta), and RANTS. J. Leukoc. Biol. 1996, 60, 147–152. [Google Scholar]
- Raport, C.J.; Gosling, J.; Schweickart, V.L.; Gray, P.W.; Charo, I.F. Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1beta, and MIP-1alpha. J. Biol. Chem. 1996, 271, 17161–17166. [Google Scholar]
- Bursill, C.A.; Channon, K.M.; Greaves, D.R. The role of chemokines in atherosclerosis: Recent evidence from experimental models and population genetics. Curr. Opin. Lipidol. 2004, 15, 145–149. [Google Scholar] [CrossRef]
- Ribeiro, S.; Horuk, R. The clinical potential of chemokine receptor antagonists. Pharmacol. Ther. 2005, 107, 44–58. [Google Scholar] [CrossRef]
- Biber, K.; Zuurman, M.W.; Dijkstra, I.M.; Boddeke, H.W. Chemokines in the brain: Neuroimmunology and beyond. Curr. Opin. Pharmacol. 2002, 2, 63–68. [Google Scholar] [CrossRef]
- Schober, A. Chemokines in vascular dysfunction and remodeling. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1950–1959. [Google Scholar] [CrossRef]
- Zernecke, A.; Liehn, E.A.; Gao, J.-L.; Kuziel, W.A.; Murphy, P.M.; Weber, C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: Involvement of IL-10. Blood 2006, 107, 4240–4243. [Google Scholar] [CrossRef]
- Zernecke, A.; Shagdarsuren, E.; Weber, C. Chemokines in atherosclerosis: An update. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1897–1908. [Google Scholar] [CrossRef]
- Kraaijeveld, A.O.; de Jager, S.C.; de Jager, W.J.; Prakken, B.J.; McColl, S.R.; Haspels, I.; Putter, H.; van Berkel, T.J.; Nagelkerken, L.; Jukema, J.W.; et al. CC chemokine ligand-5 (CCL5/RANTES) and CC chemokine ligand-18 (CCL18/PARC) are specific markers of refractory unstable angina pectoris and are transiently raised during severe ischemic symptoms. Circulation 2007, 116, 1931–1941. [Google Scholar] [CrossRef]
- Wu, L.; Paxton, W.A.; Kassam, N.; Ruffing, N.; Rottman, J.B.; Sullivan, N.; Choe, H.; Sodroski, J.; Newman, W.; Koup, R.A.; et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 1997, 185, 1681–1691. [Google Scholar] [CrossRef]
- Oswald-Richter, K.; Grill, S.M.; Leelawong, M.; Tseng, M.; Kalams, S.A.; Hulgan, T.; Haas, D.W.; Unutmaz, D. Identification of a CCR5-expressing T cell subset that is resistant to R5-tropic HIV infection. PLoS Pathog. 2007, 3, e58. [Google Scholar] [CrossRef]
- Carrington, M.; Dean, M.; Martin, M.P.; O’Brien, S.J. Genetics of HIV-1 infection: Chemokine receptor CCR5 polymorphism and its consequences. Hum. Mol. Genet. 1999, 8, 1939–1945. [Google Scholar]
- Ebert, L.M.; McColl, S.R. Up-regulation of CCR5 and CCR6 on distinct subpopulations of antigen-activated CD4+ T lymphocytes. J. Immunol. 2002, 168, 65–72. [Google Scholar]
- Mummidi, S.; Adams, L.M.; VanCompernolle, S.E.; Kalkonde, M.; Camargo, J.F.; Kulkarni, H.; Bellinger, A.S.; Bonello, G.; Tagoh, H.; Ahuja, S.S.; et al. Production of specific mRNA transcripts, usage of an alternate promoter, and octamer-binding transcription factors influence the surface expression levels of the HIV coreceptor CCR5 on primary T cells. J. Immunol. 2007, 178, 5668–5681. [Google Scholar]
- Mummidi, S.; Ahuja, S.S.; McDaniel, B.L.; Ahuja, S.K. The human CC chemokine receptor 5 (CCR5) gene. Multiple transcripts with 5'-end heterogeneity, dual promoter usage, and evidence for polymorphisms within the regulatory regions and noncoding exons. J. Biol. Chem. 1997, 272, 30662–30671. [Google Scholar]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar] [CrossRef]
- Guignard, F.; Combadiere, C.; Tiffany, H.L.; Murphy, P.M. Gene organization and promoter function for CC chemokine receptor 5 (CCR5). J. Immunol. 1998, 160, 985–992. [Google Scholar]
- Van der Merwe, P.A.; Davis, S.J. The immunological synapse—A multitasking system. Science 2002, 295, 1479–1480. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Serbina, N.V.; Jia, T.; Hohl, T.M.; Pamer, E.G. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 2008, 26, 421–452. [Google Scholar] [CrossRef]
- Schmitz, G.; Leuthauser-Jaschinski, K.; Orso, E. Are circulating monocytes as microglia orthologues appropriate biomarker targets for neuronal diseases? Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 307–330. [Google Scholar] [CrossRef]
- Chan, W.Y.; Kohsaka, S.; Rezaie, P. The origin and cell lineage of microglia: New concepts. Brain Res. Rev. 2007, 53, 344–354. [Google Scholar] [CrossRef]
- Hansson, G.K.; Robertson, A.K.L.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef]
- Fox, R.J.; Kivisakk, P.; Fisher, E.; Tucky, B.; Lee, J.C.; Rudick, R.A.; Ransohoff, R.M. Multiple sclerosis: Chemokine receptor expression on circulating lymphocytes in correlation with radiographic measures of tissue injury. Mult. Scler. 2008, 14, 1036–1043. [Google Scholar] [CrossRef]
- Trebst, C.; Konig, F.; Ransohoff, R.; Bruck, W.; Stangel, M. CCR5 expression on macrophages/microglia is associated with early remyelination in multiple sclerosis lesions. Mult. Scler. 2008, 14, 728–733. [Google Scholar] [CrossRef]
- Mummidi, S.; Bamshad, M.; Ahuja, S.S.; Gonzalez, E.; Feuillet, P.M.; Begum, K.; Galvis, M.C.; Kostecki, V.; Valente, A.J.; Murthy, K.K.; et al. Evolution of human and non-human primate CC chemokine receptor 5 gene and mRNA. Potential roles for haplotype and mRNA diversity, differential haplotype-specific transcriptional activity, and altered transcription factor binding to polymorphic nucleotides in the pathogenesis of HIV-1 and simian immunodeficiency virus. J. Biol. Chem. 2000, 275, 18946–18961. [Google Scholar]
- Wierda, R.J.; Kuipers, H.F.; van Eggermond, M.C.J.A.; Benard, A.; van Leeuwen, J.C.; Carluccio, S.; Geutskens, S.B.; Jukema, J.W.; Marquez, V.E.; Quax, P.H.A.; et al. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J. Cell. Mol. Med. 2012, 16, 1866–1877. [Google Scholar] [CrossRef]
- Liu, R.; Zhao, X.; Gurney, T.A.; Landau, N.R. Functional analysis of the proximal CCR5 promoter. AIDS Res. Hum. Retroviruses 1998, 14, 1509–1519. [Google Scholar] [CrossRef]
- Kuipers, H.F.; Biesta, P.J.; Montagne, L.J.; van Haastert, E.S.; van der Valk, P.; van den Elsen, P.J. CC chemokine receptor 5 gene promoter activation by the cyclic AMP response element binding transcription factor. Blood 2008, 112, 1610–1619. [Google Scholar] [CrossRef]
- Banerjee, A.; Pirrone, V.; Wigdahl, B.; Nonnemacher, M.R. Transcriptional regulation of the chemokine co-receptor CCR5 by the cAMP/PKA/CREB pathway. Biomed. Pharmacother. 2011, 65, 293–297. [Google Scholar] [CrossRef]
- Jin, Q.; Agrawal, L.; Meyer, L.; Tubiana, R.; Theodorou, I.; Alkhatib, G. CCR5Delta32 59537-G/A promoter polymorphism is associated with low translational efficiency and the loss of CCR5Delta32 protective effects. J. Virol. 2008, 82, 2418–2426. [Google Scholar] [CrossRef]
- Moriuchi, H.; Moriuchi, M.; Fauci, A.S. Cloning and analysis of the promoter region of CCR5, a coreceptor for HIV-1 entry. J. Immunol. 1997, 159, 5441–5449. [Google Scholar]
- Moriuchi, M.; Moriuchi, H. Octamer transcription factors up-regulate the expression of CCR5, a coreceptor for HIV-1 entry. J. Biol. Chem. 2001, 276, 8639–8642. [Google Scholar]
- Moriuchi, M.; Moriuchi, H. YY1 transcription factor down-regulates expression of CCR5, a major coreceptor for HIV-1. J. Biol. Chem. 2003, 278, 13003–13007. [Google Scholar] [CrossRef]
- Moriuchi, M.; Moriuchi, H.; Fauci, A.S. GATA-1 transcription factor transactivates the promoter for CCR5, a coreceptor for human immunodeficiency virus type 1 entry. Blood 1999, 93, 1433–1435. [Google Scholar]
- Richardson, M.W.; Jadlowsky, J.; Didigu, C.A.; Doms, R.W.; Riley, J.L. Kruppel-like Factor 2 Modulates CCR5 Expression and Susceptibility to HIV-1 Infection. J. Immunol. 2012. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG islands as gene markers in the human genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Rice, J.C.; Briggs, S.D.; Ueberheide, B.; Barber, C.M.; Shabanowitz, J.; Hunt, D.F.; Shinkai, Y.; Allis, C.D. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 2003, 12, 1591–1598. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Wang, P.; Lin, C.; Smith, E.R.; Guo, H.; Sanderson, B.W.; Wu, M.; Gogol, M.; Alexander, T.; Seidel, C.; Wiedemann, L.M.; et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 2009, 29, 6074–6085. [Google Scholar]
- Yan, C.; Boyd, D.D. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Mol. Cell. Biol. 2006, 26, 6357–6371. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Vaissiere, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef]
- Lamond, A.I.; Earnshaw, W.C. Structure and function in the nucleus. Science 1998, 280, 547–553. [Google Scholar] [CrossRef]
- Van Steensel, B. Chromatin: Constructing the big picture. EMBO J. 2011, 30, 1885–1895. [Google Scholar] [CrossRef]
- Schwartz, Y.B.; Pirrotta, V. Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 2008, 20, 266–273. [Google Scholar] [CrossRef]
- Bender, J. Chromatin-based silencing mechanisms. Curr. Opin. Plant Biol. 2004, 7, 521–526. [Google Scholar] [CrossRef]
- Ball, D.J.; Gross, D.S.; Garrard, W.T. 5-methylcytosine is localized in nucleosomes that contain histone H1. Proc. Natl. Acad. Sci. USA 1983, 80, 5490–5494. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Bapat, S.A.; Jin, V.; Berry, N.; Balch, C.; Sharma, N.; Kurrey, N.; Zhang, S.; Fang, F.; Lan, X.; Li, M.; et al. Multivalent epigenetic marks confer microenvironment-responsive epigenetic plasticity to ovarian cancer cells. Epigenetics 2010, 5, 716–729. [Google Scholar] [CrossRef]
- De Gobbi, M.; Garrick, D.; Lynch, M.; Vernimmen, D.; Hughes, J.R.; Goardon, N.; Luc, S.; Lower, K.M.; Sloane-Stanley, J.A.; Pina, C.; et al. Generation of bivalent chromatin domains during cell fate decisions. Epigenet. Chromatin 2011, 4, 9. [Google Scholar] [CrossRef]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4597. [Google Scholar]
- Hu, E.; Dul, E.; Sung, C.-M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.-F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; de Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar]
- Matalon, S.; Palmer, B.E.; Nold, M.F.; Furlan, A.; Kassu, A.; Fossati, G.; Mascagni, P.; Dinarello, C.A. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J. Acquir. Immune Defic. Syndr. 2010, 54, 1–9. [Google Scholar]
- Gerstner, T.; Bell, N.; Konig, S. Oral valproic acid for epilepsy—Long-term experience in therapy and side effects. Expert. Opin. Pharmacother. 2008, 9, 285–292. [Google Scholar] [CrossRef]
- Henry, T.R. The history of valproate in clinical neuroscience. Psychopharmacol. Bull. 2003, 37, 5–16. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Wierda, R.J.; Van den Elsen, P.J. Genetic and Epigenetic Regulation of CCR5 Transcription. Biology 2012, 1, 869-879. https://doi.org/10.3390/biology1030869
AMA Style
Wierda RJ, Van den Elsen PJ. Genetic and Epigenetic Regulation of CCR5 Transcription. Biology. 2012; 1(3):869-879. https://doi.org/10.3390/biology1030869
Chicago/Turabian StyleWierda, Rutger J., and Peter J. Van den Elsen. 2012. "Genetic and Epigenetic Regulation of CCR5 Transcription" Biology 1, no. 3: 869-879. https://doi.org/10.3390/biology1030869