
Influence of pH on Morphology and Structure during Hydrolytic Degradation of the Segmented GL-b-[GL-co-TMC-co-CL]-b-GL Copolymer

 , and
, and
Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials
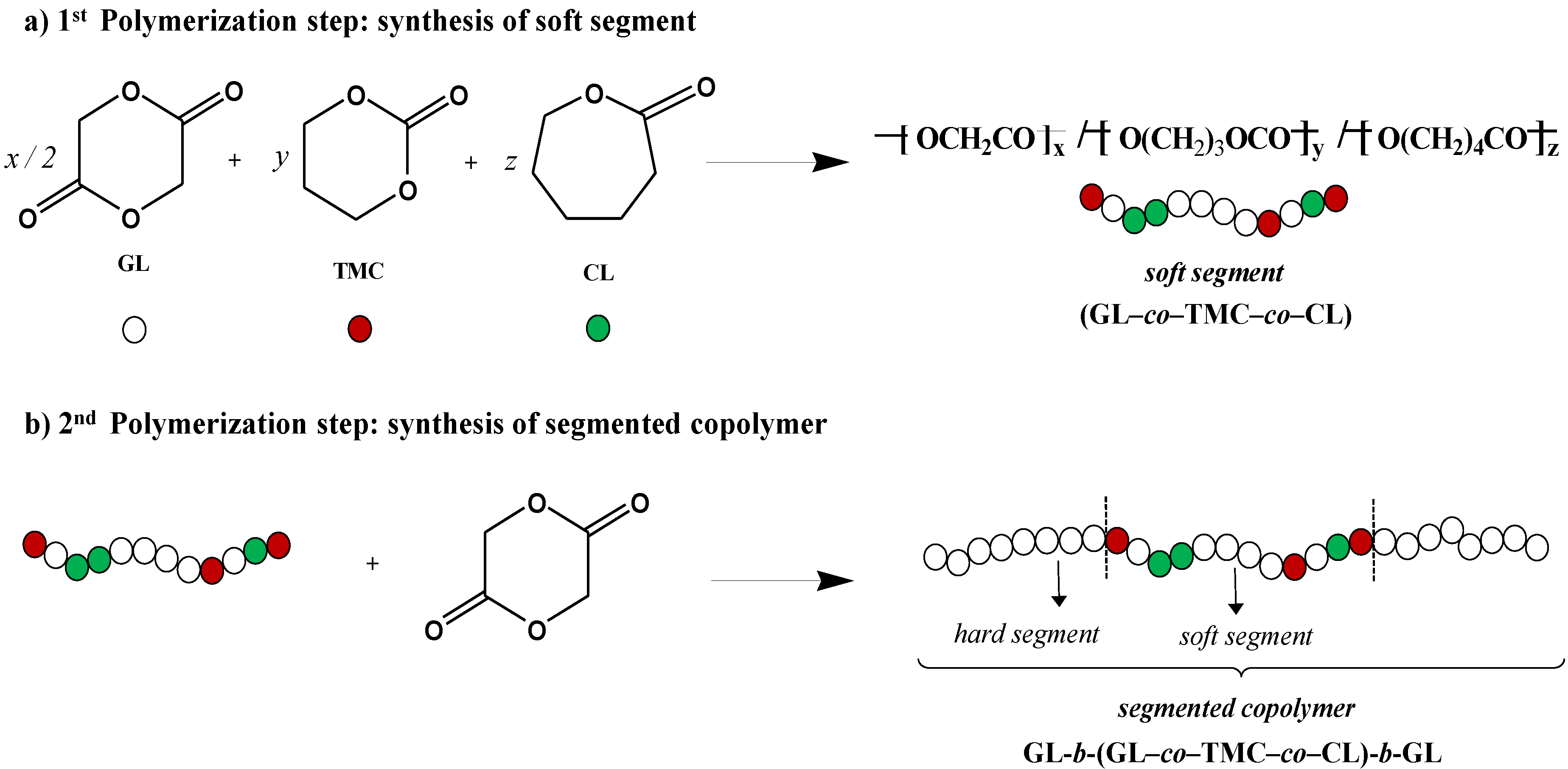
2.2. Hydrolytic Degradation
2.3. Measurements
3. Results and Discussion
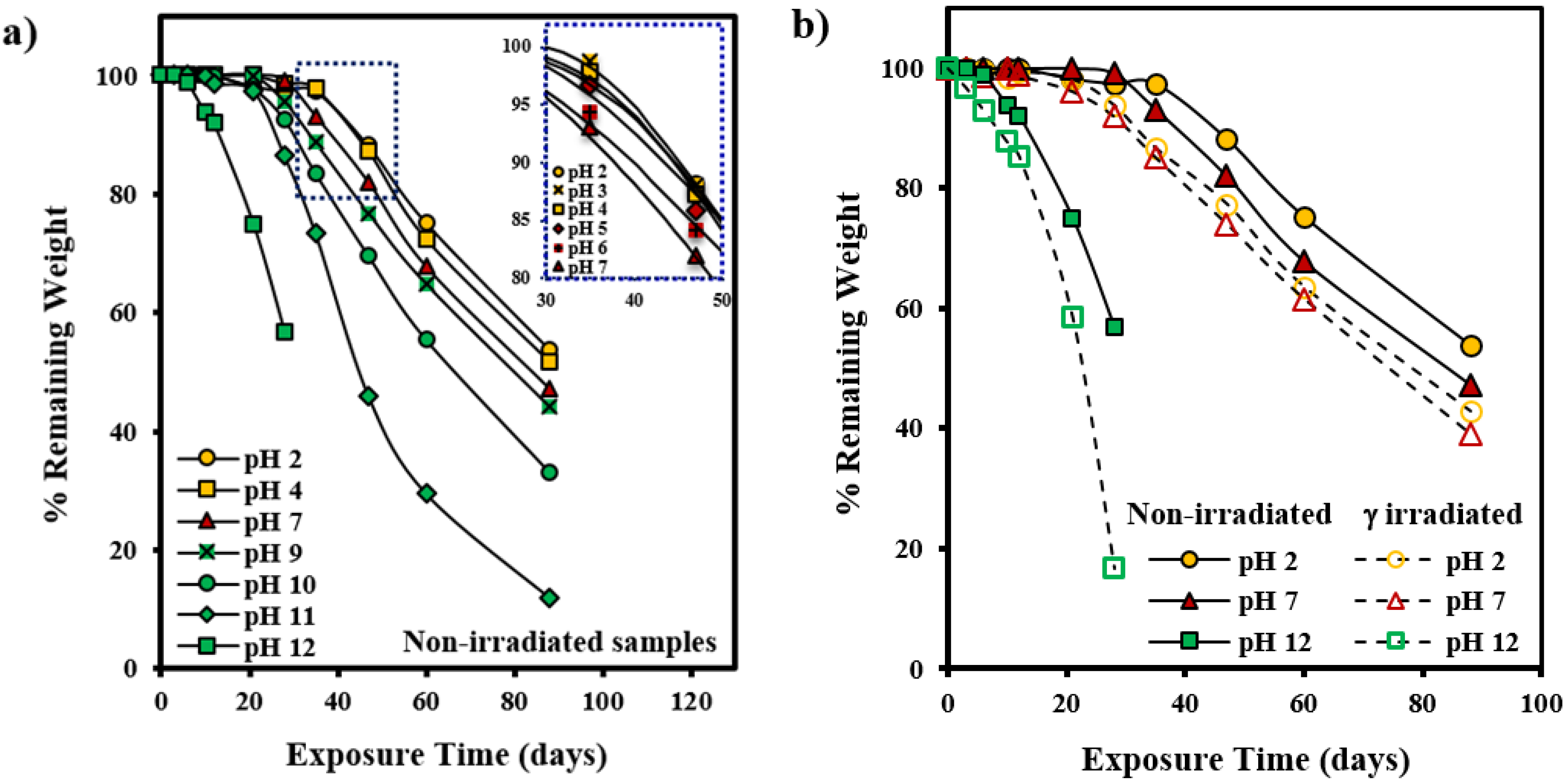
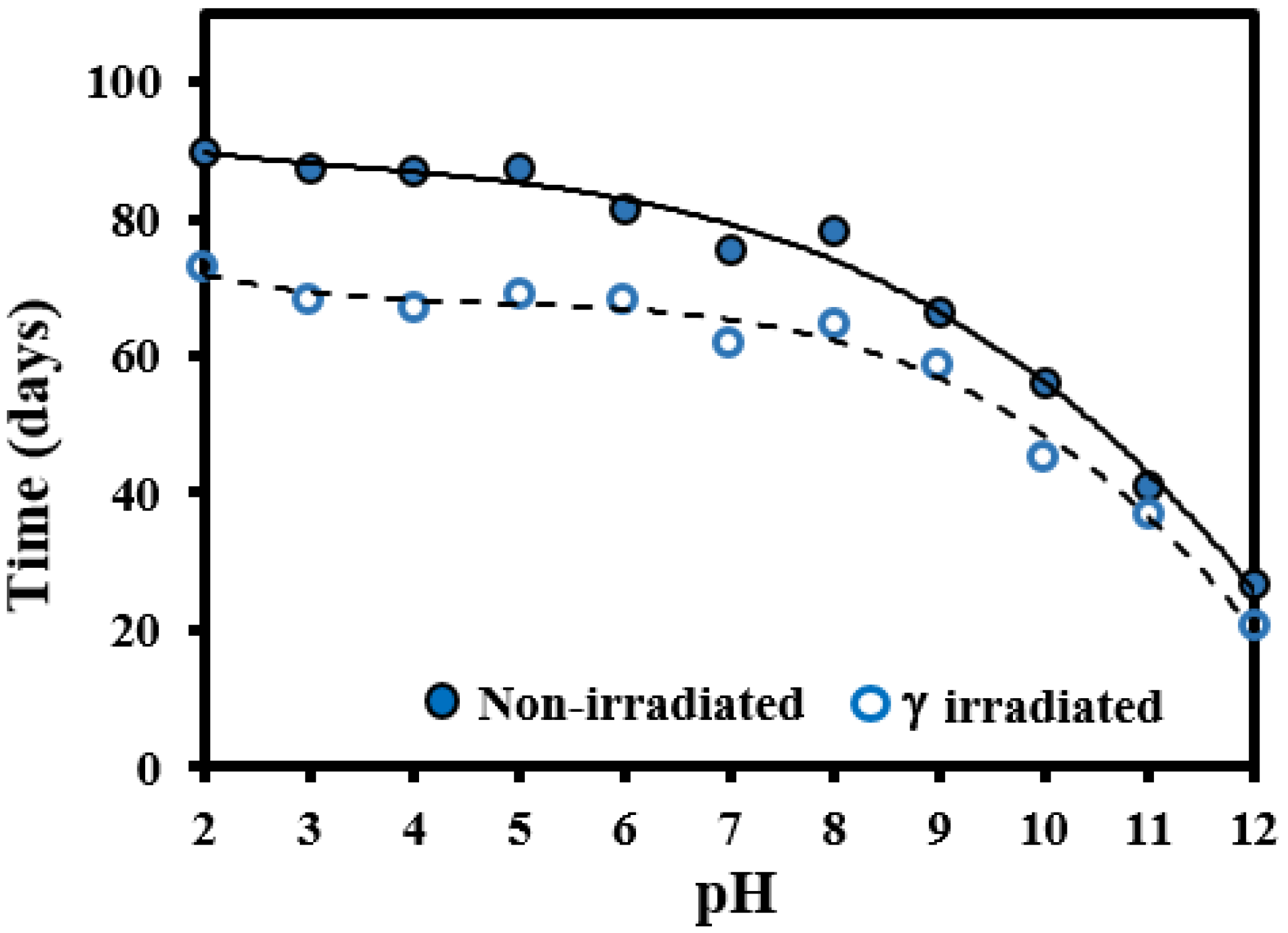
3.1. Hydrolytic Degradation of GL-b-[GL-co-TMC-co-CL]-b-GL in Different pH Media
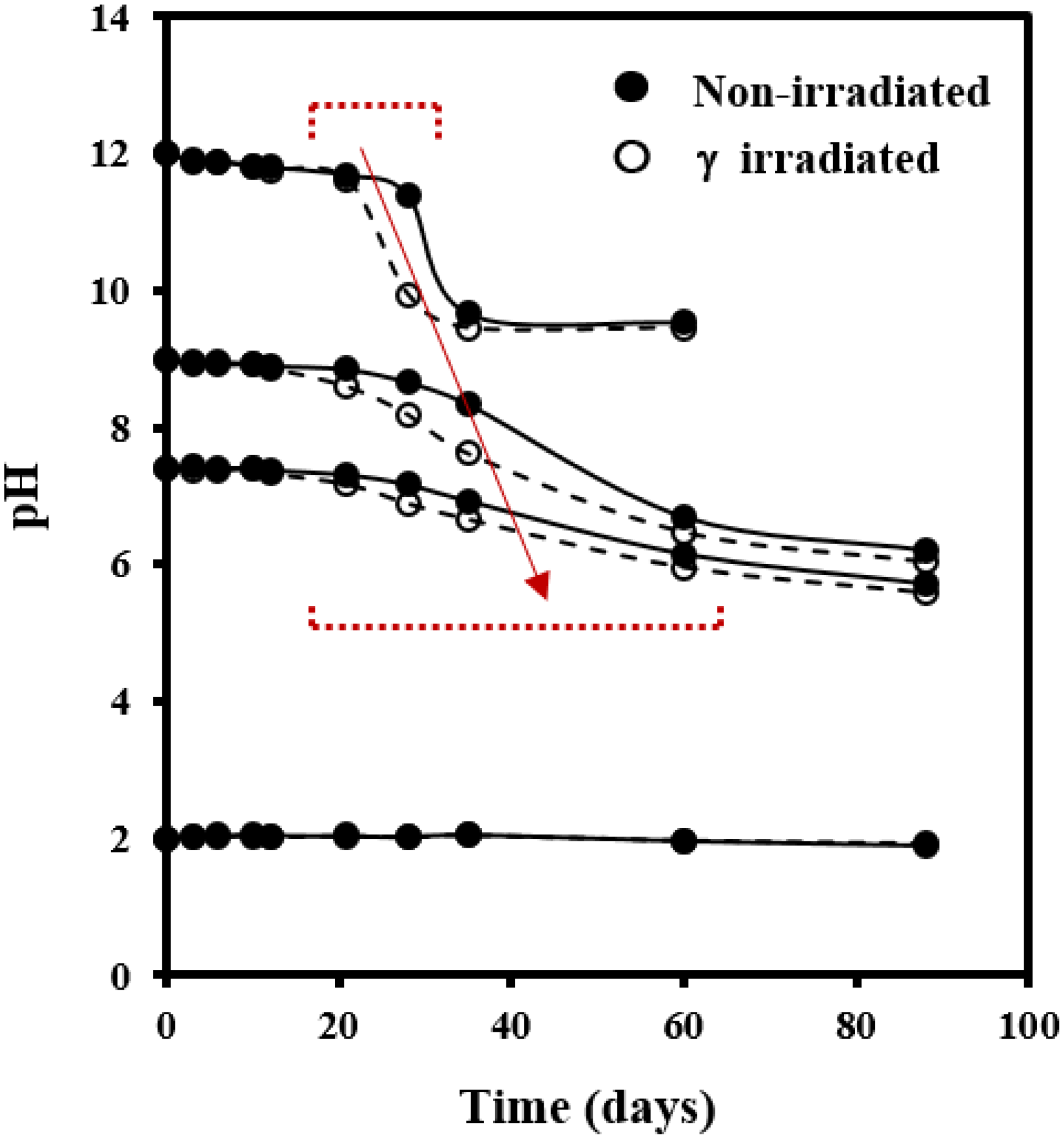
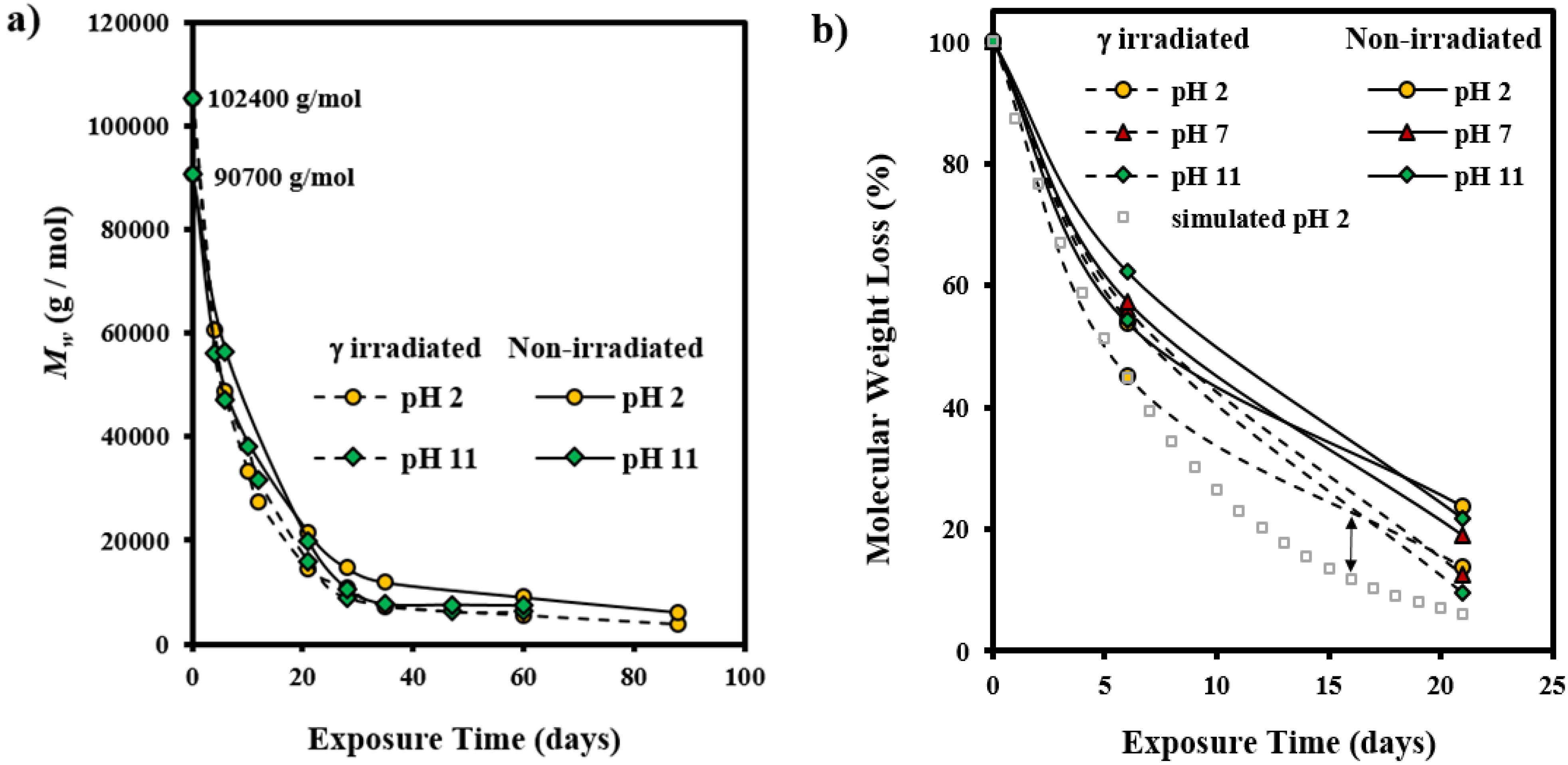

3.2. Morphological Changes during Hydrolytic Degradation of GL-b-[GL-co-TMC-co-CL]-b-GL in Different pH Media
- (1)
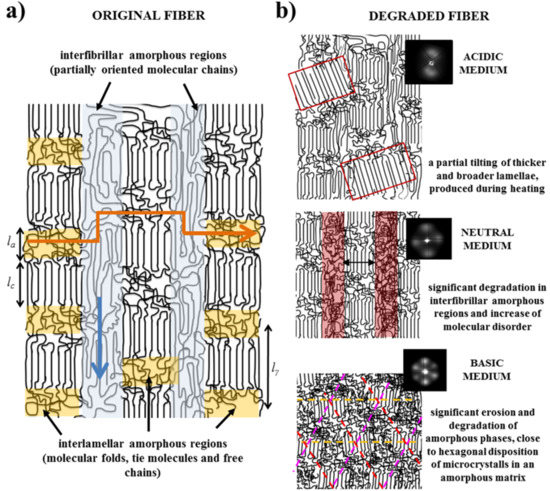
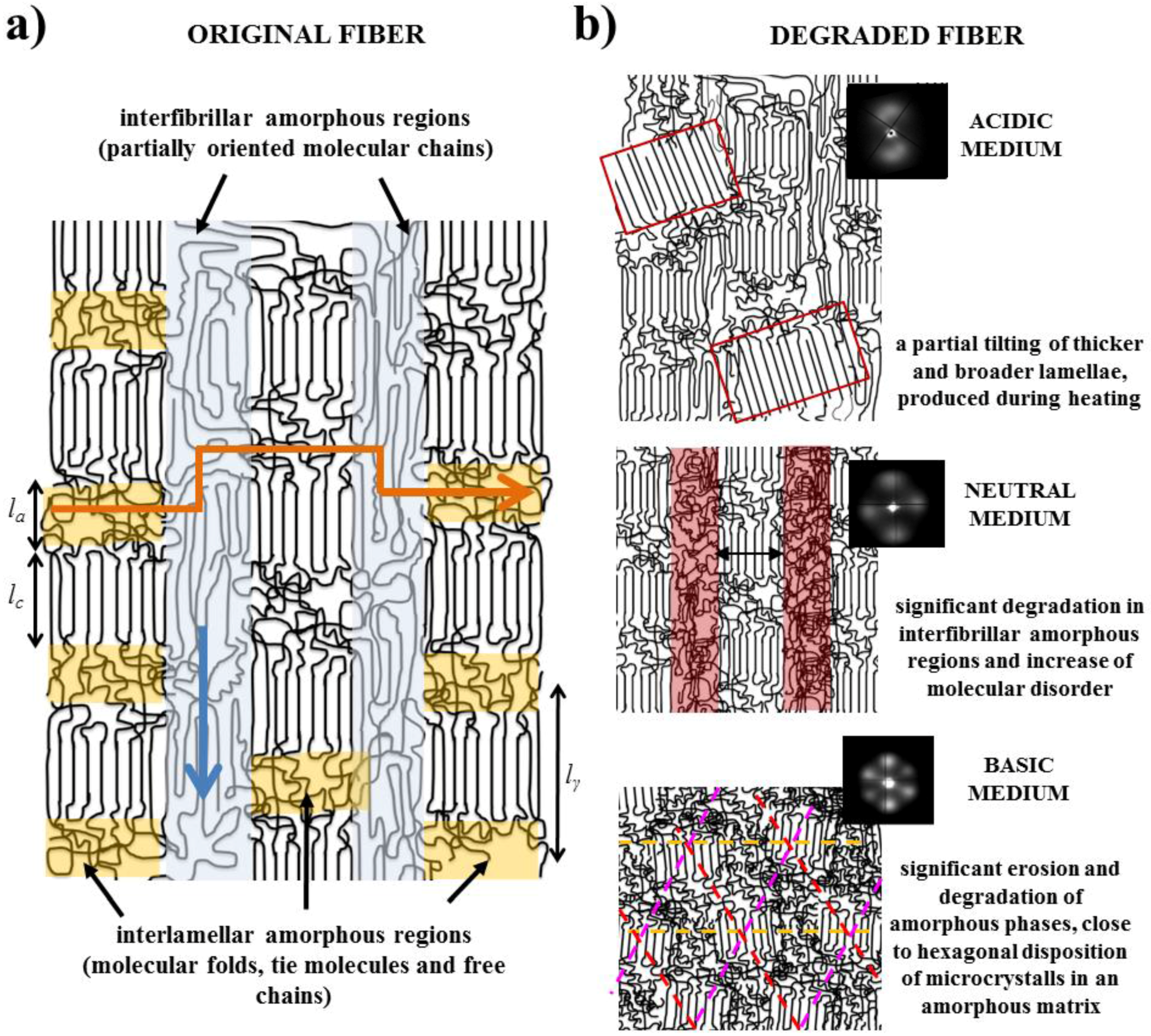
- Fibers are constituted by oriented lamellae and different amorphous regions which will be more susceptible to the hydrolysis process. In fact, oriented crystallites are embedded in an amorphous matrix, being possible to distinguish between interlamellar and interfibrilar amorphous regions (Figure 8a), according to previously postulated models [40,41]. The interlamellar domains alternate with lamellae in the direction of the fiber and possess the lowest molecular orientation and density since they are formed by molecular folds, tie chain segments between adjacent lamellar structures, and free chain ends. Therefore, these domains appear to be the most susceptible to hydrolysis. On the contrary, the interfibrilar domains may have a partial orientation and correspond to the regions placed on lateral sides of lamellae arranged in a fibrillar way. Degradation may therefore follow two different pathways: longitudinal and transversal depending on whether the hydrolysis proceeds mainly through the interfibrilar or interlamellar regions, respectively.
- (2)
- The hydrolysis mechanism is different as previously indicated for acid and basic media. Differences not only concern the kinetic mechanism but also the capability to solubilize degradation products. In this way, retention may be significant in both the interlamellar and interfibrillar regions when an acidic medium is employed and therefore lateral and longitudinal diffusion of water molecules could be hindered. On the contrary, rapid solubilization of degradation products may enhance the surface erosion of exposed fibers in a basic medium.
- (3)
- Morphology of fibers is not completely homogeneous due to a spinning process where the fiber surface is cooled faster than the core. Therefore, the shell layer should have different degree of molecular orientation and probably is constituted by a slightly different lamellar architecture (e.g. size of crystalline and amorphous domains).
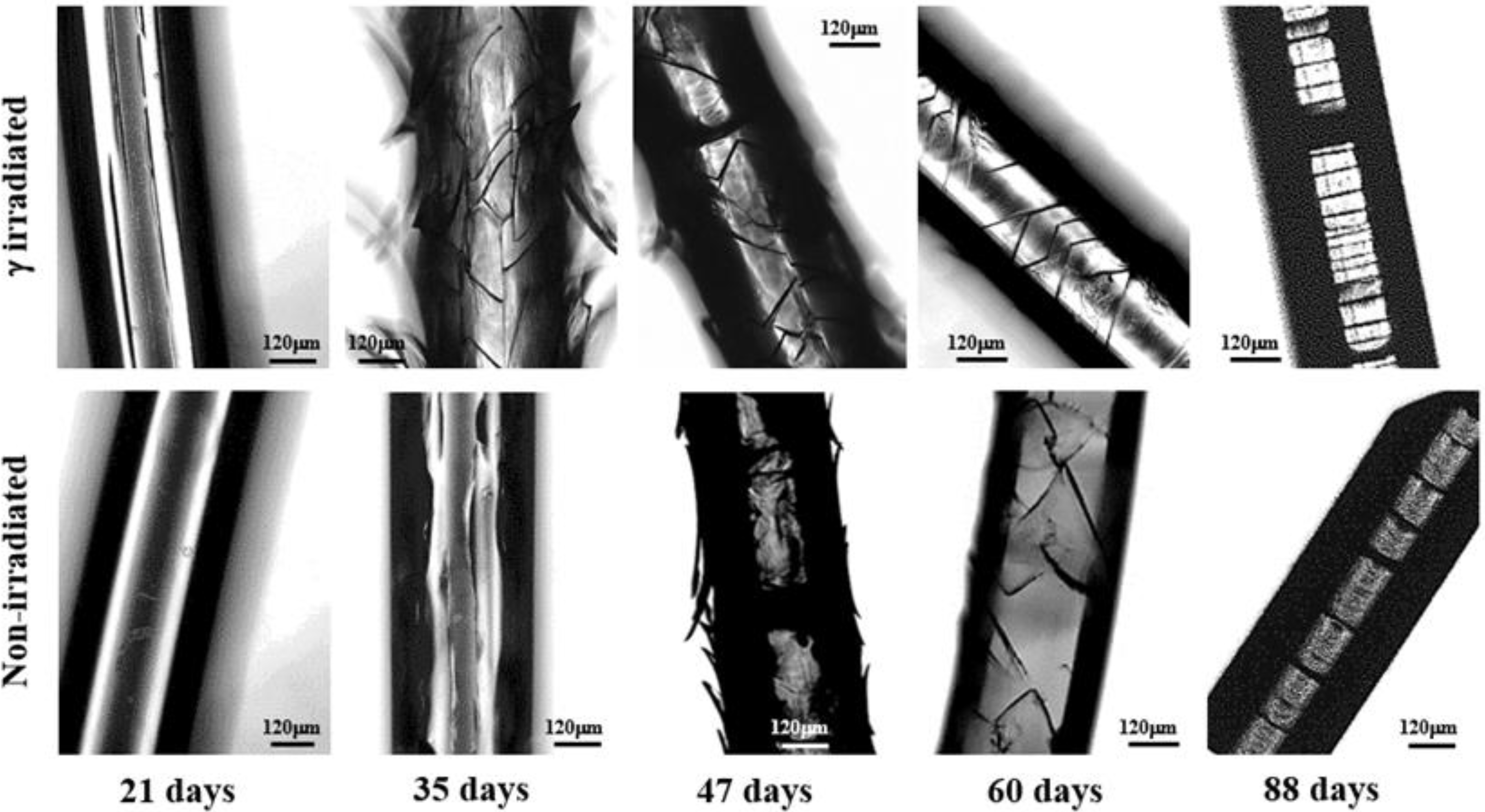
- (1)
- Degradation in a neutral pH proceeds through three well differentiated steps. The first one corresponds to the development of straight and longitudinal cracks that evolve through the detachment of the skin layer. Microcracks that propagate circumferentially around the fiber axis are characteristic of the second step although they initially contribute to the peeling out process of the outermost skin. In this step microcracks are irregularly distributed along the fiber and are not completely extended along the circumferential perimeter. In fact, optical micrographs show the presence of irregular cracks that appear inclined with respect to the fiber axis. The last step is associated to the deep propagation of the initiated circumferential cracks through the cross-sectional planes where interlamellar amorphous domains more susceptible to the hydrolysis exist. At the end of this step, regularly distributed discs perpendicular to the fiber axis are evident in the optical and SEM micrographs.
- (2)
- Peeling was not observed when the pH of the degradation medium was lower than 6 and furthermore the formation of longitudinal cracks was clearly hindered in these acidic media (Figure 10). Logically, degradation became slower as the pH decreased and in fact unaltered surfaces were observed after 21 days of exposure even for the irradiated sample. Circumferential and highly spaced cracks perpendicular to the fiber axis were detected at the first stages of degradation in the most acidic media (pHs 2 and 3) whereas irregular and inclined cracks were observed in the optical microscopy images taken at pHs between 4 and 6. Nevertheless, SEM micrographs revealed that these cracks had a zig-zag appearance. In all cases, additional cracks appeared at longer exposures, giving rise to irregular fissures that ultimately lead to the thinner and regular discs that were perfectly oriented perpendicular to the fiber axis.
- (3)
- Degradation in basic media showed the formation of deep longitudinal cracks at the beginning of exposure that lead to peeling only when pH was lower than 9. Circumferential cracks were also observed at the earlier degradation steps together with a clear erosion of the fiber surface, which can already be detected in the irradiated samples exposed for only 21 days in a pH 10 medium. SEM micrographs revealed also the formation of such circumferential cracks and the resulting wrinkled fiber surface. In fact, degradation in the high basic media was peculiar since deep transversal cracks that led to narrow discs were practically formed at the beginning of exposure. Note also that the high solubilization of degradation products caused the lineal and smooth fiber profile rapidly to evolve towards highly tortuous surfaces.



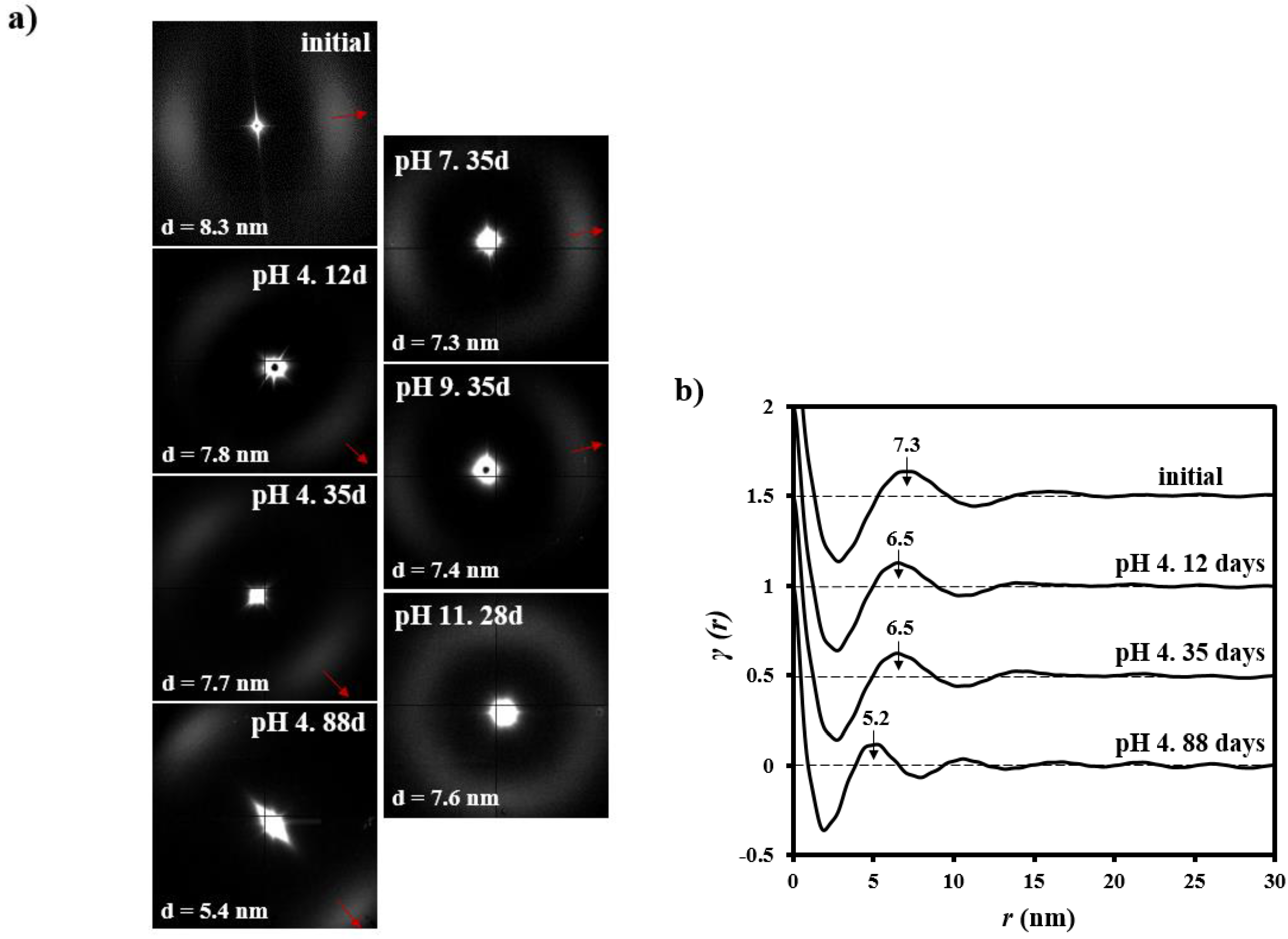
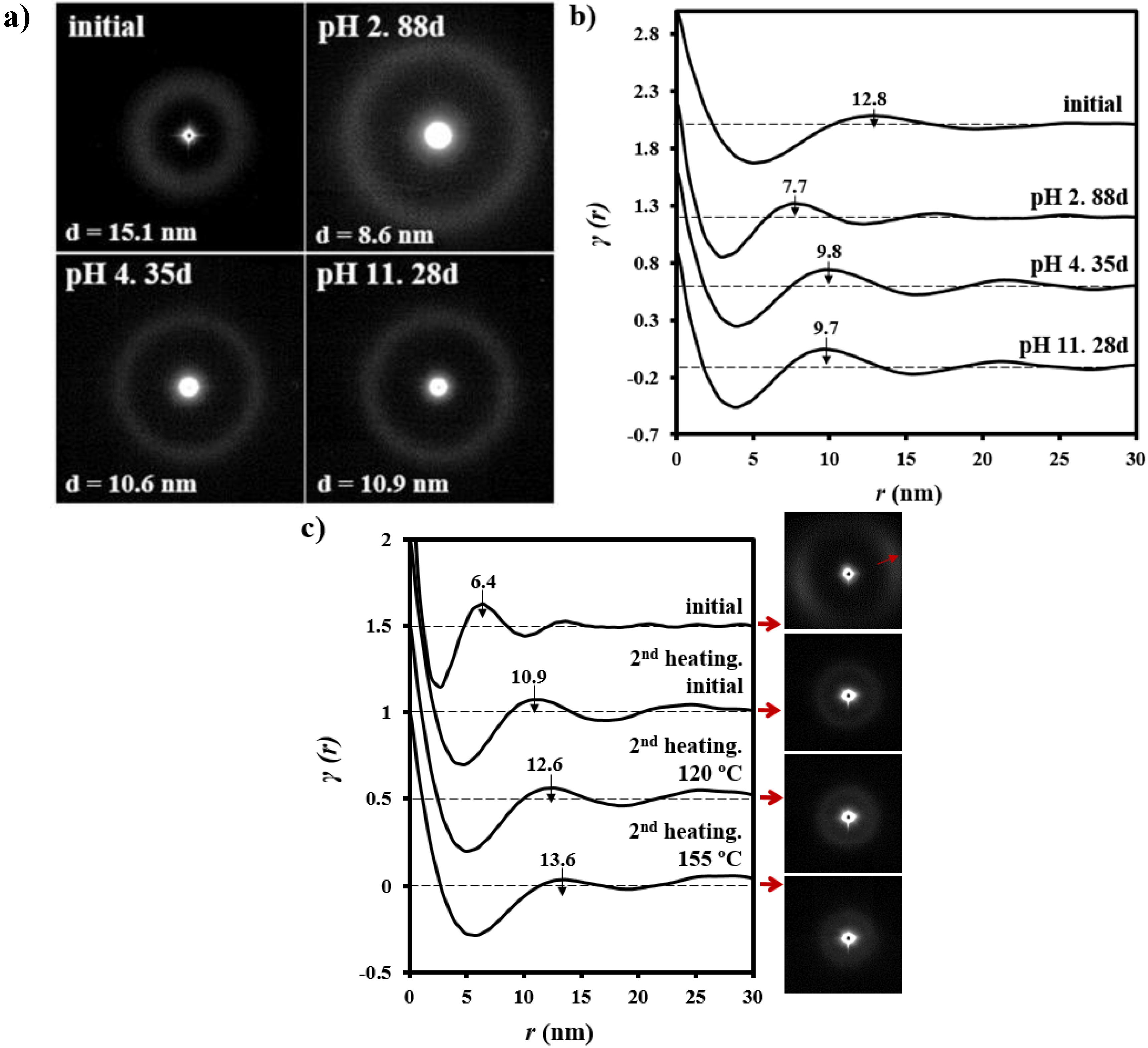
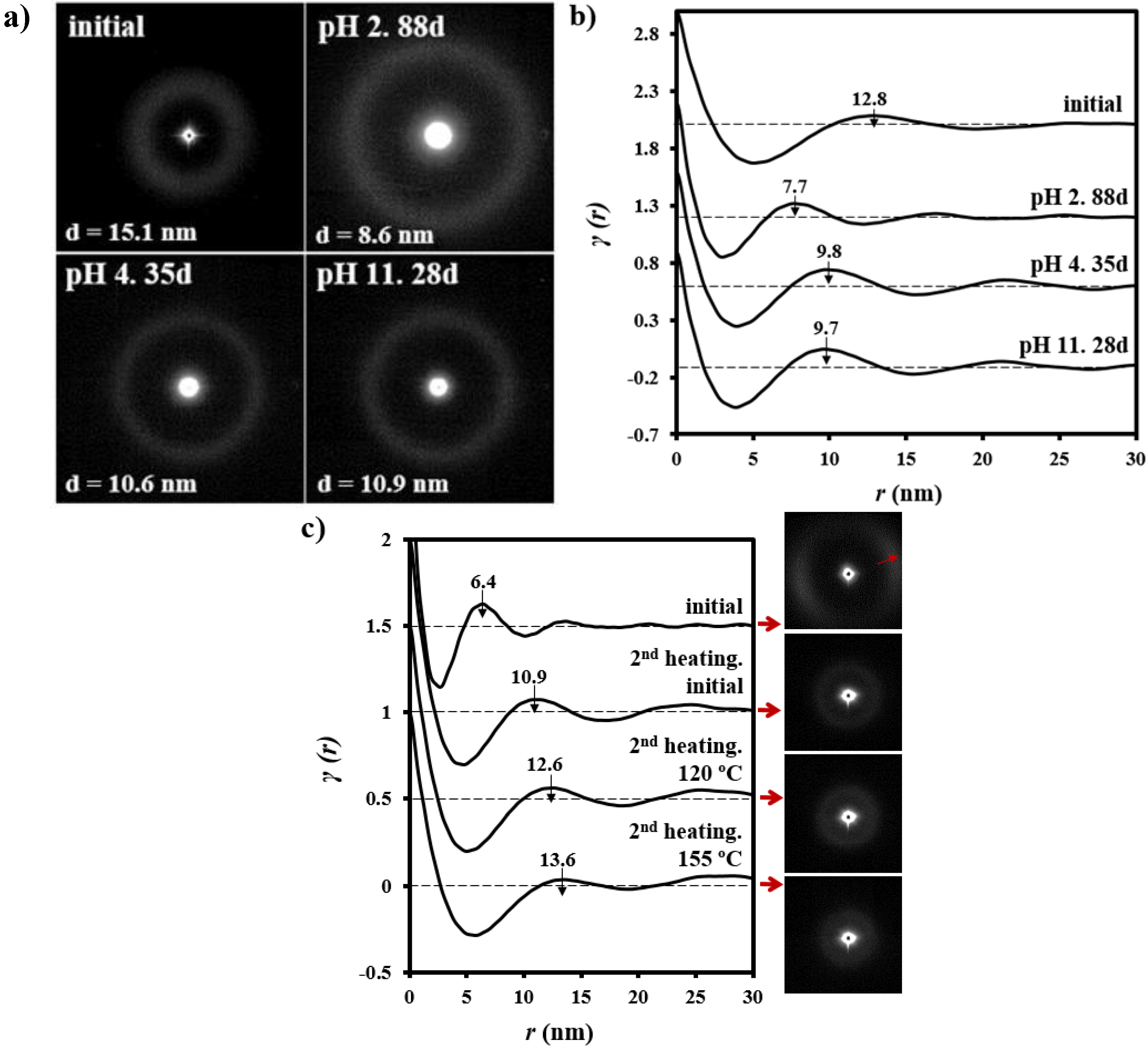
3.3. Changes on the Lamellar Parameters of GL-b-[GL-co-TMC-co-CL]-b-GL during Hydrolytic Degradation in Different pH Media

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Time (Days) | LB (nm) | Lγ (nm) | lc (nm) | la (nm) | XcSAXS |
|---|---|---|---|---|---|---|
| - | 0 | 8.3 | 7.3 | 5.3 | 2.0 | 0.73 |
| 4 | 12 | 7.8 | 6.5 | 5.2 | 1.3 | 0.80 |
| 4 | 35 | 7.7 | 6.5 | 5.2 | 1.3 | 0.80 |
| 4 | 88 | 5.4 | 5.2 | 4.2 | 1.0 | 0.79 |
| 7 | 35 | 7.3 | 6.4 | 5.1 | 1.3 | 0.80 |
| 9 | 35 | 7.4 | 6.4 | 5.1 | 1.3 | 0.80 |
| 11 | 28 | 7.6 | 6.6 | 5.2 | 1.4 | 0.80 |
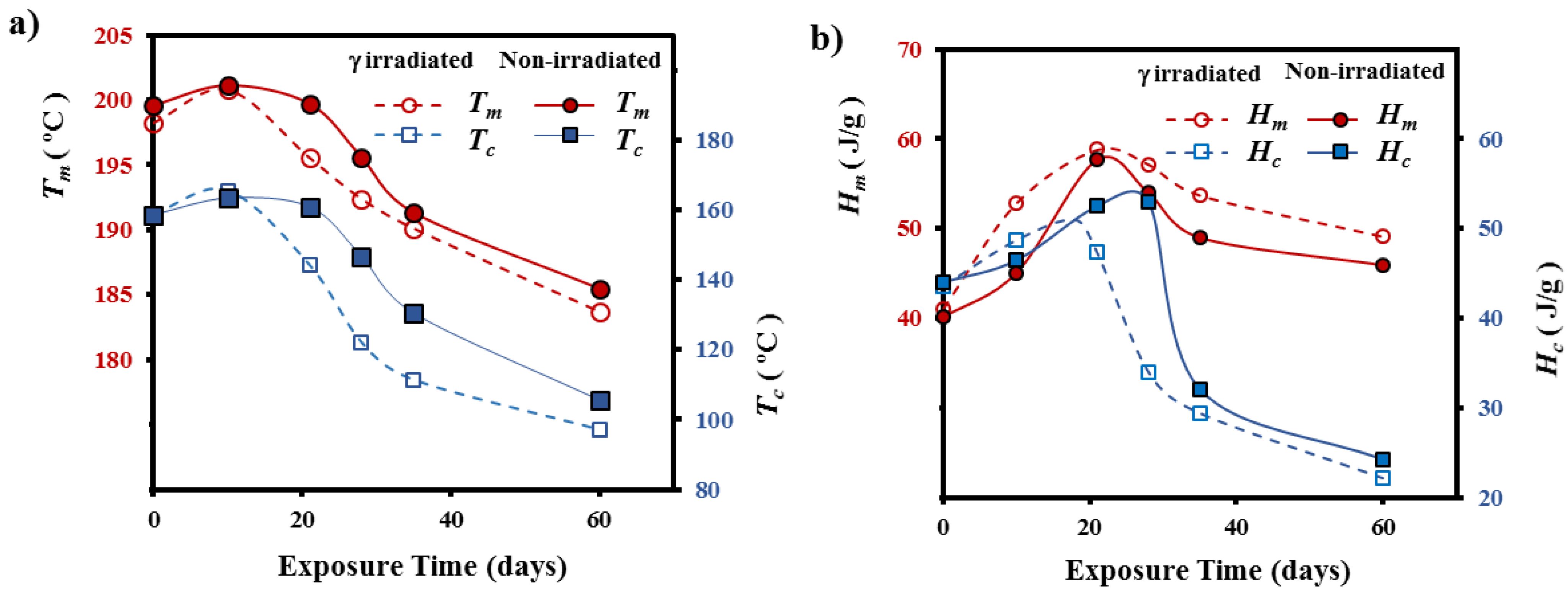
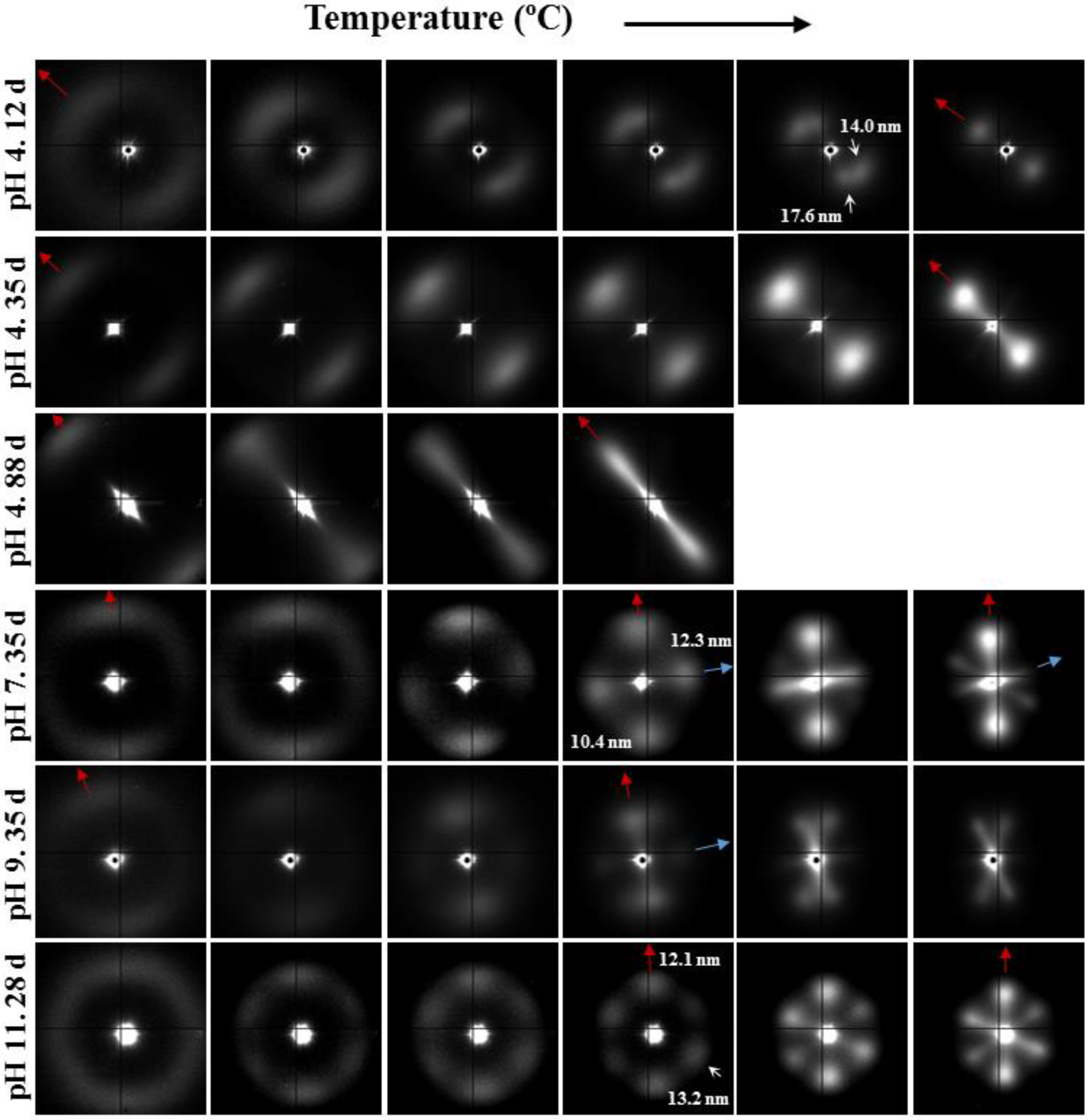
3.4. Thermal Annealing of Degraded GL-b-[GL-co-TMC-co-CL]-b-GL Samples in Different pH Media: Repercussions on the Lamellar Morphology
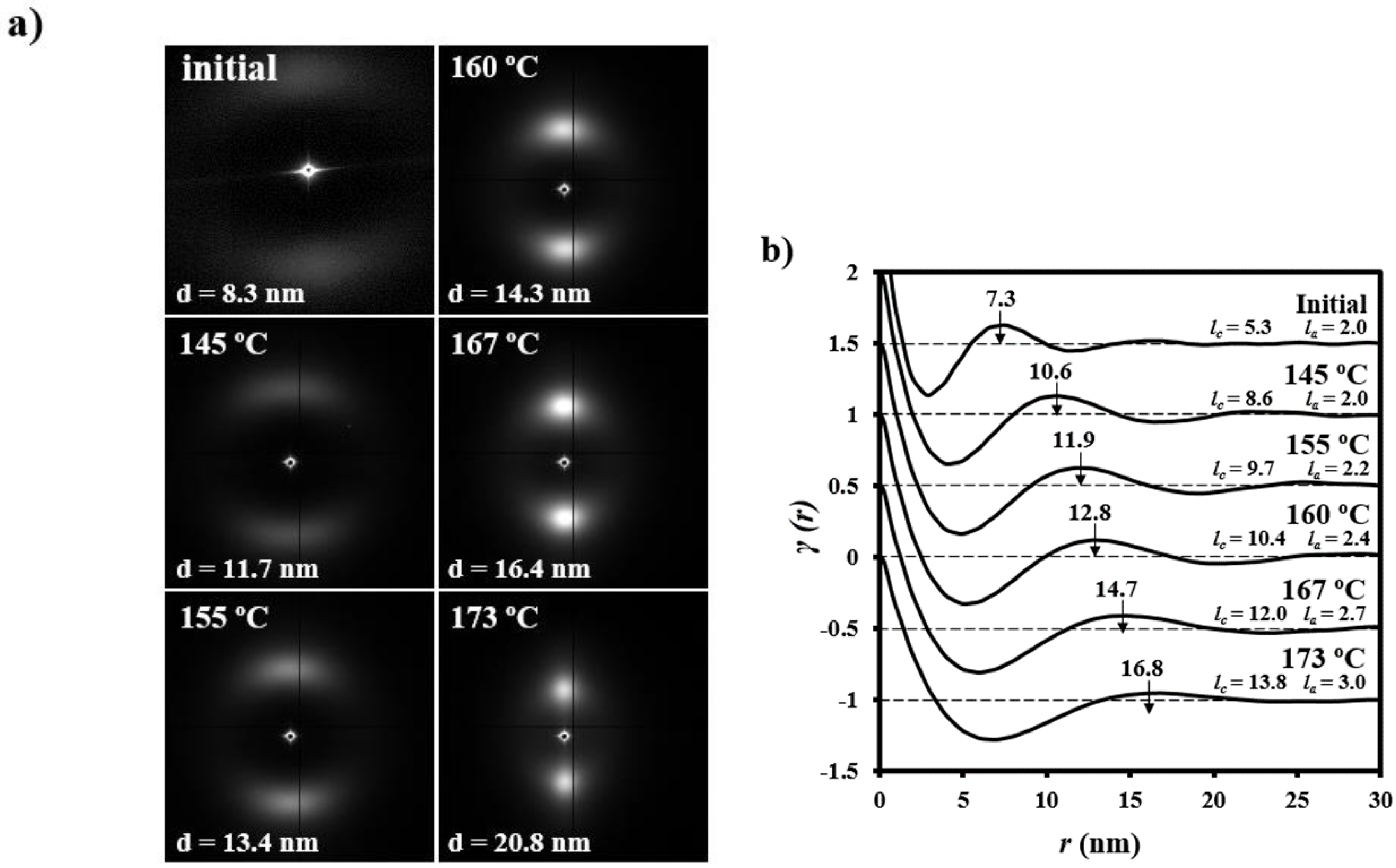
- (1)
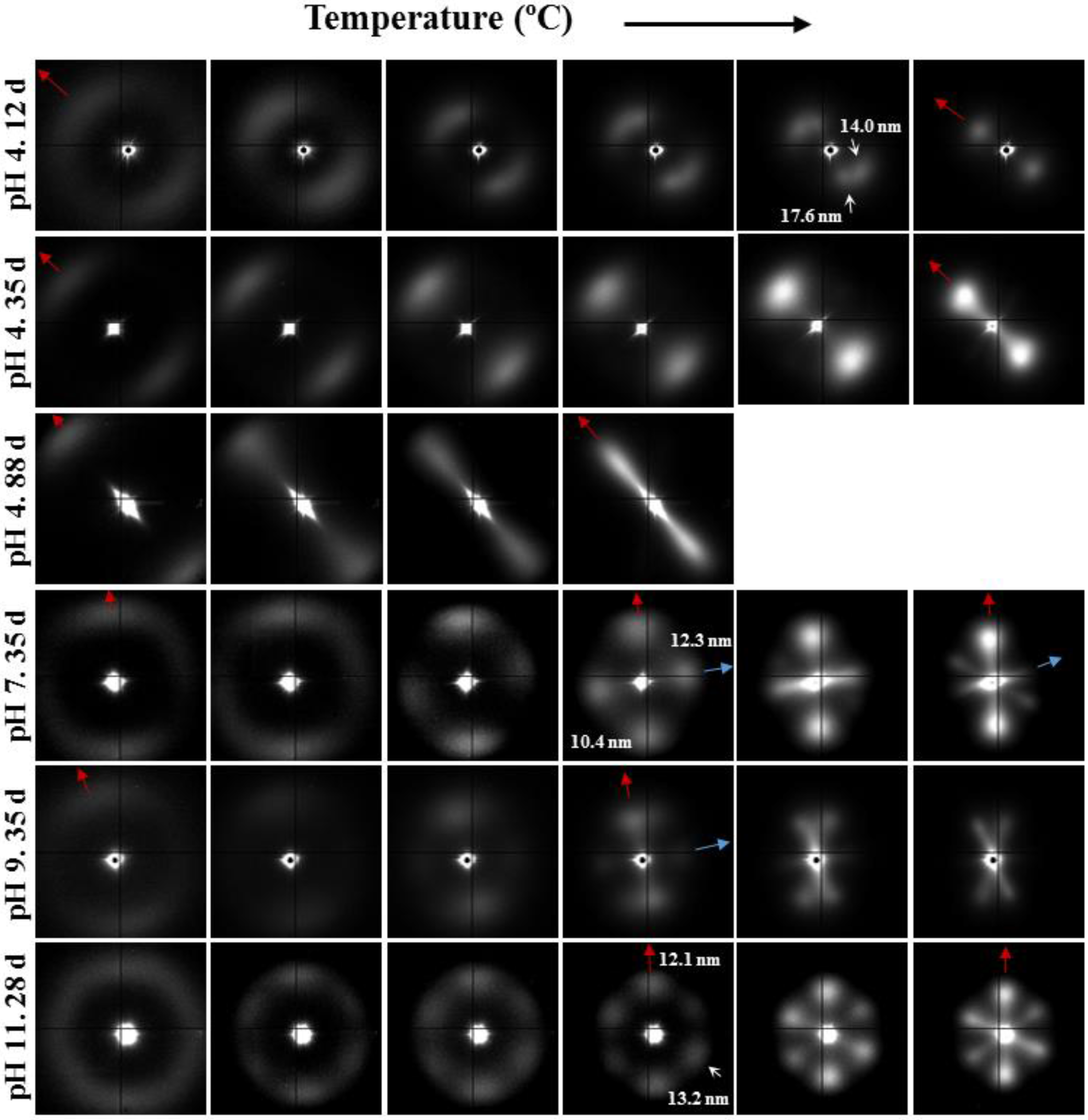
- Samples degraded in acidic media basically showed the characteristic meridional reflections associated with lamellar stacking. Firstly, these reflections had the previously indicated evolution when temperature was increased that led to an increase of both lamellar thickness and breadth. However, at the highest temperatures and especially for the most degraded samples (e.g., see patterns of samples degraded for 35 and 88 days), reflections were elongated in the meridional direction and extended towards the center of the pattern as presumable for a decrease of the crystalline domain size (i.e., the number of stacked lamellae). It is interesting to note that extrameridional reflections with a practically meridional orientation could also be detected during the heating of the less degraded samples (e.g., see arrow for the sample exposed for 12 days at pH 4). These spots suggest the sporadic formation and tilting of lamellae with a slightly greater thickness (i.e., 17.6 nm respect to 14.0 nm) that cannot be well accommodated in the surrounding and compact amorphous phase (Figure 8b). Note that solubilization of degradation products in the acidic medium was scarce at low exposure times.
- (2)
- An equatorial reflection is enhanced during heating of samples degraded at a neutral pH where the development of longitudinal cracks is characteristic. This reflection can be associated to the interfibrillar spacing (Figure 8b) which is greater than the interlamellar one (e.g., ca. 10.4 nm respect to ca. 12.3 nm as can be measured in the patterns shown in Figure 15). The regularity of interfibrillar domains was lost at the highest temperatures and consequently the corresponding reflection clearly disappeared before lamellar stacking was affected by partial fusion. Figure 15 also shows as equatorial reflections could still be detected at pH 9.
- (3)
- Patterns of samples exposed to basic pHs were more complicated since solubilization of degradation products allowed a greater readjustment/reorientation of constitutive lamellae. For example, the meridional reflection was split in the samples exposed to pH 9, suggesting a slight tilting of lamellae according to opposite directions. Note that optical micrographs revealed the development of tortuous fiber morphologies during exposure to basic pHs (Figure 11). This lamellar tilting seems not appropriate to justify the regular six spot pattern detected at higher pHs, being a plausible explanation the enhancement during degradation of a macrolattice arrangement where lamellar domains were disposed at different levels along the fiber axis as displayed in Figure 8.
3.5. Change of Lamellar Parameters of Degraded GL-b-[GL-co-TMC-co-CL]-b-GL Samples during Subsequent Non-Isothermal Crystallization and Reheating Processes
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Griffin Lewis, O.; Fabisial, W. Sutures. In Kirk-Othmer Encyclopedia of Chemical Technology, 4th ed.; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Gilding, D.K.; Reed, A.M. Biodegradable polymers for use in surgery-polyglycolic-poly(lactic acid) homopolymers and copolymers. Polymer 1979, 2, 1459–1464. [Google Scholar] [CrossRef]
- Homsy, C.A.; Mcdonald, E.R.; Akers, W.W. Surgical suture-canine tissue interaction for six common suture types. J. Biomed. Mater. Res. 1968, 2, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Blomstedt, B.; Ostenberg, B. Suture materials and wound infection. An experimental study. Acta Chir. Scand. 1978, 144, 269–274. [Google Scholar] [PubMed]
- Rodeheaver, G.T.; Thacker, J.G.; Owen, J.; Strauss, M.; Masterson, T.; Edlich, R.F. Knotting and handling characteristics of coated synthetic absorbable sutures. J. Surg. Res. 1983, 35, 525–530. [Google Scholar] [CrossRef]
- Katz, A.R.; Mukherjee, D.P.; Kaganov, A.L.; Gordon, S. A new synthetic monofilament absorbable suture made from polytrimethylene carbonate. Surgery Gynecol. Obstet. 1985, 161, 213–222. [Google Scholar]
- Zurita, R.; Puiggalí, J.; Franco, L.; Rodríguez-Galán, A. Copolymerization of glycolide and trimethylene carbonate. J. Polym. Sci. Part A Polym. Chem. 2005, 44, 993–1013. [Google Scholar] [CrossRef]
- Kasperczyk, J. Copolymerization of glycolide and ɛ-caprolactone, 1: Analysis of the copolymer microstructure by means of 1H and 13C-NMR spectroscopy. Macromol. Chem. Phys. 1999, 200, 903–910. [Google Scholar] [CrossRef]
- Díaz-Celorio, E.; Franco, L.; Rodríguez-Galán, A.; Puiggalí, J. Synthesis of glycolide/trimethylene carbonate copolymers: Influence of microstructure on properties. Eur. Polym. J. 2012, 48, 60–73. [Google Scholar] [CrossRef]
- Noorsal, K.; Mantle, M.D.; Gladden, L.F.; Cameron, R.E. Degradation and drug-release studies of a poly(glycolide-co-trimethylene carbonate) copolymer (Maxon). J. Appl. Polym. Sci. 2005, 95, 475–486. [Google Scholar] [CrossRef]
- Díaz-Celorio, E.; Franco, L.; Rodríguez-Galán, A.; Puiggalí, J. Study on the hydrolytic degradation of glycolide/trimethylene carbonate copolymers having different microstructure and composition. Polym. Degrad. STable 2013, 98, 133–143. [Google Scholar] [CrossRef]
- Freudenberg, F.; Rewerk, S.; Kaess, M.; Weiss, C.; Dorn-Beinecke, A.; Post, S. Biodegradation of absorbable sutures in body fluids and pH buffers. Eur. Surg. Res. 2004, 36, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Farrar, D.; Gillson, R. Hydrolytic degradation of polyglyconate B: the relationship between degradation time, strength and molecular weight. Biomaterials 2002, 23, 3905–3912. [Google Scholar] [CrossRef]
- Ahmed, A.H.; Goldie, B.S. Comparison of the mechanical properties of polyglycolide-trimethylene carbonate (Maxon) and polydioxanone suture (PDSII) used for flexor tendon repair and active mobilization. J. Hand Surg. Am. 2002, 27B, 329–332. [Google Scholar]
- Mäkelä, P.; Pohjonen, T.; Törmalä, P.; Waris, T.; Ashammakhi, N. Strength retention properties of self-reinforced poly l-lactide (SR-PLLA) sutures compared with polyglyconate (Maxon®) and polydioxanone (PDS) sutures. An in vitro study. Biomaterials 2002, 23, 2587–2592. [Google Scholar] [CrossRef]
- Díaz-Celorio, E.; Franco, L.; Puiggalí, J. Isothermal crystallization Study on a biodegradable segmented copolymer constituted by glycolide and trimethylene carbonate units. J. Appl. Polym. Sci. 2010, 116, 577–589. [Google Scholar] [CrossRef]
- Díaz-Celorio, E.; Franco, L.; Puiggalí, J. Nonisothermal Crystallization behavior of a biodegradable segmented copolymer constituted by glycolide and trimethylene carbonate units. J. Appl. Polym. Sci. 2011, 119, 1548–1559. [Google Scholar] [CrossRef]
- Tomihata, K.; Suzuki, M.; Ikada, Y. The pH dependence of monofilament sutures on hydrolytic degradation. J. Biomed. Mater. Res. 2001, 58, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Celorio, E.; Franco, L.; Puiggalí, J. Influence of microstructure on the crystallization of segmented copolymers constituted by glycolide and trimethylene carbonate units. Express Polym. Lett. 2013, 7, 186–198. [Google Scholar] [CrossRef]
- Oberhoffner, S.; Planck, H. Surgical Suture Material from Triblockterpolymer, Its Use in Surgery and Process for Its Preparation. EP 0835895, 3 December 2003. [Google Scholar]
- Barber, F.A. Resorbable fixation devices: A product guide. Orthop. Spec. Ed. 1998, 4, 1111–1117. [Google Scholar]
- Roby, M.S.; Bennet, S.L.; Liu, E.K. Absorbable Block Copolymers and Surgical Articles Fabricated Thereform. U.S. Patent 5.403.347, 4 April 1995. [Google Scholar]
- Pineros-Fernández, A.; David, B.; Drake, D.B.; Rodeheaver, P.A.; Moody, D.L.; Edlich, R.; Rodeheaver, G.T. CAPROSYN*, another major advance in synthetic monofilament absorbable suture. J. Long. Term. Eff. Med. Implants 2004, 14, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Barrera, G.; Brostow, W. Fiber-reinforced polymer concrete: Property improvement by gamma irradiation. In Gamma Radiation Effects on Polymeric Materials and Its Applications; Barrera, C., Ed.; Research Signpost: Kerala, India, 2009; pp. 27–44. [Google Scholar]
- D’Alelio, G.F.; Haberli, R.; Pezdirtz, G.F. Effect of ionizing radiation on a series of saturated polyester. J. Macromol. Sci. Chem. 1968, 2, 501–588. [Google Scholar] [CrossRef]
- Zhang, L.; Loh, I.H.; Chu, C.C. A combined gamma irradiation and plasma deposition treatment to achieve that ideal degradation properties of synthetic absorbable polymers. J. Biomed. Mater. Res. 1993, 27, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.C.; Williams, D.F. The effect of gamma irradiation on the enzymatic degradation of polyglycolic acid absorbable sutures. J. Biomed. Mater. Res. 1983, 17, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Márquez, Y.; Franco, L.; Turon, P.; Rodríguez-Galán, A.; Puiggalí, J. Study on the hydrolytic degradation of the segmented GL-b-[GL-co-TMC-co-CL]-b-GL copolymer with application as monofilar surgical suture. Polym. Degrad. STable 2013, 98, 2709–2721. [Google Scholar] [CrossRef]
- Theorell, T.; Stenhagen, E. Ein Universalpuffer fiir den pH-Bereich 2.0 bis 12.0. Biochem. Z. 1939, 299, 416–419. (In German) [Google Scholar]
- Lin, H.L.; Chu, C.C.; Grubb, D. Hydrolytic degradation and morphologic study of poly-p-dioxanone. J. Biomed. Mater. Res. 1993, 27, 153–166. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.E. Polyester Fibres. In Handbook of Fibre and Technology: Fibre Chemistry; Lewin, M., Pearce, E.M., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1998; pp. 1–73. [Google Scholar]
- Li, S.M. Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. J. Biomed. Mater. Res. Part B Appl. Biomater. 1999, 48B, 342–353. [Google Scholar] [CrossRef]
- Fischer, E.W.; Sterzel, H.J.; Wegner, G. Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reactions. Kolloid Z. Z. Polym. 1973, 251, 980–990. [Google Scholar] [CrossRef]
- Fredericks, R.J.; Melveger, A.J.; Dolegiewtz, L.J. Morphological and structural changes in a copolymer of glycolide and lactide occurring as a result of hydrolysis. J. Polym. Sci. Polym. Phys. Ed. 1984, 22, 57–66. [Google Scholar] [CrossRef]
- Wu, L.; Ding, J. Effects of porosity and pore size on in vitro degradation of three-dimensional porous poly(D,L-lactide-co-glycolide) scaffolds for tissue engineering. J. Biomed. Mater. Res. A 2005, 75, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Larrañaga, A.; Etxeberria, A.; Sarasua, J.R. Effects of chain microstructures and derived crystallization capability on hydrolytic degradation of poly(l-lactide/ε-caprolactone) copolymers. Polym. Degrad. Stab. 2013, 98, 481–489. [Google Scholar] [CrossRef]
- Kulkarni, A.; Reiche, J.; Lendlein, A. Hydrolytic degradation of poly(rac-lactide) and poly[(rac-lactide)-co-glycolide] at the air water interface. Surf. Interface. Anal. 2007, 39, 740–746. [Google Scholar] [CrossRef]
- Yoshioka, T.; Kawazoe, N.; Tateishi, T.; Chen, G. In vitro evaluation of biodegradation of poly(lactic-co-glycolic acid) sponges. Biomaterials 2008, 29, 3438–3443. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, A. Morphology and properties of crystalline polymers with fiber structure. Text. Res. J. 1972, 42, 20–30. [Google Scholar] [CrossRef]
- Murthy, N.S.; Reimschuessel, A.C.; Kramer, V. Changes in void content and free volume in fibers during heat setting and their influence on dye diffusion and mechanical properties. J. Appl. Polym. Sci. 1990, 40, 249–262. [Google Scholar] [CrossRef]
- Chatani, Y.; Suehiro, K.; Okita, Y.; Tadokoro, H.; Chujo, K. Structural studies of polyesters. I Crystal structure of polyglycolide. Die Makrornolekulare Chem. 1968, 113, 215–229. [Google Scholar] [CrossRef]
- Vonk, C.G.; Kortleve, G. X-ray small-angle scattering of bulk polyethylene. Kolloid Z. Z. Polym. 1967, 220, 19–24. [Google Scholar] [CrossRef]
- Vonk, C.G. A general computer program for the processing of small-angle X-ray scattering data. J. Appl. Crystallogr. 1975, 8, 340–341. [Google Scholar] [CrossRef]
- Hsiao, B.S.; Verma, R.K. A novel approach to extract morphological variables in crystalline polymers from time-resolved SAXS measurements. J. Synchrotron Radiat. 1998, 5, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Stribeck, N. Extraction of domain strucure information from small angle scattering patterns of bulk materials. J. Appl. Crystallogr. 2001, 34, 496–503. [Google Scholar] [CrossRef]
- Stribeck, N.; Fakirov, S. Three-dimensional chord distribution function SAXS analysis of the strained domain structure of a poly(ether ester) thermoplastic elastomer. Macromolecules 2001, 34, 7758–7761. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Márquez, Y.; Martínez, J.C.; Turon, P.; Franco, L.; Puiggalí, J. Influence of pH on Morphology and Structure during Hydrolytic Degradation of the Segmented GL-b-[GL-co-TMC-co-CL]-b-GL Copolymer. Fibers 2015, 3, 348-372. https://doi.org/10.3390/fib3030348
Márquez Y, Martínez JC, Turon P, Franco L, Puiggalí J. Influence of pH on Morphology and Structure during Hydrolytic Degradation of the Segmented GL-b-[GL-co-TMC-co-CL]-b-GL Copolymer. Fibers. 2015; 3(3):348-372. https://doi.org/10.3390/fib3030348
Chicago/Turabian StyleMárquez, Yolanda, Juan Carlos Martínez, Pau Turon, Lourdes Franco, and Jordi Puiggalí. 2015. "Influence of pH on Morphology and Structure during Hydrolytic Degradation of the Segmented GL-b-[GL-co-TMC-co-CL]-b-GL Copolymer" Fibers 3, no. 3: 348-372. https://doi.org/10.3390/fib3030348