
Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria

Abstract
:1. Introduction
2. Surface Remodeling
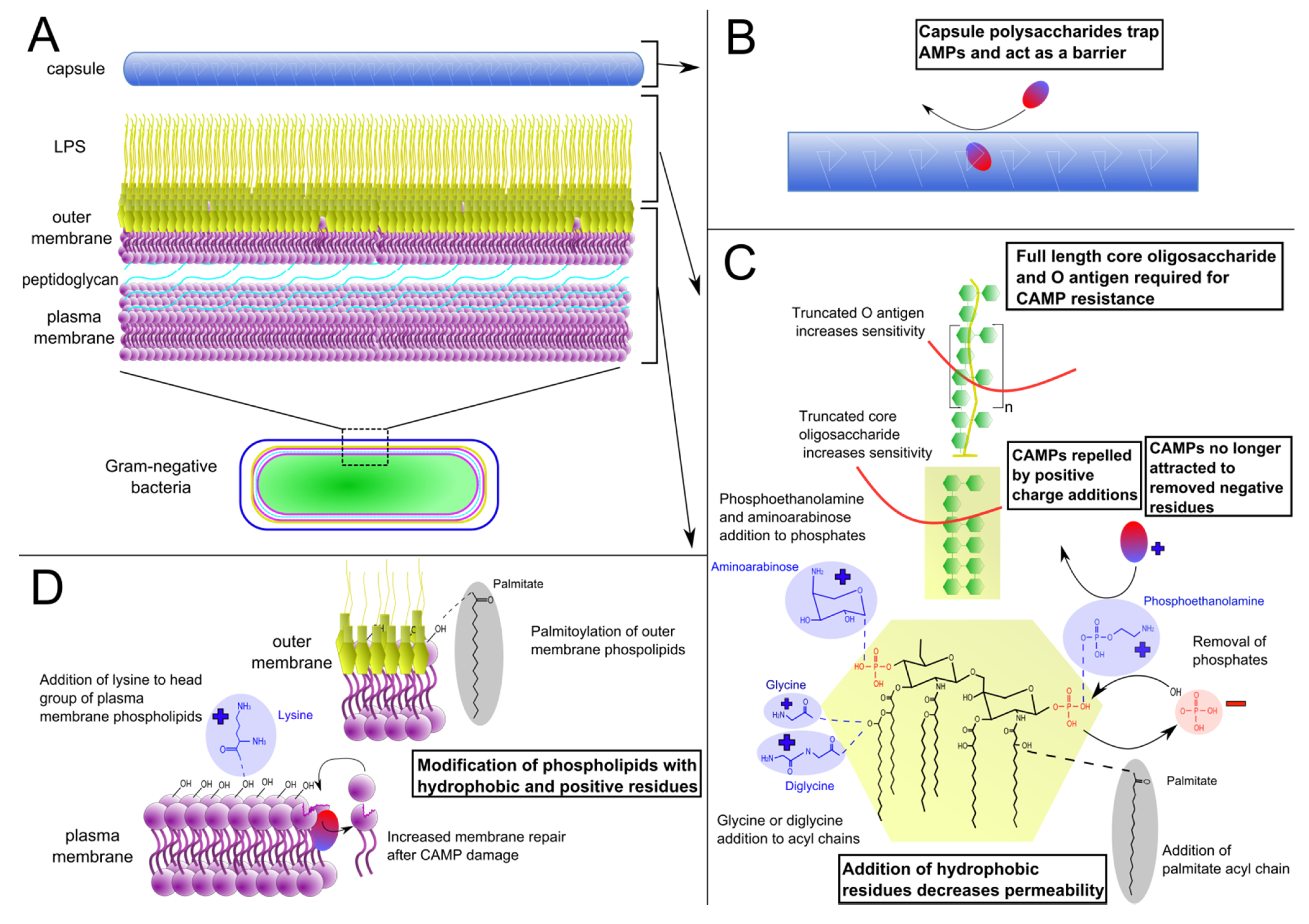
2.1. Lipopolysaccharide Modifications

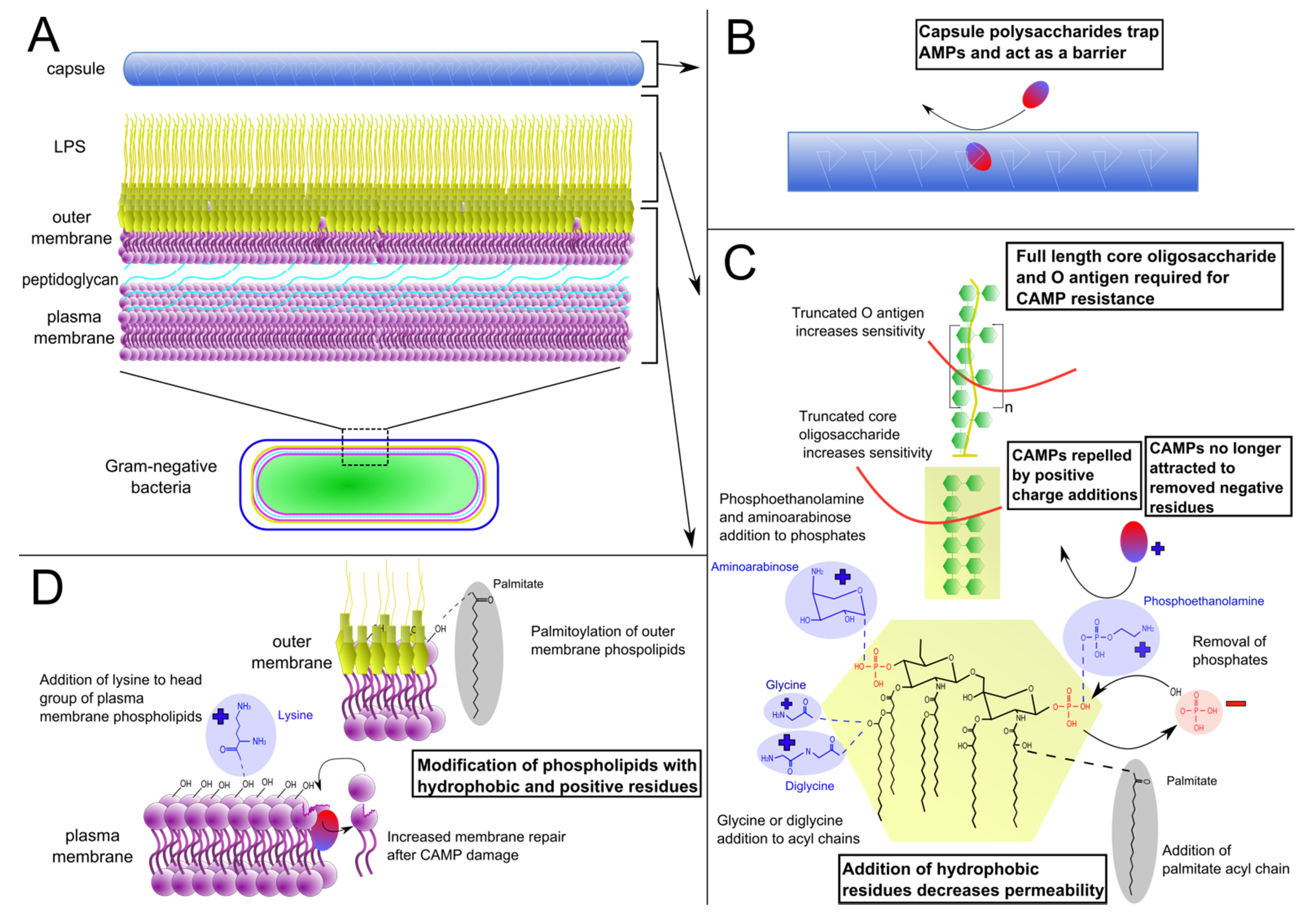{kind=link}
| Species | Modification | CAMP Resistance | Impact on Virulence | Ref. |
|---|---|---|---|---|
| Brucella abortus | LPS O antigen | Transposon mutants lacking O antigen show decreased survival to polymyxin B at 5–40 µg/mL | Transposon mutant unable to persist six weeks after mouse intraperitoneal infection | [36] |
| Burkholderia cenocepacia | LPS inner core oligosaccharide | B. cenocepacia require full length core oligosaccharide to grow in 100 µg/mL polymyxin B | Mutants with truncated core oligosaccharide were completely outcompeted by parent strain in rat lung infection model | [37] |
| Legionella pneumoniae | rcp, homolog of pagP, responsible for palmitoyl addition to lipid A | Mutants in rcp show 50% decrease in MIC to polymyxin B and synthetic CAMP C18G | Deletion mutants showed decreased survival in macrophages and were outcompeted by the parental strain in mouse lung infection | [38] |
| Neisseria gonnorhoeae | Mtr efflux pump | MICs are 8× higher for PG-1 and 30× higher for LL-37 in WT compared to mtr mutant | Deletion mutant completely outcompeted by WT after 3 day mouse genital tract infection | [39] |
| Proteus mirabilis | ZapA secreted metalloprotease | Purified ZapA readily degrades LL-37 and human beta-defensin-1 in vitro | 4 log decrease in virulence in mouse urinary tract infection with ZapA mutant | [40] |
| Pseudomonas aeruginosa | AcrAB efflux pump | Mutant in acrB 10× more susceptible to CAMP-containing BALF, as well as diminished survival in 0.1 µg/mL polymyxin B, 30 µg/mL HNP-1, and 0.1 µg/mL HBD-1 + 2 | 1–3 log decrease in virulence of deletion mutant over 72 h mouse infection using a pneumonia model | [41] |
| LasA cleavage and release of syndecan-1 from host immune cells | Shed syndecan-1 can bind Pro/Arg rich CAMPs | 3 log decrease in virulence when syndecan-1 is absent in KO mouse lung infection, with 1/3 reduction in mortality | [42,43,44] | |
| Salmonella typhimurium | Various | Transposon mutagenesis yielded 12 mutants that were susceptible to CAMP protamine at 1mg/mL | 11 of 12 mutants with high protamine susceptibility had decreased virulence in mouse intragastric infection | [45] |
| Aminoarabinose addition to lipid A through pmrF | pmrF deletion mutant unable to add aminoarabinose to lipid A, and is more sensitive to CAMPs | Mice orally infected with mutants had double the survival time as WT-infected mice. Competition infections with WT and deletion mutants show that CAMPs CRAMP and matrilysin alone not responsible for attenuation | [46] | |
| * PmrAB mediated addition of aminoarabinose to lipid A | Inactivation of pmrA results in 19× reduction in polymyxin B MIC, while overexpression results in 3× increase | pmrA deletion mutants show decreased lethality in mice by oral but not intraperitoneal infection | [8,47] | |
| * SlyA regulatory protein | slyA mutant is susceptible to 1 µg/mL polymyxin B, and SlyA protein binds to promoter of ugtL resistance gene | Deletion mutants have LD50 >4 log higher for oral infection and >5 log higher for peritoneal infection in mice | [48,49] | |
| * PhoP regulatory protein | Mutants increase sensitivity to human and rabbit neutrophil granules, as well as rabbit CAMP NP-1 | Deletion mutants in phoP show 4 log reduction in virulence in mouse peritoneal model of infection, and phoP/phoQ deletion of S. typhi was a safe vaccine candidate in humans | [28,29,50] | |
| Yersinia enterocolitica | Unspecified LPS modifications, possibly RosAB | Pathogenic Y. enterocolitica strains were more resistant to polymyxin B than non-pathogenic environmental strains when grown at 37 °C | Environmental strains not known to cause disease like the polymyxin resistant pathogenic strains | [51] |
2.2. Phospholipid Modifications
2.3. Capsule Production
3. Biofilms
4. Efflux Pumps
5. Binding and Sequestering CAMPs
6. Proteolytic Degradation
7. Modulation of CAMP Expression
8. Relevance
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- UNMC Department of Pathology and Microbiology. The antimicrobial peptide database. 2014. Available online: http://aps.unmc.edu/AP/main.php (accessed on 2 July 2014).
- Nakatsuji, T.; Gallo, R.L. Antimicrobial peptides: Old molecules with new ideas. J. Invest. Dermatol. 2012, 132, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr. Issues Mol. Biol. 2006, 8, 11–26. [Google Scholar] [PubMed]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin b for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Brunel, J.M.; Dubus, J.C.; Reynaud-Gaubert, M.; Rolain, J.M. Colistin: An update on the antibiotic of the 21st century. Expert. Rev. Anti. Infect. Ther. 2012, 10, 917–934. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, S.M.; Ernst, R.K.; Miller, S.I. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 2004, 186, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S.; Lim, K.B.; Krueger, J.; Kim, K.; Guo, L.; Hackett, M.; Miller, S.I. PmrA-pmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol. 1998, 27, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Loutet, S.A.; Valvano, M.A. Extreme antimicrobial peptide and polymyxin B resistance in the genus burkholderia. Front. Microbiol. 2011, 2, e159. [Google Scholar] [CrossRef]
- Llewellyn, A.C.; Zhao, J.; Song, F.; Parvathareddy, J.; Xu, Q.; Napier, B.A.; Laroui, H.; Merlin, D.; Bina, J.E.; Cotter, P.A.; et al. NaxD is a deacetylase required for lipid A modification and Francisella pathogenesis. Mol. Microbiol. 2012, 86, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Kanistanon, D.; Hajjar, A.M.; Pelletier, M.R.; Gallagher, L.A.; Kalhorn, T.; Shaffer, S.A.; Goodlett, D.R.; Rohmer, L.; Brittnacher, M.J.; Skerrett, S.J.; et al. A francisella mutant in lipid a carbohydrate modification elicits protective immunity. PLoS Pathog. 2008, 4, e24. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Hancock, R.E.; Fernandez, R.C. Bordetella pertussis lipid A glucosamine modification confers resistance to cationic antimicrobial peptides and increases resistance to outer membrane perturbation. Antimicrob. Agents Chemother. 2014, 58, 4931–4934. [Google Scholar] [CrossRef] [PubMed]
- Hankins, J.V.; Madsen, J.A.; Giles, D.K.; Brodbelt, J.S.; Trent, M.S. Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in gram-positive and gram-negative bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 8722–8727. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.B.; LaRocque, R.C.; Qadri, F.; Ryan, E.T.; Calderwood, S.B. Cholera. Lancet 2012, 379, 2466–2476. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.A.; Choudhury, B.; Balthazar, J.T.; Martin, L.E.; Ram, S.; Rice, P.A.; Stephens, D.S.; Carlson, R.; Shafer, W.M. Phosphoethanolamine substitution of lipid a and resistance of neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect. Immun. 2009, 77, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.A.; Shafer, W.M.; Dutta Ray, T.; Ram, S.; Rice, P.A. Phosphoethanolamine residues on the lipid a moiety of neisseria gonorrhoeae lipooligosaccharide modulate binding of complement inhibitors and resistance to complement killing. Infect. Immun. 2013, 81, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.R.; Casella, L.G.; Jones, J.W.; Adams, M.D.; Zurawski, D.V.; Hazlett, K.R.; Doi, Y.; Ernst, R.K. Unique structural modifications are present in the lipopolysaccharide from colistin-resistant strains of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2013, 57, 4831–4840. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hsu, F.F.; Turk, J.; Groisman, E.A. The pmra-regulated pmrc gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J. Bacteriol. 2004, 186, 4124–4133. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ribeiro, A.A.; Guan, Z.; Abraham, S.N.; Raetz, C.R. Attenuated virulence of a Francisella mutant lacking the lipid a 4'-phosphatase. Proc. Natl. Acad. Sci. USA 2007, 104, 4136–4141. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, E.; Perry, M.B.; Conlan, J.W. Structural analysis of francisella tularensis lipopolysaccharide. Eur. J. Biochem. 2002, 269, 6112–6118. [Google Scholar] [CrossRef] [PubMed]
- Ingram, B.O.; Masoudi, A.; Raetz, C.R. Escherichia coli mutants that synthesize dephosphorylated lipid a molecules. Biochemistry 2010, 49, 8325–8337. [Google Scholar] [CrossRef] [PubMed]
- Kumada, H.; Haishima, Y.; Umemoto, T.; Tanamoto, K. Structural study on the free lipid a isolated from lipopolysaccharide of porphyromonas gingivalis. J. Bacteriol. 1995, 177, 2098–2106. [Google Scholar] [PubMed]
- Weintraub, A.; Zähringer, U.; Wollenweber, H.W.; Seydel, U.; Rietschel, E.T. Structural characterization of the lipid a component of bacteroides fragilis strain nctc 9343 lipopolysaccharide. Eur. J. Biochem. 1989, 183, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.X.; Whittimore, J.D.; Wyrick, P.B.; McGrath, S.C.; Cotter, R.J.; Trent, M.S. The lipid A 1-phosphatase of helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J. Bacteriol. 2006, 188, 4531–4541. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.D.; Trent, M.S. Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lim, K.B.; Poduje, C.M.; Daniel, M.; Gunn, J.S.; Hackett, M.; Miller, S.I. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998, 95, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Darveau, R.P.; Blake, J.; Seachord, C.L.; Cosand, W.L.; Cunningham, M.D.; Cassiano-Clough, L.; Maloney, G. Peptides related to the carboxyl terminus of human platelet factor IV with antibacterial activity. J. Clin. Invest. 1992, 90, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Fields, P.I.; Groisman, E.A.; Heffron, F. A Salmonella locus that controls resistance to microbicidal proteins from phagocytic cells. Science 1989, 243, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.I.; Kukral, A.M.; Mekalanos, J.J. A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc. Natl. Acad. Sci. USA 1989, 86, 5054–5058. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.W.; Sanowar, S.; Daley, M.E.; Schneider, A.R.; Cho, U.; Xu, W.; Klevit, R.E.; le Moual, H.; Miller, S.I. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 2005, 122, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S.; Miller, S.I. PhoP-PhoQ activates transcription of pmrAB, encoding a two-component regulatory system involved in Salmonella typhimurium antimicrobial peptide resistance. J. Bacteriol. 1996, 178, 6857–6864. [Google Scholar] [PubMed]
- Guo, L.; Lim, K.B.; Gunn, J.S.; Bainbridge, B.; Darveau, R.P.; Hackett, M.; Miller, S.I. Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science 1997, 276, 250–253. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.Y.; Tsai, Y.L.; Liu, M.C.; Lin, W.C.; Hsueh, P.R.; Liaw, S.J. Serratia marcescens arn, a PhoP-regulated locus necessary for polymyxin B resistance. Antimicrob. Agents Chemother. 2014, 58, 5181–5190. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Nickel, G.C.; Bajaksouzian, S.; Lavender, H.; Murthy, A.R.; Jacobs, M.R.; Bonomo, R.A. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob. Agents Chemother. 2009, 53, 3628–3634. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.A.; Adams, L.G.; Ficht, T.A. Transposon-derived Brucella abortus rough mutants are attenuated and exhibit reduced intracellular survival. Infect. Immun. 1998, 66, 1008–1016. [Google Scholar] [PubMed]
- Loutet, S.A.; Flannagan, R.S.; Kooi, C.; Sokol, P.A.; Valvano, M.A. A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J. Bacteriol. 2006, 188, 2073–2080. [Google Scholar] [CrossRef] [PubMed]
- Robey, M.; O’Connell, W.; Cianciotto, N.P. Identification of Legionella pneumophila rcp, a pagP-like gene that confers resistance to cationic antimicrobial peptides and promotes intracellular infection. Infect. Immun. 2001, 69, 4276–4286. [Google Scholar] [CrossRef] [PubMed]
- Jerse, A.E.; Sharma, N.D.; Simms, A.N.; Crow, E.T.; Snyder, L.A.; Shafer, W.M. A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect. Immun. 2003, 71, 5576–5582. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.E.; Moghaddame-Jafari, S.; Lockatell, C.V.; Johnson, D.; Belas, R. ZapA, the IgA-degrading metalloprotease of Proteus mirabilis, is a virulence factor expressed specifically in swarmer cells. Mol. Microbiol. 1999, 32, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Padilla, E.; Llobet, E.; Doménech-Sánchez, A.; Martínez-Martínez, L.; Bengoechea, J.A.; Albertí, S. Klebsiella pneumoniae AcrAB efflux pump contributes to antimicrobial resistance and virulence. Antimicrob. Agents Chemother. 2010, 54, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Park, P.W.; Pier, G.B.; Preston, M.J.; Goldberger, O.; Fitzgerald, M.L.; Bernfield, M. Syndecan-1 shedding is enhanced by LasA, a secreted virulence factor of Pseudomonas aeruginosa. J. Biol. Chem. 2000, 275, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Park, P.W.; Pier, G.B.; Hinkes, M.T.; Bernfield, M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 2001, 411, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Haynes, A.; Ruda, F.; Oliver, J.; Hamood, A.N.; Griswold, J.A.; Park, P.W.; Rumbaugh, K.P. Syndecan 1 shedding contributes to Pseudomonas aeruginosa sepsis. Infect. Immun. 2005, 73, 7914–7921. [Google Scholar] [CrossRef] [PubMed]
- Groisman, E.A.; Parra-Lopez, C.; Salcedo, M.; Lipps, C.J.; Heffron, F. Resistance to host antimicrobial peptides is necessary for Salmonella virulence. Proc. Natl. Acad. Sci. USA 1992, 89, 11939–11943. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, K.L.; Richards, S.M.; Tamayo, R.; Reeves, L.T.; Gunn, J.S. An altered immune response, but not individual cationic antimicrobial peptides, is associated with the oral attenuation of Ara4N-deficient Salmonella enterica serovar typhimurium in mice. PLoS ONE 2012, 7, e49588. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S.; Ryan, S.S.; van Velkinburgh, J.C.; Ernst, R.K.; Miller, S.I. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect. Immun. 2000, 68, 6139–6146. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Latifi, T.; Cromie, M.J.; Groisman, E.A. Transcriptional control of the antimicrobial peptide resistance ugtL gene by the Salmonella PhoP and SlyA regulatory proteins. J. Biol. Chem. 2004, 279, 38618–38625. [Google Scholar] [CrossRef] [PubMed]
- Libby, S.J.; Goebel, W.; Ludwig, A.; Buchmeier, N.; Bowe, F.; Fang, F.C.; Guiney, D.G.; Songer, J.G.; Heffron, F. A cytolysin encoded by Salmonella is required for survival within macrophages. Proc. Natl. Acad. Sci. USA 1994, 91, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, E.L.; Oletta, C.A.; Killeen, K.P.; Miller, S.I. PhoP/PhoQ-deleted Salmonella typhi (ty800) is a safe and immunogenic single-dose typhoid fever vaccine in volunteers. J. Infect. Dis. 1996, 173, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea, J.A.; Díaz, R.; Moriyón, I. Outer membrane differences between pathogenic and environmental Yersinia enterocolitica biogroups probed with hydrophobic permeants and polycationic peptides. Infect. Immun. 1996, 64, 4891–4899. [Google Scholar] [PubMed]
- Raetz, C.R.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [PubMed]
- Dalebroux, Z.D.; Matamouros, S.; Whittington, D.; Bishop, R.E.; Miller, S.I. PhoPQ regulates acidic glycerophospholipid content of the Salmonella Typhimurium outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, 1963–1968. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.; Michalak, A.; Pagentine, S.; Seaton, P.; Pokorny, A. Lysylated phospholipids stabilize models of bacterial lipid bilayers and protect against antimicrobial peptides. Biochim. Biophys. Acta 2014, 1838, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Sohlenkamp, C.; Galindo-Lagunas, K.A.; Guan, Z.; Vinuesa, P.; Robinson, S.; Thomas-Oates, J.; Raetz, C.R.; Geiger, O. The lipid lysyl-phosphatidylglycerol is present in membranes of Rhizobium tropici CIAT899 and confers increased resistance to polymyxin B under acidic growth conditions. Mol. Plant Microbe Interact. 2007, 20, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.E.; Smith, J.D. Phospholipids of the differentiating bacterium Caulobacter crescentus. Can. J. Biochem. 1979, 57, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Yokum, T.S.; Hammer, R.P.; McLaughlin, M.L.; Elzer, P.H. Peptides with indirect in vivo activity against an intracellular pathogen: Selective lysis of infected macrophages. J. Pept. Res. 2002, 59, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Dorschner, R.A.; Lopez-Garcia, B.; Peschel, A.; Kraus, D.; Morikawa, K.; Nizet, V.; Gallo, R.L. The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J. 2006, 20, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Willis, L.M.; Whitfield, C. Structure, biosynthesis, and function of bacterial capsular polysaccharides synthesized by abc transporter-dependent pathways. Carbohydr. Res. 2013, 378, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.A.; Vargas, M.A.; Regueiro, V.; Llompart, C.M.; Albertí, S.; Bengoechea, J.A. Capsule polysaccharide mediates bacterial resistance to antimicrobial peptides. Infect. Immun. 2004, 72, 7107–7114. [Google Scholar] [CrossRef] [PubMed]
- Cortés, G.; Borrell, N.; de Astorza, B.; Gómez, C.; Sauleda, J.; Albertí, S. Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide o side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect. Immun. 2002, 70, 2583–2590. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Geörg, M.; Maudsdotter, L.; Jonsson, A.B. Endotoxin, capsule, and bacterial attachment contribute to Neisseria meningitidis resistance to the human antimicrobial peptide LL-37. J. Bacteriol. 2009, 191, 3861–3868. [Google Scholar] [CrossRef] [PubMed]
- Llobet, E.; Tomás, J.M.; Bengoechea, J.A. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 2008, 154, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Jolivet-Gougeon, A.; Bonnaure-Mallet, M. Biofilms as a mechanism of bacterial resistance. Drug Discov. Today Technol. 2014, 11, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Wimpenny, J.; Manz, W.; Szewzyk, U. Heterogeneity in biofilms. FEMS Microbiol. Rev. 2000, 24, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.C.; Ruseska, I.; Wright, J.B.; Costerton, J.W. Tobramycin resistance of Pseudomonas aeruginosa cells growing as a biofilm on urinary catheter material. Antimicrob. Agents Chemother. 1985, 27, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Hentzer, M.; Teitzel, G.M.; Balzer, G.J.; Heydorn, A.; Molin, S.; Givskov, M.; Parsek, M.R. Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J. Bacteriol. 2001, 183, 5395–5401. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, A.; Haagensen, J.A.; Zampaloni, C.; Sternberg, C.; Molin, S. Biofilm induced tolerance towards antimicrobial peptides. PLoS ONE 2008, 3, e1891. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Burrows, L.L.; Deber, C.M. Helix induction in antimicrobial peptides by alginate in biofilms. J. Biol. Chem. 2004, 279, 38749–38754. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Burrows, L.L.; Deber, C.M. Alginate as an auxiliary bacterial membrane: Binding of membrane-active peptides by polysaccharides. J. Pept. Res. 2005, 65, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, M.; Mattiuzzo, M.; Herasimenka, Y.; Cescutti, P.; Rizzo, R.; Gennaro, R. Activity of antimicrobial peptides in the presence of polysaccharides produced by pulmonary pathogens. J. Pept. Sci. 2009, 15, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Horsman, S.R.; Charron-Mazenod, L.; Turnbull, A.L.; Mulcahy, H.; Surette, M.G.; Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2013, 13, e115. [Google Scholar] [CrossRef]
- Gooderham, W.J.; Bains, M.; McPhee, J.B.; Wiegand, I.; Hancock, R.E. Induction by cationic antimicrobial peptides and involvement in intrinsic polymyxin and antimicrobial peptide resistance, biofilm formation, and swarming motility of PsrA in Pseudomonas aeruginosa. J. Bacteriol. 2008, 190, 5624–5634. [Google Scholar] [CrossRef] [PubMed]
- Pamp, S.J.; Gjermansen, M.; Johansen, H.K.; Tolker-Nielsen, T. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 2008, 68, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Poole, K. Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 2007, 39, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Piddock, L.J. Multidrug-resistance efflux pumps—Not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.M.; Webber, M.A.; Cooles, S.; Randall, L.P.; La Ragione, R.M.; Woodward, M.J.; Piddock, L.J. The acrab-tolc efflux system of Salmonella enterica serovar Typhimurium plays a role in pathogenesis. Cell. Microbiol. 2006, 8, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Latifi, T.; Groisman, E.A. Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 2006, 59, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Stone, B.J.; Miller, V.L. Salmonella enteritidis has a homologue of tolC that is required for virulence in BALB/c mice. Mol. Microbiol. 1995, 17, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.; Poza, M.; Fernández, A.; Fernández, M.C.; Mallo, S.; Merino, M.; Rumbo-Feal, S.; Cabral, M.P.; Bou, G. Involvement of the AcrAB-TolC efflux pump in the resistance, fitness, and virulence of Enterobacter cloacae. Antimicrob. Agents Chemother. 2012, 56, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Bunikis, I.; Denker, K.; Ostberg, Y.; Andersen, C.; Benz, R.; Bergström, S. An RND-type efflux system in Borrelia burgdorferi is involved in virulence and resistance to antimicrobial compounds. PLoS Pathog. 2008, 4, e1000009. [Google Scholar] [CrossRef] [PubMed]
- Hirakata, Y.; Srikumar, R.; Poole, K.; Gotoh, N.; Suematsu, T.; Kohno, S.; Kamihira, S.; Hancock, R.E.; Speert, D.P. Multidrug efflux systems play an important role in the invasiveness of Pseudomonas aeruginosa. J. Exp. Med. 2002, 196, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Bina, J.E.; Mekalanos, J.J. Vibrio cholerae tolC is required for bile resistance and colonization. Infect. Immun. 2001, 69, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea, J.A.; Skurnik, M. Temperature-regulated efflux pump/potassium antiporter system mediates resistance to cationic antimicrobial peptides in Yersinia. Mol. Microbiol. 2000, 37, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Shafer, W.M.; Qu, X.; Waring, A.J.; Lehrer, R.I. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. USA 1998, 95, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.M.; Shafer, W.M.; Jerse, A.E. Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol. Microbiol. 2008, 70, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.L.; Ambrose, K.D.; Zughaier, S.; Zhou, X.; Miller, Y.K.; Shafer, W.M.; Stephens, D.S. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 2005, 187, 5387–5396. [Google Scholar] [CrossRef] [PubMed]
- Bina, X.R.; Provenzano, D.; Nguyen, N.; Bina, J.E. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect. Immun. 2008, 76, 3595–3605. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chuang, Y.C.; Chang, C.C.; Jeang, C.L.; Chang, M.C. A K+ uptake protein, TrkA, is required for serum, protamine, and polymyxin B resistance in Vibrio vulnificus. Infect. Immun. 2004, 72, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kourtesi, C.; Ball, A.R.; Huang, Y.Y.; Jachak, S.M.; Vera, D.M.; Khondkar, P.; Gibbons, S.; Hamblin, M.R.; Tegos, G.P. Microbial efflux systems and inhibitors: Approaches to drug discovery and the challenge of clinical implementation. Open Microbiol. J. 2013, 7, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Zamfir, A.; Seidler, D.G.; Kresse, H.; Peter-Katalinić, J. Structural investigation of chondroitin/dermatan sulfate oligosaccharides from human skin fibroblast decorin. Glycobiology 2003, 13, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, A.; Frick, I.M.; Björck, L. Dermatan sulphate is released by proteinases of common pathogenic bacteria and inactivates antibacterial alpha-defensin. Mol. Microbiol. 2001, 39, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- McBroom, A.J.; Johnson, A.P.; Vemulapalli, S.; Kuehn, M.J. Outer membrane vesicle production by Escherichia coli is independent of membrane instability. J. Bacteriol. 2006, 188, 5385–5392. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, M.J.; Kesty, N.C. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005, 19, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- McBroom, A.J.; Kuehn, M.J. Release of outer membrane vesicles by gram-negative bacteria is a novel envelope stress response. Mol. Microbiol. 2007, 63, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Duperthuy, M.; Sjöström, A.E.; Sabharwal, D.; Damghani, F.; Uhlin, B.E.; Wai, S.N. Role of the Vibrio cholerae matrix protein Bap1 in cross-resistance to antimicrobial peptides. PLoS Pathog. 2013, 9, e1003620. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Sahl, H.G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006, 4, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, A.; Frick, I.M.; Andersson, E.; Tapper, H.; Björck, L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 2002, 46, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Guina, T.; Yi, E.C.; Wang, H.; Hackett, M.; Miller, S.I. A PhoP-regulated outer membrane protease of Salmonella enterica serovar Typhimurium promotes resistance to alpha-helical antimicrobial peptides. J. Bacteriol. 2000, 182, 4077–4086. [Google Scholar] [CrossRef] [PubMed]
- Sol, A.; Skvirsky, Y.; Nashef, R.; Zelentsova, K.; Burstyn-Cohen, T.; Blotnick, E.; Muhlrad, A.; Bachrach, G. Actin enables the antimicrobial action of LL-37 peptide in the presence of microbial proteases. J. Biol. Chem. 2014, 289, 22926–22941. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Harwig, S.S. Determination of the disulfide array in the human defensin HNP-2. A covalently cyclized peptide. J. Biol. Chem. 1989, 264, 4003–4007. [Google Scholar] [PubMed]
- Maemoto, A.; Qu, X.; Rosengren, K.J.; Tanabe, H.; Henschen-Edman, A.; Craik, D.J.; Ouellette, A.J. Functional analysis of the alpha-defensin disulfide array in mouse cryptdin-4. J. Biol. Chem. 2004, 279, 44188–44196. [Google Scholar] [CrossRef] [PubMed]
- Campopiano, D.J.; Clarke, D.J.; Polfer, N.C.; Barran, P.E.; Langley, R.J.; Govan, J.R.; Maxwell, A.; Dorin, J.R. Structure-activity relationships in defensin dimers: A novel link between beta-defensin tertiary structure and antimicrobial activity. J. Biol. Chem. 2004, 279, 48671–48679. [Google Scholar] [CrossRef] [PubMed]
- Stumpe, S.; Schmid, R.; Stephens, D.L.; Georgiou, G.; Bakker, E.P. Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli. J. Bacteriol. 1998, 180, 4002–4006. [Google Scholar] [PubMed]
- Biegeleisen, K. The probable structure of the protamine-DNA complex. J. Theor. Biol. 2006, 241, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, M.; Korhonen, T.K. The omptin family of enterobacterial surface proteases/adhesins: From housekeeping in Escherichia coli to systemic spread of Yersinia pestis. Int. J. Med. Microbiol. 2004, 294, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kooi, C.; Sokol, P.A. Burkholderia cenocepacia zinc metalloproteases influence resistance to antimicrobial peptides. Microbiology 2009, 155, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Corbett, C.R.; Burtnick, M.N.; Kooi, C.; Woods, D.E.; Sokol, P.A. An extracellular zinc metalloprotease gene of Burkholderia cepacia. Microbiology 2003, 149, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Kooi, C.; Subsin, B.; Chen, R.; Pohorelic, B.; Sokol, P.A. Burkholderia cenocepacia ZmpB is a broad-specificity zinc metalloprotease involved in virulence. Infect. Immun. 2006, 74, 4083–4093. [Google Scholar] [CrossRef] [PubMed]
- Belas, R.; Manos, J.; Suvanasuthi, R. Proteus mirabilis ZapA metalloprotease degrades a broad spectrum of substrates, including antimicrobial peptides. Infect. Immun. 2004, 72, 5159–5167. [Google Scholar] [CrossRef] [PubMed]
- Taggart, C.C.; Greene, C.M.; Smith, S.G.; Levine, R.L.; McCray, P.B.; O’Neill, S.; McElvaney, N.G. Inactivation of human beta-defensins 2 and 3 by elastolytic cathepsins. J. Immunol. 2003, 171, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.R.; To, T.T.; Jain, S.; Braham, P.H.; Darveau, R.P. Porphyromonas gingivalis resistance to polymyxin B is determined by the lipid A 4'-phosphatase, PGN_0524. Int. J. Oral Sci. 2009, 1, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.R.; Jones, J.W.; Do, C.T.; Braham, P.H.; Bainbridge, B.W.; To, T.T.; Goodlett, D.R.; Ernst, R.K.; Darveau, R.P. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4'-phosphatase activities. Cell Microbiol. 2009, 11, 1587–1599. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.E. The lipid A palmitoyltransferase PagP: Molecular mechanisms and role in bacterial pathogenesis. Mol. Microbiol. 2005, 57, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, N.; Fernandez, R.C. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 2013, 3, e3. [Google Scholar] [CrossRef]
- Ernst, R.K.; Yi, E.C.; Guo, L.; Lim, K.B.; Burns, J.L.; Hackett, M.; Miller, S.I. Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science 1999, 286, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Saroj, S.D.; Llewellyn, A.C.; Tzeng, Y.L.; Weiss, D.S. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 2013, 497, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Napier, B.A.; Schroeder, M.R.; Louwen, R.; Zhao, J.; Chin, C.Y.; Ratner, H.K.; Llewellyn, A.C.; Jones, C.L.; Laroui, H.; et al. A CRISPR-Cas system enhances envelope integrity mediating antibiotic resistance and inflammasome evasion. Proc. Natl. Acad. Sci. USA 2014, 111, 11163–11168. [Google Scholar] [CrossRef] [PubMed]
- Moranta, D.; Regueiro, V.; March, C.; Llobet, E.; Margareto, J.; Larrarte, E.; Larrate, E.; Garmendia, J.; Bengoechea, J.A. Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells. Infect. Immun. 2010, 78, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Islam, D.; Bandholtz, L.; Nilsson, J.; Wigzell, H.; Christensson, B.; Agerberth, B.; Gudmundsson, G. Downregulation of bactericidal peptides in enteric infections: A novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 2001, 7, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, B.; Regnault, B.; Guo, J.; Zhang, Z.; Stanley, S.L.; Sansonetti, P.J.; Pédron, T. Virulent Shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. J. Exp. Med. 2008, 205, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Hornef, M.W.; Wick, M.J.; Rhen, M.; Normark, S. Bacterial strategies for overcoming host innate and adaptive immune responses. Nat. Immunol. 2002, 3, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Napier, B.A.; Burd, E.M.; Satola, S.W.; Cagle, S.M.; Ray, S.M.; McGann, P.; Pohl, J.; Lesho, E.P.; Weiss, D.S. Clinical use of colistin induces cross-resistance to host antimicrobials in Acinetobacter baumannii. MBio 2013, 4, e00021-13. [Google Scholar] [CrossRef] [PubMed]
- Napier, B.A.; Band, V.; Burd, E.M.; Weiss, D.S. Colistin heteroresistance in Enterobacter cloacae is associated with cross-resistance to the host antimicrobial lysozyme. Antimicrob. Agents Chemother. 2014, 58, 5594–5597. [Google Scholar] [CrossRef] [PubMed]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Band, V.I.; Weiss, D.S. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics 2015, 4, 18-41. https://doi.org/10.3390/antibiotics4010018
Band VI, Weiss DS. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics. 2015; 4(1):18-41. https://doi.org/10.3390/antibiotics4010018
Chicago/Turabian StyleBand, Victor I., and David S. Weiss. 2015. "Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria" Antibiotics 4, no. 1: 18-41. https://doi.org/10.3390/antibiotics4010018