Utility of the Biosynthetic Folate Pathway for Targets in Antimicrobial Discovery

Department of Veterinary Pathobiology, Oklahoma State University, 250 McElroy Hall, Stillwater, OK 74078, USA
†
Current address: Department of Chemistry and Biochemistry, University of Oklahoma, Norman, OK 73019, USA.
Antibiotics 2014, 3(1), 1-28; https://doi.org/10.3390/antibiotics3010001
Submission received: 1 November 2013
/
Revised: 8 January 2014
/
Accepted: 9 January 2014
/
Published: 21 January 2014
(This article belongs to the Special Issue Feature Paper 2013)
Abstract
:The need for new antimicrobials is great in face of a growing pool of resistant pathogenic organisms. This review will address the potential for antimicrobial therapy based on polypharmacological activities within the currently utilized bacterial biosynthetic folate pathway. The folate metabolic pathway leads to synthesis of required precursors for cellular function and contains a critical node, dihydrofolate reductase (DHFR), which is shared between prokaryotes and eukaryotes. The DHFR enzyme is currently targeted by methotrexate in anti-cancer therapies, by trimethoprim for antibacterial uses, and by pyrimethamine for anti-protozoal applications. An additional anti-folate target is dihyropteroate synthase (DHPS), which is unique to prokaryotes as they cannot acquire folate through dietary means. It has been demonstrated as a primary target for the longest standing antibiotic class, the sulfonamides, which act synergistically with DHFR inhibitors. Investigations have revealed most DHPS enzymes possess the ability to utilize sulfa drugs metabolically, producing alternate products that presumably inhibit downstream enzymes requiring the produced dihydropteroate. Recent work has established an off-target effect of sulfonamide antibiotics on a eukaryotic enzyme, sepiapterin reductase, causing alterations in neurotransmitter synthesis. Given that inhibitors of both DHFR and DHPS are designed to mimic their cognate substrate, which contain shared substructures, it is reasonable to expect such “off-target” effects. These inhibitors are also likely to interact with the enzymatic neighbors in the folate pathway that bind products of the DHFR or DHPS enzymes and/or substrates of similar substructure. Computational studies designed to assess polypharmacology reiterate these conclusions. This leads to hypotheses exploring the vast utility of multiple members of the folate pathway for modulating cellular metabolism, and includes an appealing capacity for prokaryotic-specific polypharmacology for antimicrobial applications.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The application of antibiotic treatments has revolutionized health care, but the antimicrobial development pipeline is quickly becoming depleted and new therapeutics are badly needed [1,2,3,4]. During this timeframe, healthcare has also experienced an explosive growth in the number of strains of drug-resistant bacteria, exacerbating the need for new antimicrobials [5,6]. Antibiotic drug development was stalled for almost two decades, in large part due to unforeseen problems with newer and innovative approaches [7]. The advent of large-scale genomics prompted target-based efforts, where specific proteins essential to bacterial growth were employed in high-throughput screening efforts. While this did produce “hits”, the translation to whole cells screens resulted in a lack of permeability or solubility in almost every case [3,4,8].
It is increasingly recognized that the paradigm of a single drug inhibiting a single target is oversimplified, and in reality most compounds exhibit a spectrum of pharmacology [9,10,11,12]. Efforts for identifying targets have led investigators to explore metabolic pathways, and computational efforts have delineated a predominance of a select few chemotype scaffolds that also display previously unappreciated polypharmacology [10,13,14,15,16]. The recognition of multiple mechanisms of action (MOAs) is being taken advantage of by re-purposing of existing therapeutics for so-called “off label” or “secondary” effects [17,18]. To date, however, efforts to re-purpose U.S. Federal Drug Administration approved drugs to leverage discovered off-target effects have not progressed quickly enough to meet current challenges. The application of sub-minimum inhibitory concentrations (MIC) levels of antibiotic is known to affect cellular targets not necessarily related to the therapeutic MOA, consistent with a polypharmacologic approach [19,20,21]. With the computational studies of metabolic pathways has come the recognition that many members of a pathway bind to structurally similar endogenous substrates and products [22,23,24,25]. When an inhibitor to one of these members is designed to mimic the endogenous ligand, the probability of cross-reactions with other pathway members increases dramatically. By targeting multiple cellular systems the efficiency of bacterial resistance mechanisms would be greatly diminished [11,24,26].
A typical example of polypharmacology can be found in the folate pathway [22,26]. While there has been much work within the human folate pathway, little is published regarding polypharmacology of the bacterial folate metabolism pathways. Current approaches employing anti-folates rely on inhibiting dihydrofolate reductase (DHFR), which displays sequence variations allowing selective inhibition, and dihydropteroate synthase (DHPS), an enzyme lacking in higher eukaryotes [27,28,29,30,31,32,33,34]. Extensive work on chemo-centric groupings has highlighted the overlap among folate pathway members with respect to ligand structures [35,36,37,38]. However, much of this work is focused on pathway members with known inhibitors, such as DHFR and DHPS, and, thus, other members in the folate pathway remain uncharacterized with regards to their potential for overlapping inhibition. Computational studies have found that ligands for one particular folate enzyme are likely to also bind to other folate compounds. In addition, inhibitors for a given enzyme in the folate pathway have a high probability of binding to other folate pathway members [22]. While specific examples of anti-folate inhibitors binding multiple proteins in the pathway exist (for example, Pemetrexed [39]), there remains a gap in systematic experimental evidence to support these activities. Specific links have been made between inhibitors of dihydrofolate reductase (DHFR) and other downstream enzymes in eukaryotes: phosphoribosyl-glycinamide formyl-transferase (GART), phosphoribosyl-aminoimidazole-carboxamide formyl-transferase (AICART), and thymidine synthase (TS) [35,39,40,41]. The folate biosynthetic pathway found upstream of DHFR is unique to prokaryotes and lower eukaryotes, which cannot acquire folate from their environment. There are efforts being made to target multiple binding sites within the same folate pathway enzyme (“bi-substrate” inhibitors) [42,43,44,45]; however, efforts to target multiple individual binding sites of enzymes upstream of prokaryotic DHFR are limited [46].
Numerous previous reviews of the folate biosynthesis pathway are available [27,34,47,48,49,50,51]. The current review will set out to characterize the antibacterial therapeutic potential of targets in folate metabolism upstream of DHFR with an emphasis on the overlap of binding potentials of individual targets. This is to address the hypothesis that, armed with the current state of knowledge, a minimalistic inhibitor could be designed to modulate multiple but specific targets within this pathway in pathogens. The main hurdle will be species selectivity, such that bacterial pathogens can be targeted while minimizing deleterious actions against the eukaryotic host. Homology between prokaryotes and eukaryotes is limited by focusing on the biosynthetic pathway upstream of DHFR, which is not present in organisms such as eukaryotes. A table listing the available entries for bacterial folate biosynthesis enzymes from the Protein Databank (as of October 27, 2013), as discussed further below, is included for reference purposes as Supplementary Table S1 [52]. These listings include the organism used as the source of the enzyme, the complexed ligands and the primary citations in efforts to facilitate further studies.
Pathways for Folate Production and Utilization
The DHFR enzyme is a central player in the folate pathway that links divergent folate synthesis or acquisition mechanisms to the production of tetrahydrofolate (THF). The THF product serves as a critical co-factor by shuttling methyl and formyl groups utilized in one-carbon transfer reactions, which are required for synthesis of purines, amino acids, S-adenosylmethionine, and formyl-methionine [51,53]. As there is no redundancy in the reaction carried out by DHFR, it is an obligate and critical metabolic node. Inhibition of DHFR directly halts cellular replication by starving the cell of needed cellular precursors; its effectiveness has been proven in anti-cancer, anti-plasmodial, and anti-bacterial treatments [27,34,50].
Most of the one-carbon transfer reactions downstream of DHFR are redundant or highly conserved, but the upstream pathways are divergent between eukaryotes and prokaryotes. Included in the upstream biosynthesis are reactions that form the pterin component of folate. Pterin synthesis in eukaryotes utilizes, among others, the enzyme sepiapterin reductase to produce tetrahydrobiopterin [30]. This product is critical for proper signaling in nerves and has an important role in the homeostasis of brain chemistry [54]; mutations in this protein are associated with phenylketonuria and related metabolic syndromes [55,56]. Prokaryotes lack this pathway, but instead synthesize the pterin portion of folate de novo from guanosine triphosphate (GTP), p-aminobenzoic acid (pABA) and glutamate. This flux passes through a series of seven enzymes unique to bacteria, protozoa, and some lower eukaryotes [27,34,51]. The folate pathway is heavily pursued for drug targets due to its obligate nature and to the very selective profile of the enzymes. Of note is the reliance on the shikimate pathway for production of GTP, which contains the target of the herbicide glyphosate [57,58,59,60,61]. In some organisms, including yeast and plants, there also exists an overlapping pathway that can salvage folate, such as malarial parasite from the serum of the infected host [48,62].
2. Results and Discussion
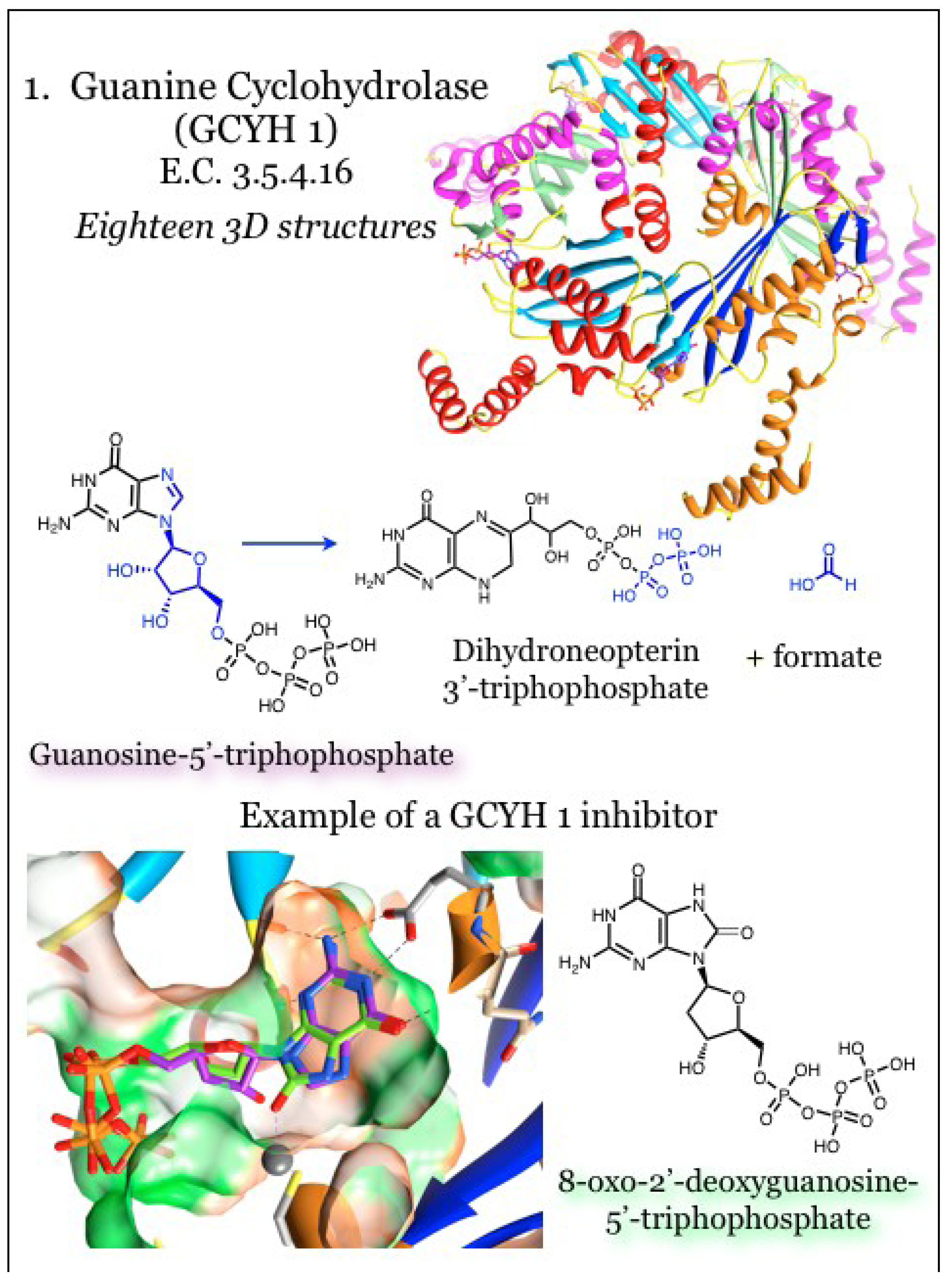
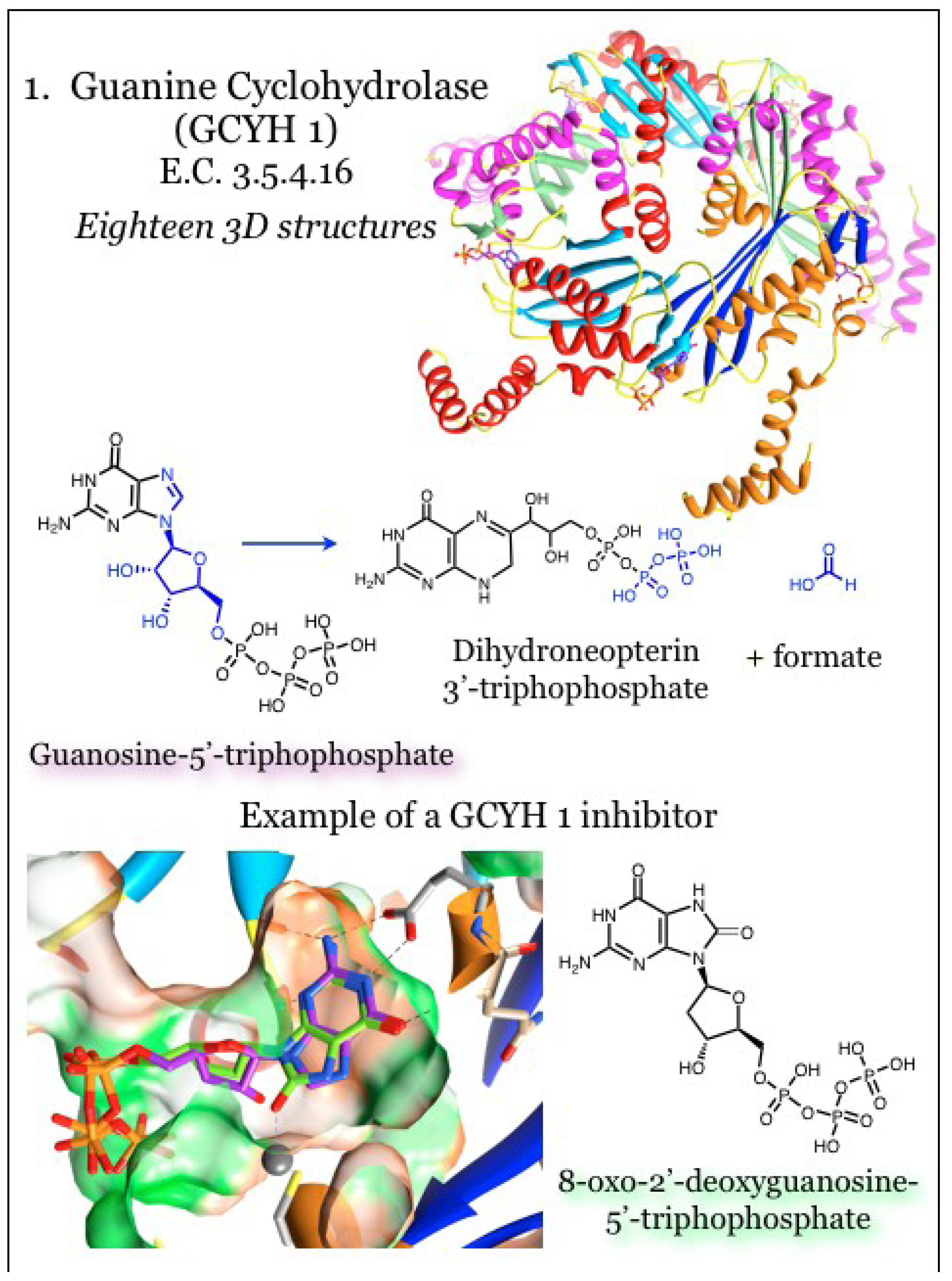
2.1. Enzymes for the Biosynthesis of Folate: 1. Guanine Cyclohydrolase (GCYH I)
In bacteria, that pathway to pterin synthesis is initiated with GTP, which undergoes ring opening, rearranging and reclosing to form dihydroneopterin 3'-triphosphate via guanine cyclohydrolase I (GCYH I, E.C. 3.5.4.16, Figure 1), encoded by the folE gene [51,56,63,64,65,66]. The protein is composed of a pentameric beta-barrel surrounded by alpha helices, and the active site requires contributions from three subunits [56,63,64,65,66]. The enzyme requires a zinc cation, which is coordinated by two cysteine residues, one histidine residue, and ordered water molecules [56,63]. Derivatives of 8-oxoguanine competitively inhibit the bacterial enzyme in vitro by mimicking the transition state configuration [56]. The guanine moiety of 8-oxo-GTP was found inserted into a cavity of the protein utilizing interactions similarly to pterin-binding proteins. Polar groups within guanine ring are hydrogen bonded within the binding site, and in particular interact in a characteristic fashion with an acidic residue [56,63]. The triphosphate group was coordinated at the periphery of the binding site, which is basic in nature to complement the charged phosphate moieties [56].
Another enzyme possessing GCYH I activity has been described, and its gene has been named folE2 [67]. This enzyme has no shared sequence with GCYH I described above, is prokaryotic specific, and is present in most Archaea and in bacteria that frequently lack the canonical GCYH I [68]. This highlights a potential Achilles heel for exploitation in antibacterial-specific targets. The overall T-type protein fold is maintained in a tetrameric arrangement, with two eight-stranded antiparallel beta-sheets pairing to form a 16-stranded beta barrel that is surrounded on each side with alpha helices. The tetramer further oligomerizes by forming a homodimer, yielding an octameric arrangement with D4 symmetry. The extrapolated binding cavity for the guanine moiety within this GCYH Ib enzyme is more shallow than in GCYH I, but the acidic Glu residue that coordinates the nitrogen groups is present. The basic patch to neutralize the phosphate groups is maintained, although the residues themselves are contributed by different subunits between GCYH I and GCYH Ib. While there are differences noted in the specific interactions, the overall pose of the guanine is the same between the two GCYH enzymes [68].
Figure 1.
Guanine cyclohydrolase (GCYH I) catalyzes a reaction utilizing GTP and producing dihydroneopterin 3'-triphosphate. Coordinates for GCYH I were taken from PDB ID 1WUR [56] and 4DU6 [69] with GTP (purple). Pentameric subunits can be distinguished by altered colors for beta-strands (blue, dodger blue, cyan) and alpha-helices (orange, red, magenta). A catalytic zinc atom is represented by a larger grey sphere, and is coordinated with a conserved Cys residue. The guanine base resides in a relatively hydrophobic pocket (orange surface) with polarity limited to the nitrogen atoms of the heterocycle. Conserved interactions, including between the guanine base and an acidic residue, are indicated.
Figure 1.
Guanine cyclohydrolase (GCYH I) catalyzes a reaction utilizing GTP and producing dihydroneopterin 3'-triphosphate. Coordinates for GCYH I were taken from PDB ID 1WUR [56] and 4DU6 [69] with GTP (purple). Pentameric subunits can be distinguished by altered colors for beta-strands (blue, dodger blue, cyan) and alpha-helices (orange, red, magenta). A catalytic zinc atom is represented by a larger grey sphere, and is coordinated with a conserved Cys residue. The guanine base resides in a relatively hydrophobic pocket (orange surface) with polarity limited to the nitrogen atoms of the heterocycle. Conserved interactions, including between the guanine base and an acidic residue, are indicated.
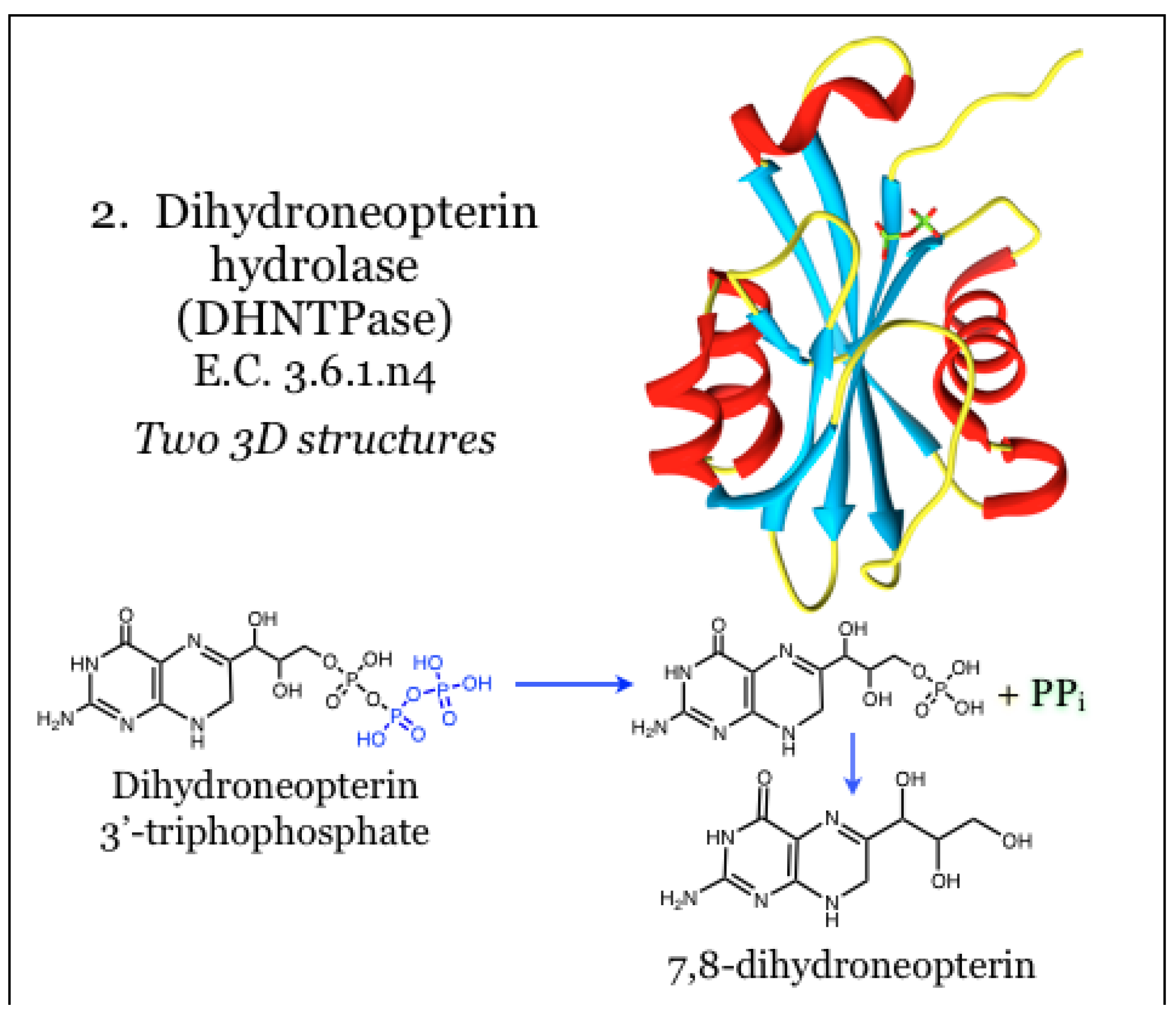
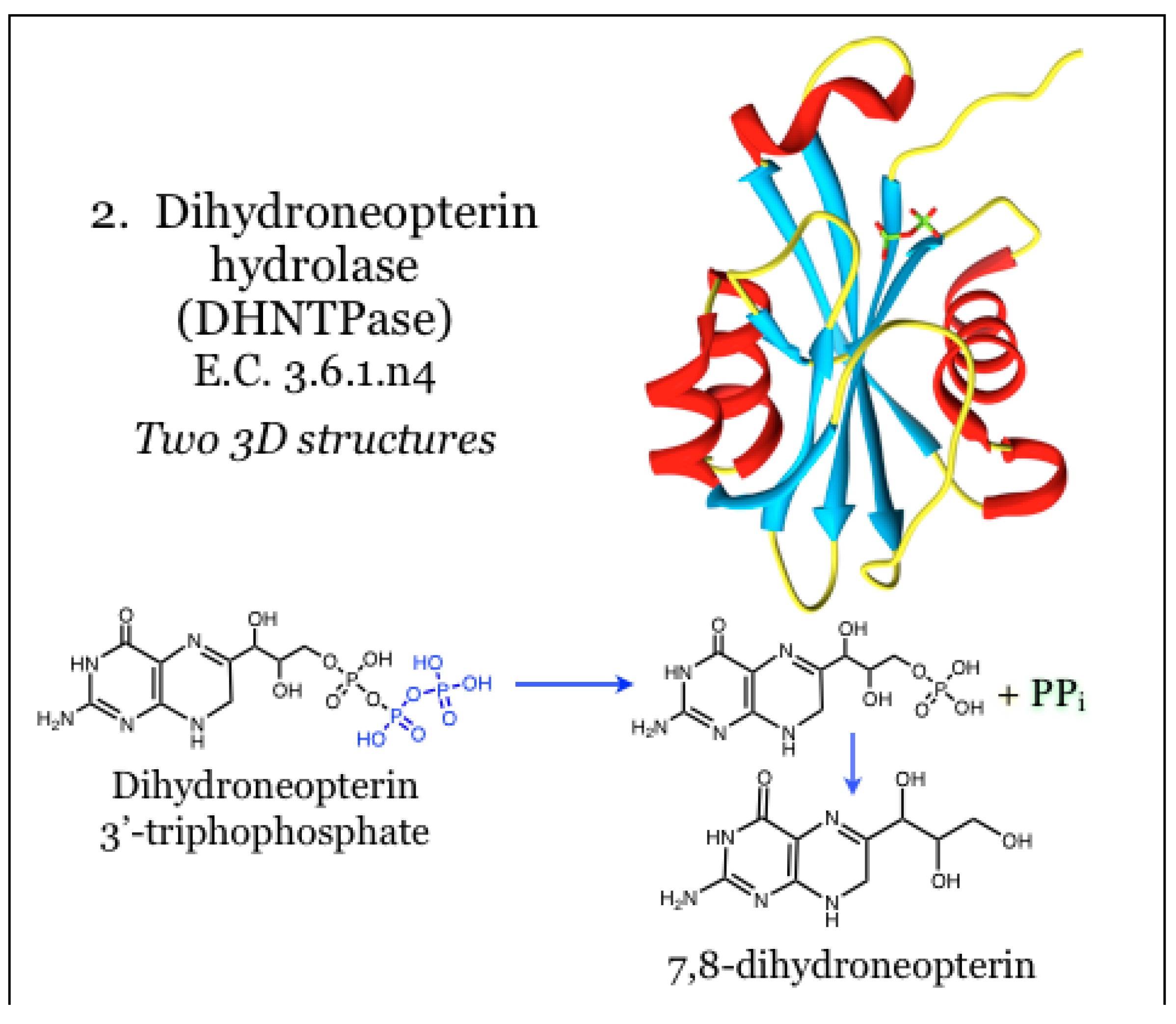
2.2. Enzymes for the Biosynthesis of Folate: 2. Dihydroneopterin Hydrolase (DHNTPase)
The product of GCYH, 7,8-dihydroneopterin triphosphate, is then acted upon by a poorly characterized pyrophosphatase, called dihydroneopterin hydrolase (DHNTPase, E.C. 3.6.1.n4), to yield 7,8-dihydroneopterin. The identity of this required enzymatic activity has been questioned, and as such it has generally been excluded from therapeutic targeting efforts [27,34,51]. The gene for this activity has been identified as nudB in bacteria [48,70] and folQ in Lactobacillus and plants [48,70]. The product of the nudB gene, Nudix NTP hydrolase, catalyzes the removal of pyrophosphate from dihydroneopterin triphosphate (Figure 2) [70]. The site of catalysis is housed on a mixed beta-sheet surrounded by alpha helices, and the catalytic mechanism requires coordination with divalent metal ions, such as magnesium. Some evidence indicates that loops overhanging the catalytic site are mobile and control access, likely in concert with catalytic events. A structural model of dihydroneopterin within the active site was generated and reportedly predicts an aromatic stack of the pterin heterocycle between two Phe residues. In addition, hydrogen bonding potential of the pterin was satisfied by interaction with mainchain atoms, as well as Ser and Glu residues. To date this is the only structural information available for nudB gene product [70].
Figure 2.
Dihydroneopterin hydrolase possesses pyrophosphatase activity for dihydroneopterin 3'-triphosphate, and in bacteria is carried out by a Nudix NTP hydrolase. Coordinates for DHNTPase were taken from PDB ID 2O1C, as complexed with pyrophosphate (green), which yields feedback inhibition [70]. No structural models with substrate or information on inhibitors are available.
Figure 2.
Dihydroneopterin hydrolase possesses pyrophosphatase activity for dihydroneopterin 3'-triphosphate, and in bacteria is carried out by a Nudix NTP hydrolase. Coordinates for DHNTPase were taken from PDB ID 2O1C, as complexed with pyrophosphate (green), which yields feedback inhibition [70]. No structural models with substrate or information on inhibitors are available.
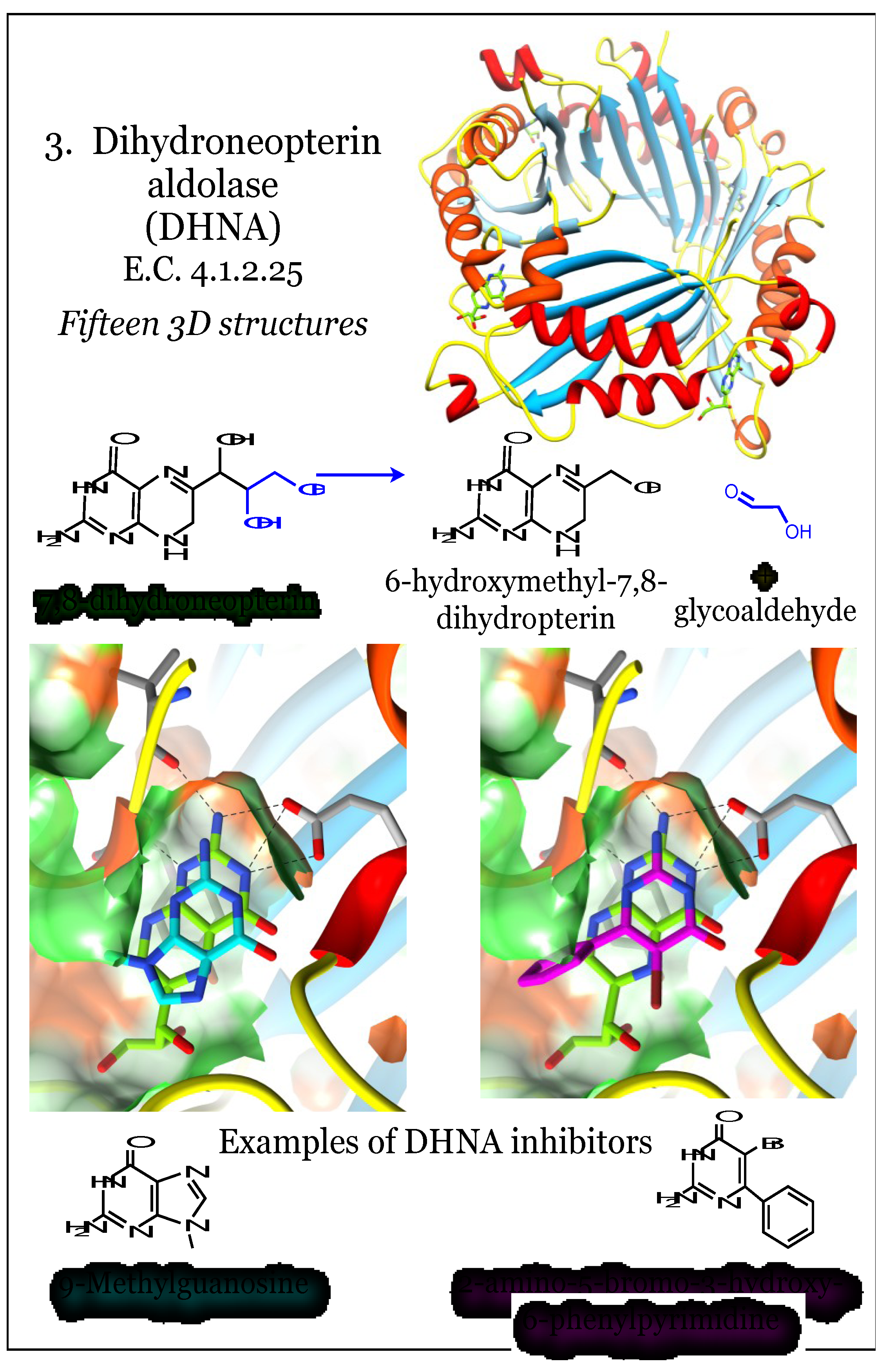
2.3. Enzymes for the Biosynthesis of Folate: 3. Dihydroneopterin Aldolase (DHNA)
When folate synthesis reactions are evaluated for conservation among bacterial species, the first pathway member that readily qualifies is the enzyme encoded by the folB gene, dihydroneopterin aldolase (DHNA, E.C. 4.1.2.25, Figure 3) [27]. This enzyme uses 7,8-dihydroneopterin as a substrate and cleaves a carbon-carbon bond in a stereospecific manner to produce 6-hydroxymethyl-7,8-dihydropterin and glycoaldehyde [71,72,73]. This is a unique aldolase enzyme, requiring neither Schiff base formation within the enzyme (Class I aldolases) or a zinc ion (Class II aldolases) for catalysis [71,73]. Further, it is reported to exhibit reversible epimerase activity, converting 7,8-dihydromonapterin to its own substrate 7,8-dihydroneopterin [74]. Its structure is composed of a four-stranded antiparallel beta sheet with two long helices along one side of each monomer (Figure 3). The protein oligomerizes into a tetramer, resulting in a 16-stranded beta barrel, with the longer alpha helices surrounding the outside. In some bacteria, such as M. tuberculosis, these tetramers dimerize to yield an allosterically regulated active octamer [72]. In other bacteria, such as Streptococcus pneumonia, the active site is fully formed even as a tetramer [74]. There has been some suggestion that the octamerization is artifactual and a result of high protein concentration [75]. There are known variations in the organization of DHNA; for example, this activity is encoded by two different domains (fasA, fasB) in Pneumocystis carinii, and these are fused to the 2-amino4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase (HPPK) and DHPS enzymes [76]. There is a report of a FolX protein from E. coli that is a paralog of DHNA [48].
Figure 3.
Dihydroneopterin aldolase (DHNA) utilizes 7,8-dihydroneopterin to produce 6-hydroxymethyl-7,8-dihydroneopterin and glycoaldehyde. Coordinates for DHNA were taken from PDB ID 2NM2, as complexed with a substrate neopterin compound (green) [71]. Additional structures were taken from PDB ID 1RRW, complexed with 9-methylguanosine (cyan), and 1RSI, complexed with a brominated pyrimidine (magenta) [77]. The four subunits comprising the tetrametric protein assembly can be distinguished by altered colors for beta-strands (dodger blue, sky blue) and alpha-helices (red, orange). The pterin moiety is bound in a relatively polar (green surface) cleft between two subunits. Conserved interactions, including between the aminopterin moiety and an acidic residue, are indicated.
Figure 3.
Dihydroneopterin aldolase (DHNA) utilizes 7,8-dihydroneopterin to produce 6-hydroxymethyl-7,8-dihydroneopterin and glycoaldehyde. Coordinates for DHNA were taken from PDB ID 2NM2, as complexed with a substrate neopterin compound (green) [71]. Additional structures were taken from PDB ID 1RRW, complexed with 9-methylguanosine (cyan), and 1RSI, complexed with a brominated pyrimidine (magenta) [77]. The four subunits comprising the tetrametric protein assembly can be distinguished by altered colors for beta-strands (dodger blue, sky blue) and alpha-helices (red, orange). The pterin moiety is bound in a relatively polar (green surface) cleft between two subunits. Conserved interactions, including between the aminopterin moiety and an acidic residue, are indicated.
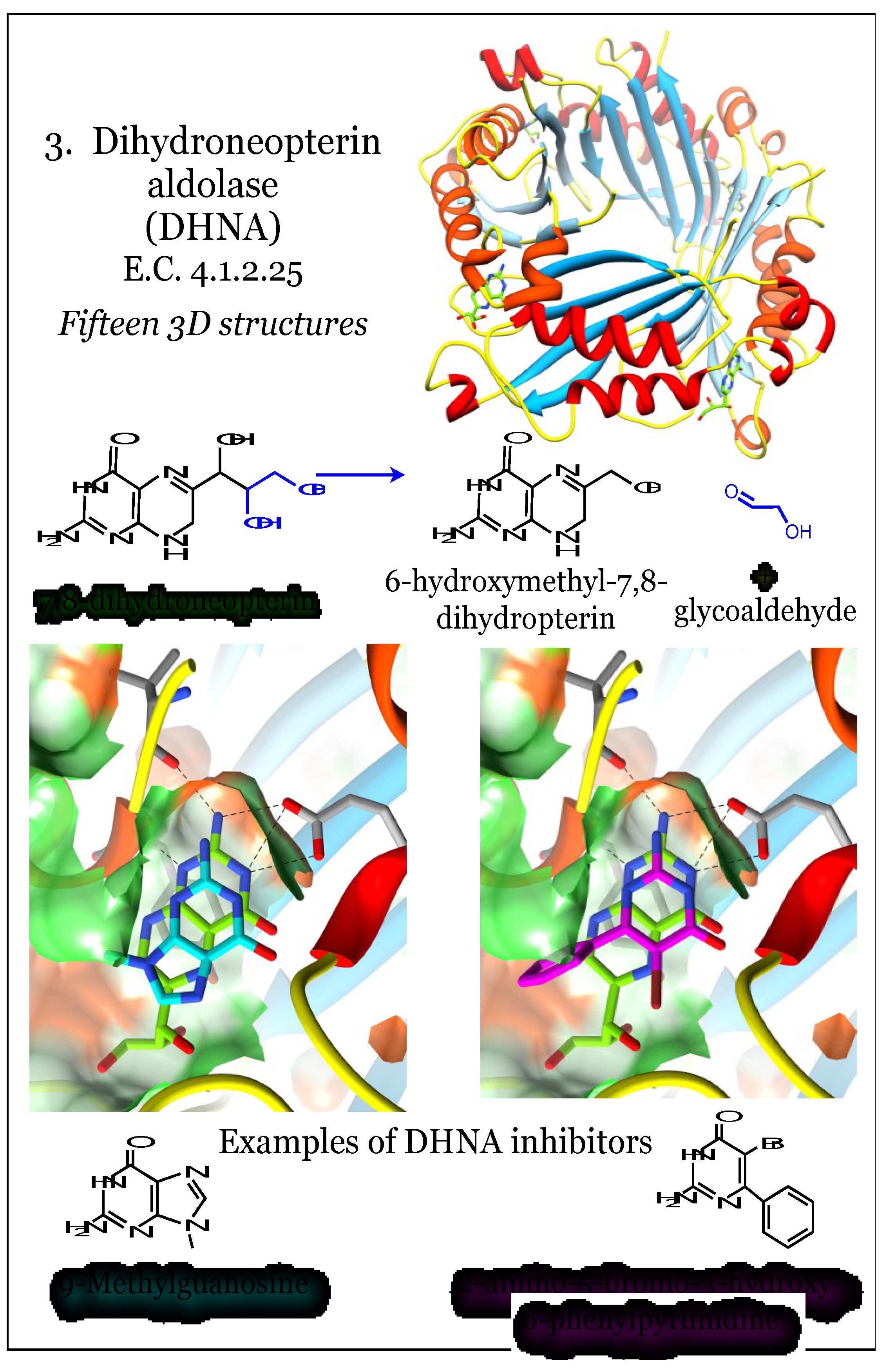
The DHNA active site is composed of residues from two different monomers and located external to the beta barrel [71,72,73,74,77,78]. The binding of the pterin substrate is driven by at least three specific hydrogen bonds contributed by main chain polar atoms of the protein and by acidic Glu and/or Asp residues. These anchor the pyrimidine ring, and in addition the pocket can be heavily hydrated [73,74,77]. The hydrophobic faces of the heterocycle are sandwiched between hydrophobic residues from each monomer, including Val, Leu, Pro, and a conserved and critical Tyr residue. The catalytic portion of the site is distal from the pterin-docking cavity, and catalysis utilizes a highly conserved acidic residue (usually a Glu) and a Lys residue [74,77]. The DHNA enzyme has been pursued as an antimicrobial target, as well as potential herbicide target in plants [78]. Of identified inhibitors, they are exclusively based on pterin or pyrimidine analogs that can fulfill a defined spatial pattern of hydrogen bonds within the binding site [77]. In particular, a medicinal chemistry undertaking demonstrated the ability to extend the structure of the analogs beyond an anchoring pterin-like group, although their most potent in vitro inhibitor failed to elicit inhibition of bacterial growth [77]. At least one group has noted the conservation of interactions between DHNA with the pterin ligand and of DHFR and its pterin ligand [73].
The architecture of the DHNA active site is homologous to other pterin-binding proteins, and, in particular, with pyruvoyltetrahydropterin synthase (PTPS, E.C. 4.2.3.12), an apparently redundant enzyme that is present in some bacteria and in Archaea [49,79,80]. PTPS is also present in eukaryotes, although sequence homology is distinctly low [81]. Organisms that rely on PTPS for folate synthesis include many important mediators of pathogenesis, including Leishmania species [81], Plasmodium falciparum [79] and Toxoplasma gondii [81]. The organization of this enzyme is as a dimer of trimers to form an active hexamer. The pterin-binding site has contributions from three monomers, and the overall orientation is reminiscent of DHNA as the trimers compose a 12-stranded barrel that then undergo dimerization [82].
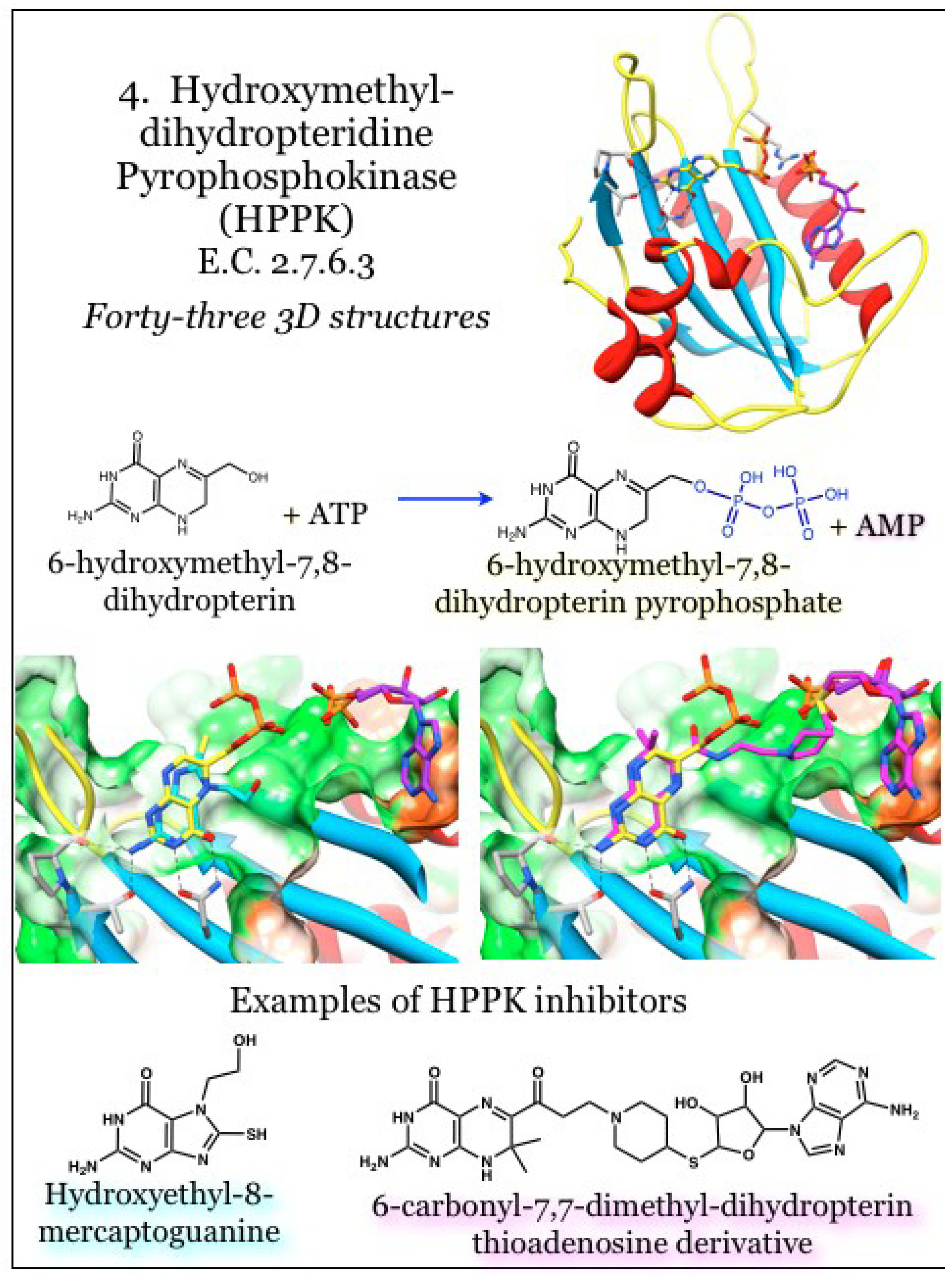
2.4. Enzymes for the Biosynthesis of Folate: 4. 2-Amino-4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase (HPPK)
The enzyme 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase, or HPPK (E.C. 2.7.6.3), catalyzes the transfer of a pyrophosphate group from an ATP donor onto 6-hydroxylmethyl-7,8-dihydropterin (HMDP), the product of the DHNA enzyme (Figure 4). This enzyme is among the most well studied pyrophosphate kinases, transferring a pyrophosphate unit at the beta phosphate rather than more typical kinase reaction utilizing the gamma phosphate. HPPK has been pursued as an antimicrobial target; however, identification of in vitro inhibitors has not translated into microbial growth inhibitors to date [43,44,51,83,84,85,86,87].
The monomeric protein contains a thioredoxin-like fold [86] composed of a four-stranded anti-parallel beta sheet with helices appended to each face. Connections between the elements of secondary structure provide three loops that undergo major structural transitions in response to binding and catalysis [88,89,90], although these changes are attenuated due to sequence differences between E. coli and other bacteria, such as S. pneumoniae [74] and Y. pestis [91]. The enzyme requires ordered binding, with a Mg2+-ATP unit binding first and triggering movement of a loop (L3) to more than 20 Å away from the binding site. This movement triggers ordering of critical Arg residues and also completes the formation of the substrate HMDP site [91,92]. The bound pterin moiety triggers a stabilization of the binding site and subsequent closure of loops L1 and L3 around the site; a Trp residue at the tip of L3 effectively seals the catalytic site from the bulk solvent [93]. The rate-limiting step is product release, which requires movement of L3 again away from the site. The entire catalytic cycle is believed to take six steps, including a transition state [94].
Figure 4.
2-amino-4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase (HPPK) transfers a pyrophosphate unit from ATP onto 6-hydroxymethyl-7,8-dihydropterin. Coordinates for HPPK were taken from PDB ID 1RAO, complexed with the product 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (yellow) and AMP (purple) [92]. The complexes with inhibitors were taken from PDB ID 4AD6 (hydroxyethyl-8-mercaptoguanine, cyan) and 3UDV (a bisubstrate linked dihydropterin thioadenosine, magenta) [84,95]. The pterin moiety is bound in a shallow and neutral (white surface) to polar (green surface) crevice. Conserved interactions with the aminopterin moiety are indicated.
Figure 4.
2-amino-4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase (HPPK) transfers a pyrophosphate unit from ATP onto 6-hydroxymethyl-7,8-dihydropterin. Coordinates for HPPK were taken from PDB ID 1RAO, complexed with the product 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (yellow) and AMP (purple) [92]. The complexes with inhibitors were taken from PDB ID 4AD6 (hydroxyethyl-8-mercaptoguanine, cyan) and 3UDV (a bisubstrate linked dihydropterin thioadenosine, magenta) [84,95]. The pterin moiety is bound in a shallow and neutral (white surface) to polar (green surface) crevice. Conserved interactions with the aminopterin moiety are indicated.

As with other pterin-binding enzymes from the biosynthetic folate pathway, the pterin moiety is bound within a cavity, with the catalytically active portion situated in a groove along the enzyme surface proximal to the ATP binding site [86,87]. The interactions between the protein and the pterin moiety include extensive hydrogen bonding and aromatic residues stacking on either face of the pterin heterocycle [86,94]. Investigations into inhibition revealed the most effective method was a to occupy both the pterin substrate and the adenosine co-factor sites, thus generating a “bisubstrate analog” [43,44,85,86]. Demonstrations have highlighted the requirement for the pterin moiety; a motif shared among the folate pathway members, such as in 8-mercaptoguanine [86]. The bridging phosphate groups could be replaced with other physiochemically desirable groups and the adenine base also had flexibility in docking orientation [92]. It is of note that 8-mercaptoguanine also shows competitive inhibition against DHPS, the next enzyme in the biosynthetic folate pathway [96]. Similarly, investigations with Saccharomyces cerevisiae HPPK-DHPS-fused enzyme revealed occupancy of both enzymes’ active sites by the same inhibitory pterin monophosphate analog compound [97]. These observations strongly support the supposition of inhibiting multiple enzymes from the folate biosynthetic pathway with a single therapeutic agent [86].
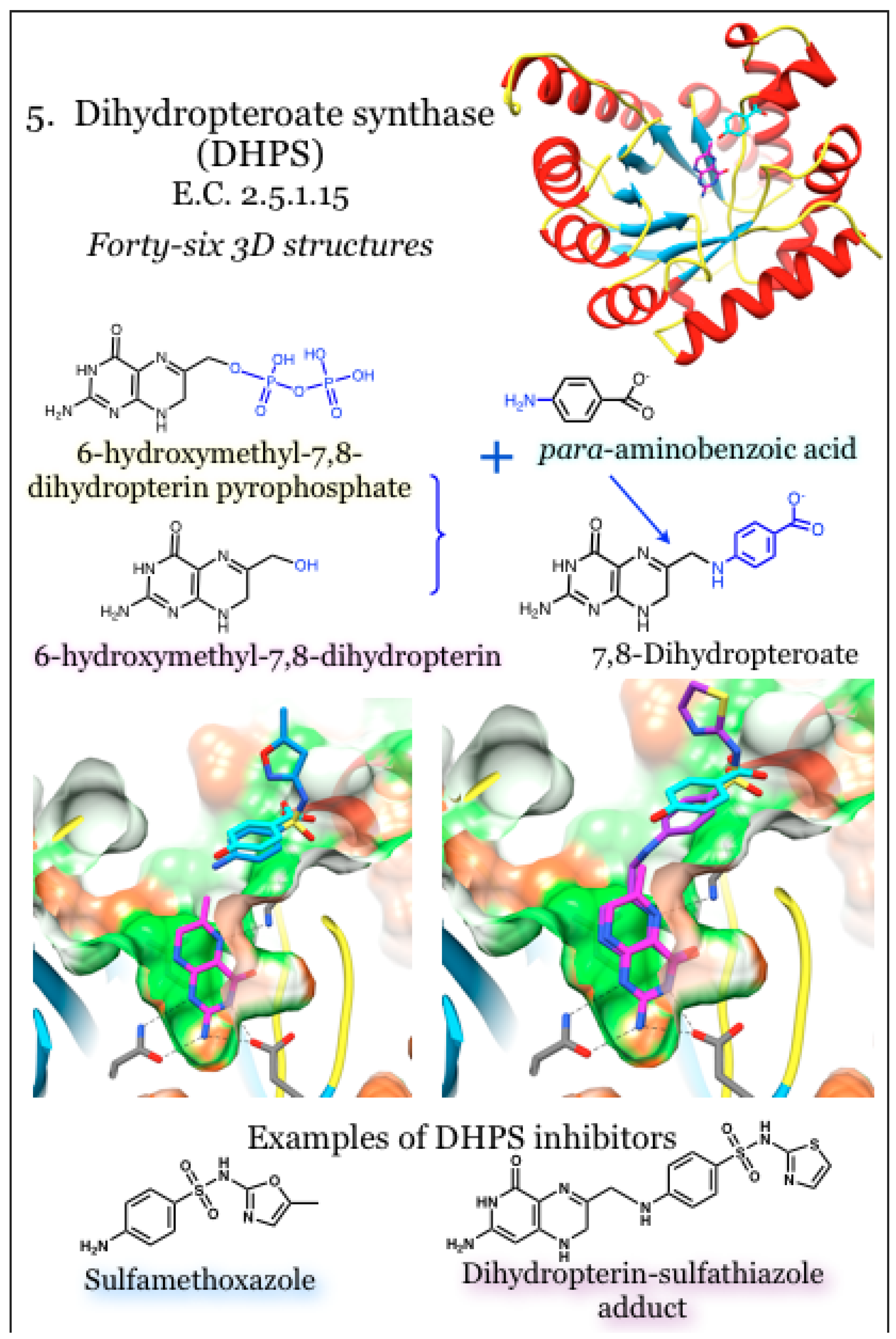
2.5. Enzymes for the Biosynthesis of Folate: 5. Dihydropteroate Synthase (DHPS)
The enzyme dihydropteroate synthase (DHPS) is a dimeric triosephosphate isomerase-type (TIM)-barrel protein with eight parallel beta strands surrounded by eight alpha helices [42,98,99,100]. It catalyzes the condensation of 6-hydroxymethyl-7,8-dihydropterin pyrophosphate with para-aminobenzoic acid (pABA) to yield 7,8-dihydropteroate (Figure 5). DHPS is the bacterial target for one of the earliest introduced classes of synthetic antimicrobials, the sulfonamides [99,101]. Sulfonamide compounds are pABA analogs that in many, if not all, cases serve as an alternative substrate and yield a dead-end product [101,102,103,104,105]. Among the numerous off-target effects of sulfa drugs is the inhibition of mammalian sepiapterin reductase, critical to nerve cells for the production of tetrahydrobiopterin, which serves as a co-factor for neurotransmitter synthesis [54]. Mutations in the DHPS sequence are responsible for conferring resistance to sulfonamide therapeutics, and structurally these are located around the pABA binding site [42,96,100,106]. There is a required order of addition such that binding of the pterin substrates must precede pABA (or inhibitory analog) binding [99]. Early structures revealed high mobility for two loops known to be involved in binding to pABA and thus in conferring resistant to sulfa drugs. Pioneering studies using crystallized and catalytically active DHPS recently revealed a novel SN1-type mechanism that first bound the pterin pyrophosphate substrate, anchoring a mobile loop, which in turn formed the pABA site [101]. In the absence of the pterin substrate, the pterin-binding site is occupied by an Arg side chain. This Arg residue is displaced upon pterin binding and then re-locates to form part of the pABA site; as such, it is hypothesized to increase stability and provide for regulation of enzymatic activity [42]. The reaction is initiated by removing the pyrophosphate moiety independently from the condensation reaction, resulting in occupancy of unphosphorylated pterin substrates in the absence of pABA or an analog thereof [99,101]; magnesium is needed for stabilization and release of the pyrophosphate [101]. Free pyrophosphate can be used to enhance binding in the pABA pocket, such as the binding and inhibition by sulfonamide compounds [99].
Figure 5.
Dihydropteroate synthase (DHPS) joins 6-hydroxymethyl-7,8-dihydropterin pyrophosphate with para-aminobenzoic acid to produce 7,8-dihydropteroate. Coordinates for DHPS were taken from PDB ID 3TYB, complexed with the substrates 6-hydroxymehtyl-7,8-dihydropterin (magenta) and para-aminobenzoic acid acid (cyan) [101]. Structures with inhibitors are PDB ID 3TZF with sulfamethoxazole (blue) and 3TYE with a dihydropterin-sulfathiazole adduct (magenta) [101]. The pterin binding pocket is very polar (green surface); conserved interactions with the aminopterin moiety are indicated.
Figure 5.
Dihydropteroate synthase (DHPS) joins 6-hydroxymethyl-7,8-dihydropterin pyrophosphate with para-aminobenzoic acid to produce 7,8-dihydropteroate. Coordinates for DHPS were taken from PDB ID 3TYB, complexed with the substrates 6-hydroxymehtyl-7,8-dihydropterin (magenta) and para-aminobenzoic acid acid (cyan) [101]. Structures with inhibitors are PDB ID 3TZF with sulfamethoxazole (blue) and 3TYE with a dihydropterin-sulfathiazole adduct (magenta) [101]. The pterin binding pocket is very polar (green surface); conserved interactions with the aminopterin moiety are indicated.
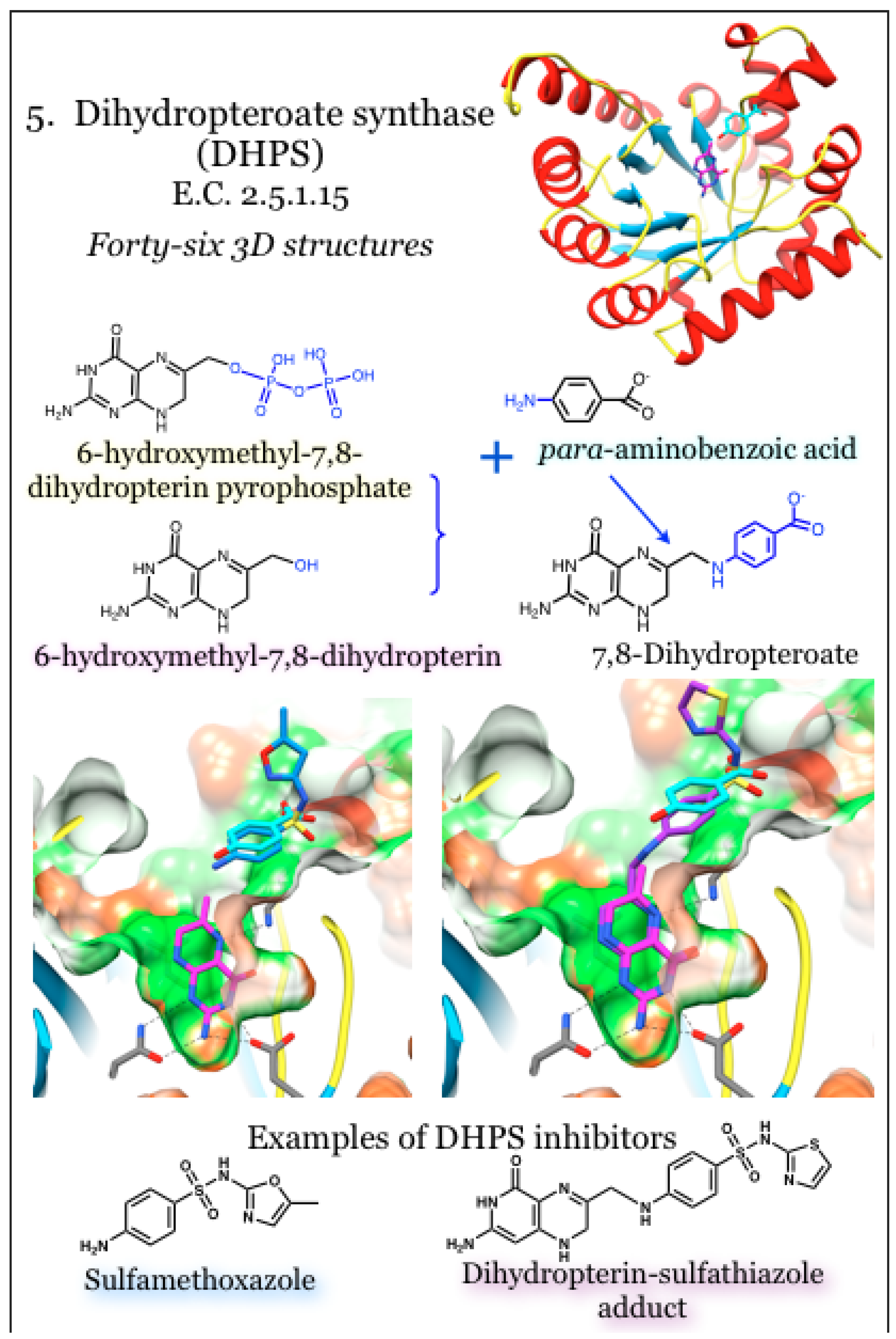
In contrast to the sulfonamide-targeted pABA site, which is not preformed and requires ordering of mobile loops, the pterin site is buried and directed down within the core beta-barrel of the protein [98]. The sequence of the pterin-binding pocket is highly conserved among bacterial DHPS enzymes, as are the pterin-sites within enzymes involved in one-carbon transfers that use methyltetrahydrofolate as a co-factor [96,99,107]. Non-phosphorylated pterins, such as 6-(methylamino)-5-nitroisocytosine, are competitive inhibitors of DHPS in vitro [42]. Additionally, the basic pterin structure can provide weak inhibition that is improved by extension away from the pterin group and towards the surface of the beta barrel, which is into the pABA pocket [96,107]. Not surprisingly, a guanine-like compound was also found to be an inhibitor of DHPS by mimicking the pterin substrate [96]. Interactions with pterin groups, or analogs thereof, utilize hydrogen bonds formed with Asn, Lys and Asp residues, as well as polar interactions bridged by water molecules. Hydrophobic stacking interactions satisfy the heterocycle face with the guanidinium group of an Arg on one side and a cluster of small hydrophobic and aromatic residues on the other face [98,100,101,107]. A conserved Lys residue undergoes a conformational change to form hydrogen bonds with the pterin and, in so doing, comprises the base of the pABA site [96].
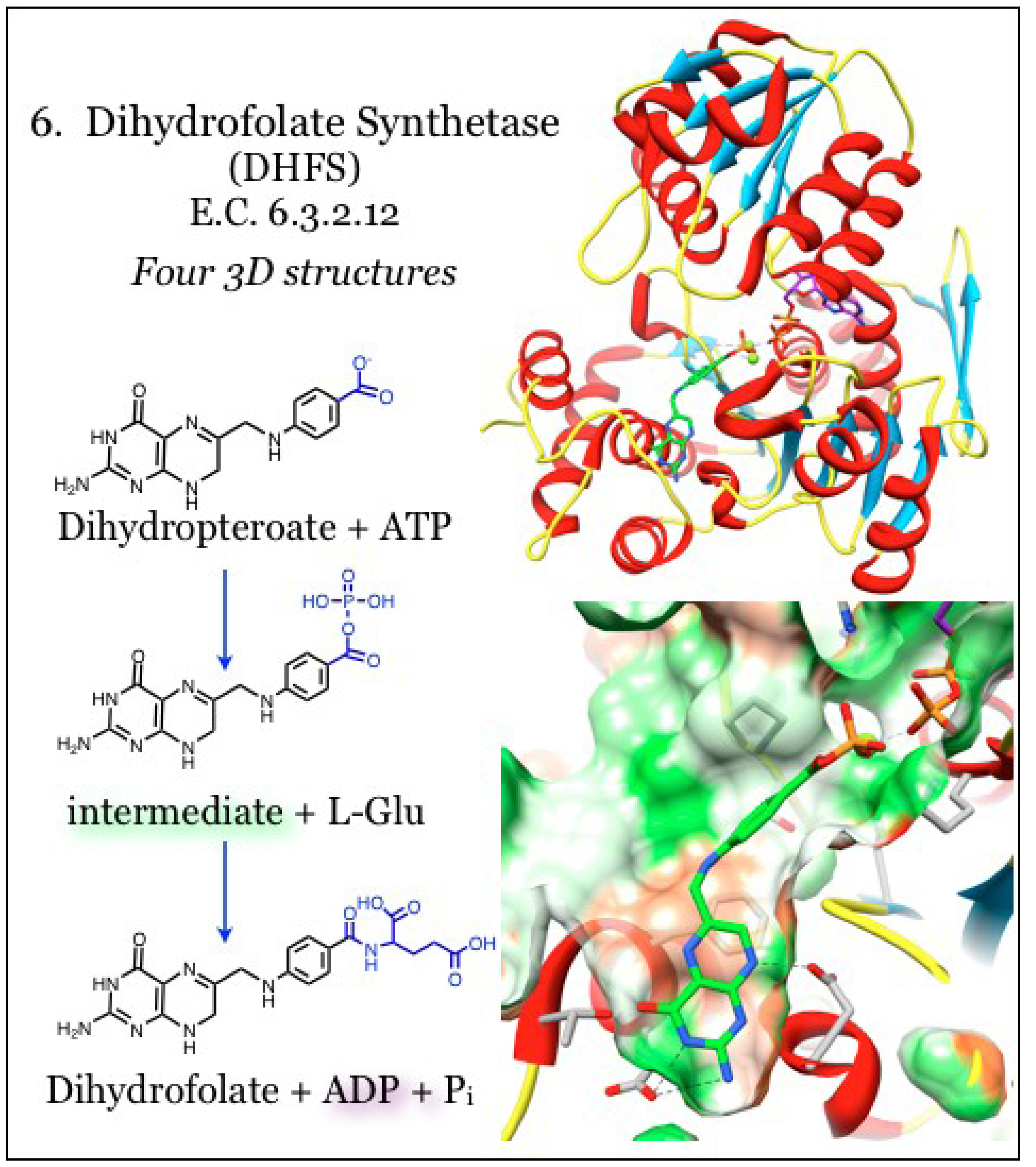
2.6. Enzymes for the Biosynthesis of Folate: 6. Dihydrofolate Synthetase (DHFS)
The substrate for dihydrofolate reductase (DHFR) enzyme is generated by the activity of dihydrofolate synthetase (DHFS, E.C. 6.3.2.12, Figure 6), which appends a single L-glutamate tail to the product of the DHPS enzyme, dihydropteroate, in an ATP-dependent reaction [51,108]. Further polyglutamylation can occur after reduction of the substrate by DHFR to tetrahydrofolate, and this polyglutamation activity is carried out by a more ubiquitous folylpolyglutamate synthetase activity (FPGS, E.C. 6.3.2.17) [109]. The appendage of negatively charged glutamate structures to the folate component prevents diffusion of the folate compound through cell membranes. This polar charge is also an important determinant for binding with enzymes downstream of DHFR, i.e., the one-carbon transfer enzymes, making polyglutamation strategies particularly useful in anti-cancer therapeutics [53]. Despite the almost identical enzymatic activities of monoglutamation by DHFS and polyglutamation by FPGS, DHFS activity is found only in the folate biosynthesis pathway upstream of DHFR while the FPGS activity is only found downstream; however, in many bacteria both activities are encoded in the same folC gene [53,108,110,111,112].
The fold of either DHFS or FPGS conforms to the Mur synthetase superfamily and contains an ATPase domain and a Rossmann-fold domain, with the pterin binding site located between the two [113,114]. The ATP-binding site is conserved in a narrow channel between these domains and the binding is stabilized by interaction with water molecules [108]. The phosphate groups are coordinated with conserved residues and with two Mg2+ ions that neutralize the negative charges. In many structures, the ATP is already partially hydrolyzed even in the absence of the substrate. The pterin site is poorly ordered in the absence of substrate, but a mobile loop becomes ordered when the site is occupied [108]. The heterocycle of the pterin is secluded within a cavity and stacks between hydrophobic residues including Phe, Ala, Ile, and Leu. When the DHFS and FPGS activity is shared within a single protein, there is a separate inhibitory profile for each of the two enzymatic specificities [53,108,115]. The FPGS substrate contains additional functional groups on the pterin that require a larger binding pocket than what is found in DHFS enzymes. However, in bacterial species that share dual functions within the same enzyme, the larger pterin substrate is accommodated at the same site by an induced fit mechanism [113]. The distinction between substrates also arises from a specific hydrogen bond between the pterin heterocycle, which is functionalized in the FPFG substrate, with a conserved Asp residue in the DHFS site. The DHFS enzymes have a highly conserved loop obstructing the functionalized pterin, preserving the specificity for monoglutamation [108]. The benzoyl portion of the dihydropteroate substrate is positioned at the periphery of the pterin site, which is distinctive but shared between DHFS and FPFG [108].
Figure 6.
Dihydrofolate synthetase (DHFS) carries out glutamation of folate substrates. Coordinates for DHFS were taken from PDB ID 1W78, which is in complex with a phosphorylated intermediate of dihydropteroate (green) and ADP (purple) [108]. The polarity of the pocket coincides with the nitrogen atoms of the aminopterin (green surface), while the heterocycle face is paired with hydrophobic portions of the pocket (orange surface). Conserved interactions with the aminopterin moiety are indicated.
Figure 6.
Dihydrofolate synthetase (DHFS) carries out glutamation of folate substrates. Coordinates for DHFS were taken from PDB ID 1W78, which is in complex with a phosphorylated intermediate of dihydropteroate (green) and ADP (purple) [108]. The polarity of the pocket coincides with the nitrogen atoms of the aminopterin (green surface), while the heterocycle face is paired with hydrophobic portions of the pocket (orange surface). Conserved interactions with the aminopterin moiety are indicated.
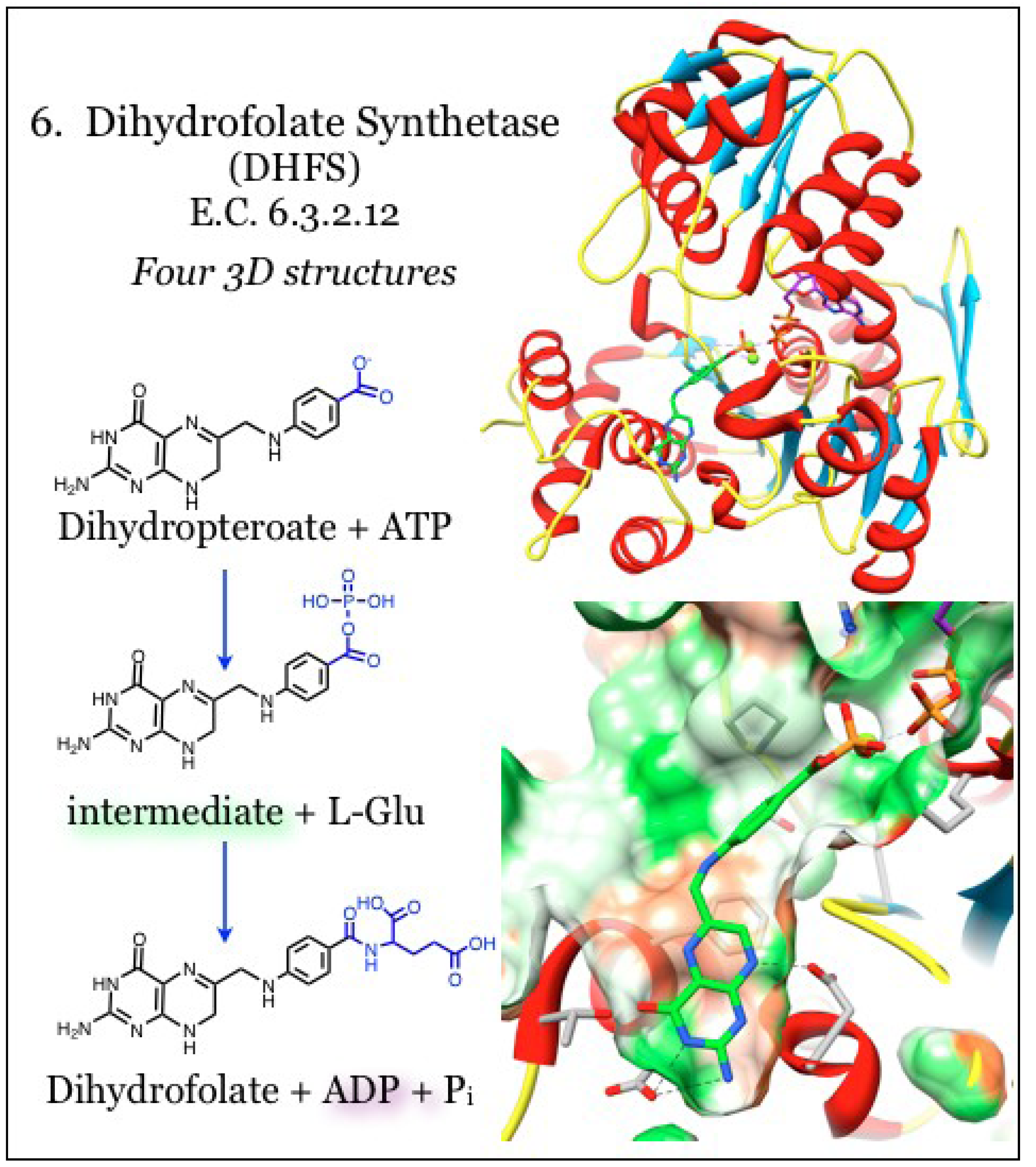
The reaction generates a tetrahedral intermediate by first transferring a phosphate moiety from ATP onto dihydropteroate, and that is subsequently reacted with L-glutamate. This intermediate is resolved, generating glutamylated folate and freeing an inorganic phosphate unit [108]. It was demonstrated that methotrexate, the universal DHFR inhibitor, does not inhibit DHFS [116]. A feedback mechanism has been identified, whereby buildup of the DHFR product (DHFR substrate) dihydrofolate causes inhibition of DHFS, such as in response to DHFR inhibitors [117]. Novel folate analogs that mimic the tetrahedral intermediate with a phosphinate linkage were identified [118] and the structural basis for inhibition has been validated [116].
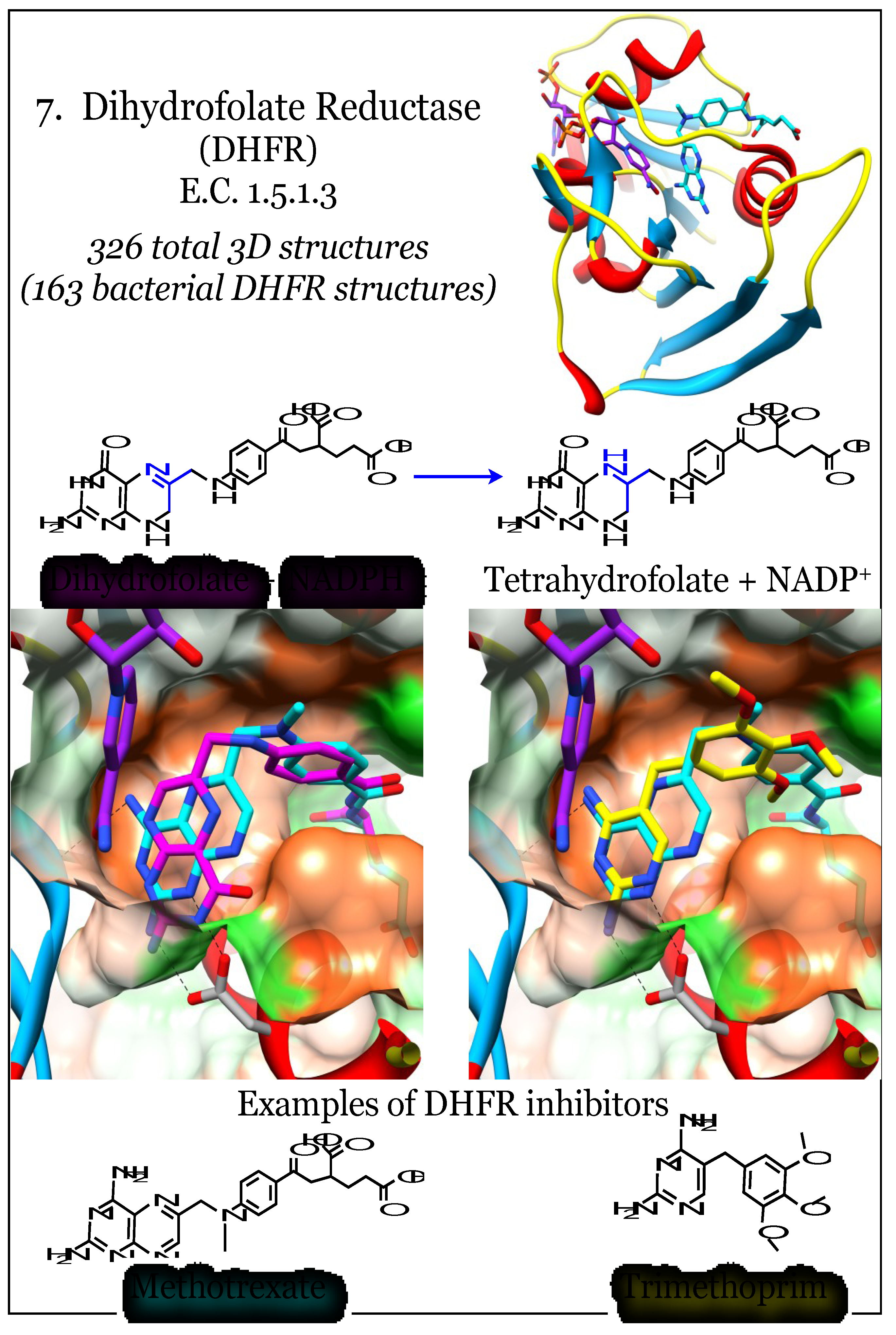
2.7. Enzymes for the Biosynthesis of Folate: 7. Dihydrofolate Reductase (DHFR)
Dihydrofolate reductase (DHFR, E.C. 1.5.1.3), the most targeted member in folate metabolism, uses dihydrofolate as a substrate and reduces it to tetrahydrofolate in an NADPH-dependent reaction. Its effectiveness as a target mediating anti-proliferation effects arises from its absolute requirement for cellular metabolism. It produces tetrahydrofolate, the co-factor and donor for one-carbon transfer reactions (Figure 7). The NADP(H) co-factor wraps around the outside of the molecule, while the dihydrofolate substrate and catalytically involved nicotinamide are held adjacent in a less accessible pocket. Early work with the DHFR from E. coli characterized the catalytic mechanism of the enzyme [119,120]. This work highlighted a flexible loop centered on residue Met20 (E. coli numbering) that is responsive to co-factor binding and catalysis, and is strongly implicated in product release. Bacterial DHFR enzymes can be effectively targeted while sparing the eukaryotic DHFR due to these sequence changes [47,121]. Recent work examined the evolution of DHFR sequences and identified three crucial areas of human DHFR sequence variation and delineated their impacted on the catalytic mechanism [122]. This study revealed mutations arose to remove the dependency of the Met20 loop’s movement, thus limiting the flexibility of eukaryotic DHFR. Compensatory mutations in the eukaryotic DHFR binding site proximal to the glutamyl-tail binding site, particularly the “PEKN” loop insertion, restore the catalytic efficiency.
Inhibitors of DHFR are folate substrate mimetics, and their properties have been extensively reviewed; there are currently eight anti-folates in clinical use [34]. They can be divided into “classical” inhibitors and “non-classical” inhibitors based upon the presence of the poly-glutamate moiety. Glutamation is present in the “classical” type and is typical in anti-cancer agents as it allows active uptake of compounds into cells. Eukaryotic DHFR is a target for anti-cancer therapies such as methotrexate, which works so remarkably well that non-cancerous cells must sometimes be rescued by leucovorin (a tetrahydrofolate analog) [123,124]. Methotrexate, a substrate mimetic, inhibits all known DHFR enzymes. Its potency derives from its structural similarity to the natural substrate, dihydrofolate, with only an oxygen to nitrogen substitution at the pterin moiety (Figure 7). The pterin substituent, which is almost identical to that of folate, docks deep within the binding site and satisfies the same hydrogen bonding relationships as found with the natural substrate [28,45,50,119,125]. Methotrexate is known to interact with enzymes downstream of DHFR, but it is ineffective with the upstream enzymes needed for the biosynthesis of folate.
Antimicrobials that inhibit the DHFR enzyme belong to the “non-classical” category and rely on cell entry via diffusion [111]. However, they are readily susceptible to the action of efflux pumps in many Gram-negative bacteria [126,127,128,129]. Anti-folates that target fungal pathogens have been heavily pursued, particularly with regard to Acquired Immunodeficiency Syndrome (AIDS)-related complications [47,130,131,132,133,134]. Trimethoprim (TMP) is a very effective antimicrobial and its structure contains a 2,4-diaminopyrimidine moiety rather than a pterin heterocycle. The diaminopyrimidine ring is able to satisfy the hydrogen bonding criteria implicit in the folate substrate site. In general, the most promising inhibitors, as assessed by potency, maintain this pattern of hydrogen bonding, and these interactions are needed for effective inhibition [28,50]. TMP was introduced to the clinic in 1962 and resistance identified by 1968 [135]. Many more inhibitors have been extended from the 2,4-diaminopyrimidine base, including some that have entered clinical trials [136,137].
Figure 7.
Dihydrofolate reductase (DHFR) acts on dihydrofolate to produce tetrahydrofolate using the reducing power of NADPH. Coordinates for DHFR complexed with the dihydrofolate substrate (magenta) and the NADPH co-factor (purple) were taken from PDB ID 1RF7 [120]. Coordinates for the complex with methotrexate (cyan) are from 3DAU, and those with trimethoprim (yellow) are from 3FRE [45,138]. The pterin binding pocket is fairly hydrophobic (orange surface) with limited polar areas (green surface). Conserved interactions of the aminopterin moiety, and mimics thereof, with a conserved acidic residue are indicated.
Figure 7.
Dihydrofolate reductase (DHFR) acts on dihydrofolate to produce tetrahydrofolate using the reducing power of NADPH. Coordinates for DHFR complexed with the dihydrofolate substrate (magenta) and the NADPH co-factor (purple) were taken from PDB ID 1RF7 [120]. Coordinates for the complex with methotrexate (cyan) are from 3DAU, and those with trimethoprim (yellow) are from 3FRE [45,138]. The pterin binding pocket is fairly hydrophobic (orange surface) with limited polar areas (green surface). Conserved interactions of the aminopterin moiety, and mimics thereof, with a conserved acidic residue are indicated.

The DHFR enzymes from a variety of bacteria have been analyzed and as of 27 October 2013, more than 160 X-ray crystal structures have been deposited with the Protein Databank (see Supplemental Table S1). These have revealed a highly conserved deeper pocket with optimum hydrogen bond donors and acceptors to accommodate a pterin heterocycle or dihydropyrimidine ring, although the orientation of the di-amino moieties can be flipped within the pocket [50,139]. Mutations around this deep pocket have been demonstrated to infer resistance to trimethoprim, such as the Phe to Tyr mutation at position 98 of the S. aureus DHFR [140,141,142,143,144,145,146,147]. Isolates of many bacteria encode for anti-folate resistant DHFR enzymes, and these mutations cluster around and just proximal to the pterin-docking site. Further exploration of the remaining large volume in the substrate site of bacterial DHFR enzymes has revealed species-specific variations that should be useful to develop narrow-spectrum targeting strategies [131,146,148,149,150,151]. In addition, those studies highlighted the generally more hydrophobic character of the DHFR site relative to the other (upstream) members of the biosynthetic folate pathway.
3. Conclusions
The similarity of the natural ligands can directly inform on the structure for a similar, possibly minimalistic, inhibitor structure that can target multiple points in the pathway with the same single compound. The idea of successfully inhibiting a series of highly related enzymes in a species-specific manner seems difficult. Such a compound must maintain the specific hydrogen-bonding pattern observed with the pterin-like moiety of the folate substrates (Figure 8), and in addition should be presented in the context of a relatively planar and hydrophobic unit. Among the seven enzymes utilized for biosynthesis of folate, the first four (GCYH I, DHNTPase, DHNA, and HPPK) contain binding sites that are more polar or proximal to the protein’s surface. The latter three enzymes (DHPS, DHFS, DHFR) have deeper and more hydrophobic pockets. While these properties may distinguish the inhibitory profiles into two subsets, it may be possible to accommodate each of these using carbohydrates such as ribose sugars (Figure 8). These maintain a high polarity but are constrained within a volume or shape that would complement even the deeper binding sites. The spacing between such a moiety relatively to the hydrogen-bonded end is a variable that can only be properly addressed through experimentation. The high similarity of pterin-binding pockets within the biosynthetic folate pathway, particularly upstream of the DHFR enzyme, shows great promise for establishing proof-of-principle of this concept.
Interestingly, although anti-folates are synthetic, there is a large pool of bacteria with natural resistance to these therapies [147,152,153]. It has been proposed that antibiotic resistance arises due to selective pressure; however, recovery of resistant populations from “untouched” ecological sites either refutes this or indicates unknown pressures from folate-like compounds. If such folate-like compounds exist they would provide a new template for design of therapeutics.
Figure 8.
Overlap of enzyme product sub-structures provides a template pharmacophore for multi-pronged compounds. The structure of the pterin-like heterocycle is conserved in the ligands for each of the seven enzymes, and each enzyme makes similar hydrogen bonding interactions. A potential inhibitor that targets more than one of these seven enzymes should include a relatively planar and hydrophobic framework with hydrogen-bonding potential at the periphery. The ligand sub-structures from the first four enzymes maintain a relatively polar character, while the latter three ligand sub-structures are hydrophobic at the core and polar at the distal end. This can possibly be accommodated in an inhibitory compound using carbohydrate units, and its spacing from the first component should be varied by 1–3 bond lengths of neutral character.
Figure 8.
Overlap of enzyme product sub-structures provides a template pharmacophore for multi-pronged compounds. The structure of the pterin-like heterocycle is conserved in the ligands for each of the seven enzymes, and each enzyme makes similar hydrogen bonding interactions. A potential inhibitor that targets more than one of these seven enzymes should include a relatively planar and hydrophobic framework with hydrogen-bonding potential at the periphery. The ligand sub-structures from the first four enzymes maintain a relatively polar character, while the latter three ligand sub-structures are hydrophobic at the core and polar at the distal end. This can possibly be accommodated in an inhibitory compound using carbohydrate units, and its spacing from the first component should be varied by 1–3 bond lengths of neutral character.

Acknowledgments
The author wishes to thank colleagues in the Center for Veterinary Health Sciences for suggestions and support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Coates, A.R.; Hu, Y. Novel approaches to developing new antibiotics for bacterial infections. Br. J. Pharmacol. 2007, 152, 1147–1154. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Mills, S.D. When will the genomics investment pay off for antibacterial discovery? Biochem. Pharmacol. 2006, 71, 1096–1102. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef]
- Torres, C. Up against the wall. Nat. Med. 2010, 16, 628–631. [Google Scholar] [CrossRef]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef]
- Munos, B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 2009, 8, 959–968. [Google Scholar] [CrossRef]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef]
- Peters, J.U.; Schnider, P.; Mattei, P.; Kansy, M. Pharmacological promiscuity: Dependence on compound properties and target specificity in a set of recent Roche compounds. ChemMedChem 2009, 4, 680–686. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Mason, J.S.; Overington, J.P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006, 16, 127–136. [Google Scholar] [CrossRef]
- Hamad, B. The antibiotics market. Nat. Rev. Drug Discov. 2010, 9, 675–676. [Google Scholar] [CrossRef]
- Pathania, R.; Brown, E.D. Small and lethal: Searching for new antibacterial compounds with novel modes of action. Biochem. Cell. Biol. 2008, 86, 111–115. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, J.; Estiu, G.; Isin, B.; Ahn, Y.Y.; Lee, D.S.; Barabasi, A.L.; Kapatral, V.; Wiest, O.; Oltvai, Z.N. Blueprint for antimicrobial hit discovery targeting metabolic networks. Proc. Natl. Acad. Sci. USA 2010, 107, 1082–1087. [Google Scholar] [CrossRef]
- Hu, Y.; Wassermann, A.M.; Lounkine, E.; Bajorath, J. Systematic analysis of public domain compound potency data identifies selective molecular scaffolds across druggable target families. J. Med. Chem. 2010, 53, 752–758. [Google Scholar] [CrossRef]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef]
- Swamidass, S.J. Mining small-molecule screens to repurpose drugs. Brief. Bioinform. 2011, 12, 327–335. [Google Scholar] [CrossRef]
- Yim, G.; Wang, H.H.; Davies, J. Antibiotics as signalling molecules. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1195–1200. [Google Scholar] [CrossRef]
- Dantas, G.; Sommer, M.O.; Oluwasegun, R.D.; Church, G.M. Bacteria subsisting on antibiotics. Science 2008, 320, 100–103. [Google Scholar] [CrossRef]
- Wei, J.R.; Krishnamoorthy, V.; Murphy, K.; Kim, J.H.; Schnappinger, D.; Alber, T.; Sassetti, C.M.; Rhee, K.Y.; Rubin, E.J. Depletion of antibiotic targets has widely varying effects on growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4176–4181. [Google Scholar] [CrossRef]
- Adams, J.C.; Keiser, M.J.; Basuino, L.; Chambers, H.F.; Lee, D.S.; Wiest, O.G.; Babbitt, P.C. A mapping of drug space from the viewpoint of small molecule metabolism. PLoS Comput. Biol. 2009, 5, e1000474. [Google Scholar] [CrossRef]
- Kinnings, S.L.; Xie, L.; Fung, K.H.; Jackson, R.M.; Bourne, P.E. The Mycobacterium tuberculosis drugome and its polypharmacological implications. PLoS Comput. Biol. 2010, 6, e1000976. [Google Scholar] [CrossRef]
- Maggiora, G.M. Is there a future for computational chemistry in drug research? J. Comput. Aided Mol. Des. 2011. [Google Scholar] [CrossRef]
- Phelan, V.V.; Liu, W.T.; Pogliano, K.; Dorrestein, P.C. Microbial metabolic exchange—The chemotype-to-phenotype link. Nat. Chem. Biol. 2012, 8, 26–35. [Google Scholar]
- Keiser, M.J.; Irwin, J.J.; Shoichet, B.K. The chemical basis of pharmacology. Biochemistry 2010, 49, 10267–10276. [Google Scholar] [CrossRef]
- Bermingham, A.; Derrick, J.P. The folic acid biosynthesis pathway in bacteria: Evaluation of potential for antibacterial drug discovery. Bioessays 2002, 24, 637–648. [Google Scholar] [CrossRef]
- Cody, V.; Schwalbe, C.H. Structural characteristics of antifolate dihydrofolate reductase enzyme interactions. Crystallogr. Rev. 2006, 12, 301–333. [Google Scholar] [CrossRef]
- Hyde, J.E. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr. Drug Targets 2007, 8, 31–47. [Google Scholar] [CrossRef]
- Jabrin, S.; Ravanel, S.; Gambonnet, B.; Douce, R.; Rebeille, F. One-carbon metabolism in plants. Regulation of tetrahydrofolate synthesis during germination and seedling development. Plant. Physiol. 2003, 131, 1431–1439. [Google Scholar] [CrossRef]
- Nzila, A. Inhibitors of de novo folate enzymes in Plasmodium falciparum. Drug Discov. Today 2006, 11, 939–944. [Google Scholar] [CrossRef]
- Van Hoek, A.H.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J. Acquired antibiotic resistance genes: An overview. Front. Microbiol. 2011, 2, e203. [Google Scholar]
- Volpato, J.P.; Pelletier, J.N. Mutational “hot-spots’” in mammalian, bacterial and protozoal dihydrofolate reductases associated with antifolate resistance: Sequence and structural comparison. Drug Resist. Updat. 2009, 12, 28–41. [Google Scholar] [CrossRef]
- Wright, D.L.; Anderson, A.C. Antifolate agents: A patent review (2006–2010). Expert Opin. Ther. Pat. 2011, 21, 1293–1308. [Google Scholar] [CrossRef]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef]
- Brown, J.B.; Okuno, Y. Systems biology and systems chemistry: New directions for drug discovery. Chem. Biol. 2012, 19, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Paolini, G.V.; Shapland, R.H.; van Hoorn, W.P.; Mason, J.S.; Hopkins, A.L. Global mapping of pharmacological space. Nat. Biotechnol. 2006, 24, 805–815. [Google Scholar] [CrossRef]
- Schreiber, S.L. Small molecules: The missing link in the central dogma. Nat. Chem. Biol. 2005, 1, 64–66. [Google Scholar] [CrossRef]
- Li, C.; Xu, L.; Wolan, D.W.; Wilson, I.A.; Olson, A.J. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J. Med. Chem. 2004, 47, 6681–6690. [Google Scholar] [CrossRef]
- Baggott, J.E.; Morgan, S.L.; Ha, T.; Vaughn, W.H.; Hine, R.J. Inhibition of folate-dependent enzymes by non-steroidal anti-inflammatory drugs. Biochem. J. 1992, 282, 197–202. [Google Scholar]
- Cheong, C.G.; Wolan, D.W.; Greasley, S.E.; Horton, P.A.; Beardsley, G.P.; Wilson, I.A. Crystal structures of human bifunctional enzyme aminoimidazole-4-carboxamide ribonucleotide transformylase/IMP cyclohydrolase in complex with potent sulfonyl-containing antifolates. J. Biol. Chem. 2004, 279, 18034–18045. [Google Scholar]
- Babaoglu, K.; Qi, J.; Lee, R.E.; White, S.W. Crystal structure of 7,8-dihydropteroate synthase from Bacillus anthracis; Mechanism and novel inhibitor design. Structure 2004, 12, 1705–1717. [Google Scholar] [CrossRef]
- Shi, G.; Blaszczyk, J.; Ji, X.; Yan, H. Bisubstrate analogue inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: Synthesis and biochemical and crystallographic studies. J. Med. Chem. 2001, 44, 1364–1371. [Google Scholar] [CrossRef]
- Shi, G.; Shaw, G.; Li, Y.; Wu, Y.; Yan, H.; Ji, X. Bisubstrate analog inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: New lead exhibits a distinct binding mode. Bioorg. Med. Chem. 2012, 20, 4303–4309. [Google Scholar] [CrossRef]
- Bennett, B.C.; Wan, Q.; Ahmad, M.F.; Langan, P.; Dealwis, C.G. X-ray structure of the ternary MTX-NADPH complex of the anthrax dihydrofolate reductase: A pharmacophore for dual-site inhibitor design. J. Struct. Biol. 2009, 166, 162–171. [Google Scholar] [CrossRef]
- Pemble, C.W.T.; Mehta, P.K.; Mehra, S.; Li, Z.; Nourse, A.; Lee, R.E.; White, S.W. Crystal structure of the 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase*dihydropteroate synthase bifunctional enzyme from Francisella tularensis. PLoS One 2010, 5, e14165. [Google Scholar]
- Chan, D.C.; Anderson, A.C. Towards species-specific antifolates. Curr. Med. Chem. 2006, 13, 377–398. [Google Scholar] [CrossRef]
- De Crecy-Lagard, V.; El Yacoubi, B.; de la Garza, R.D.; Noiriel, A.; Hanson, A.D. Comparative genomics of bacterial and plant folate synthesis and salvage: Predictions and validations. BMC Genomics 2007, 8, e245. [Google Scholar] [CrossRef]
- Levin, I.; Giladi, M.; Altman-Price, N.; Ortenberg, R.; Mevarech, M. An alternative pathway for reduced folate biosynthesis in bacteria and halophilic archaea. Mol. Microbiol. 2004, 54, 1307–1318. [Google Scholar] [CrossRef]
- Schwalbe, C.H.; Cody, V. Structural characteristics of small-molecule antifolate compounds. Crystallogr. Rev. 2006, 12, 267–300. [Google Scholar] [CrossRef]
- Swarbrick, J.; Iliades, P.; Simpson, J.S.; Macreadie, I. Folate biosynthesis-Reappraisal of old and novel targets in the search for new antimicrobials. Open Enzyme Inhib. J. 2008, 1, 12–33. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Young, P.G.; Smith, C.A.; Metcalf, P.; Baker, E.N. Structures of Mycobacterium tuberculosis folylpolyglutamate synthase complexed with ADP and AMPPCP. Acta Crystallogr. D Biol. Crystallogr. 2008, D64, 745–753. [Google Scholar]
- Haruki, H.; Pedersen, M.G.; Gorska, K.I.; Pojer, F.; Johnsson, K. Tetrahydrobiopterin biosynthesis as an off-target of sulfa drugs. Science 2013, 340, 987–991. [Google Scholar] [CrossRef]
- Ichinose, H.; Ohye, T.; Matsuda, Y.; Hori, T.; Blau, N.; Burlina, A.; Rouse, B.; Matalon, R.; Fujita, K.; Nagatsu, T. Characterization of mouse and human GTP cyclohydrolase I genes. Mutations in patients with GTP cyclohydrolase I deficiency. J. Biol. Chem. 1995, 270, 10062–10071. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakagawa, N.; Kuramitsu, S.; Yokoyama, S.; Masui, R. Novel reaction mechanism of GTP cyclohydrolase I. High-resolution X-ray crystallography of Thermus thermophilus HB8 enzyme complexed with a transition state analogue, the 8-oxoguanine derivative. J. Biochem. 2005, 138, 263–275. [Google Scholar] [CrossRef]
- Cheng, W.C.; Chen, Y.F.; Wang, H.J.; Hsu, K.C.; Lin, S.C.; Chen, T.J.; Yang, J.M.; Wang, W.C. Structures of Helicobacter pylori shikimate kinase reveal a selective inhibitor-induced-fit mechanism. PLoS One 2012, 7, e33481. [Google Scholar]
- Ducati, R.G.; Basso, L.A.; Santos, D.S. Mycobacterial shikimate pathway enzymes as targets for drug design. Curr. Drug Targets 2007, 8, 423–435. [Google Scholar] [CrossRef]
- Funke, T.; Han, H.; Healy-Fried, M.L.; Fischer, M.; Schonbrunn, E. Molecular basis for the herbicide resistance of Roundup Ready crops. Proc. Natl. Acad. Sci. USA 2006, 103, 13010–13015. [Google Scholar]
- Pitchandi, P.; Hopper, W.; Rao, R. Comprehensive database of Chorismate synthase enzyme from shikimate pathway in pathogenic bacteria. BMC Pharmacol. Toxicol. 2013, 14, e29. [Google Scholar] [CrossRef]
- Tapas, S.; Kumar, A.; Dhindwal, S.; Preeti; Kumar, P. Structural analysis of chorismate synthase from Plasmodium falciparum: A novel target for antimalaria drug discovery. Int. J. Biol. Macromol. 2011, 49, 767–777. [Google Scholar] [CrossRef]
- Christensen, K.E.; MacKenzie, R.E. Mitochondrial one-carbon metabolism is adapted to the specific needs of yeast, plants and mammals. Bioessays 2006, 28, 595–605. [Google Scholar] [CrossRef]
- Rebelo, J.; Auerbach, G.; Bader, G.; Bracher, A.; Nar, H.; Hosl, C.; Schramek, N.; Kaiser, J.; Bacher, A.; Huber, R.; et al. Biosynthesis of pteridines. Reaction mechanism of GTP cyclohydrolase I. J. Mol. Biol. 2003, 326, 503–516. [Google Scholar] [CrossRef]
- Nar, H.; Huber, R.; Meining, W.; Schmid, C.; Weinkauf, S.; Bacher, A. Atomic structure of GTP cyclohydrolase I. Structure 1995, 3, 459–466. [Google Scholar] [CrossRef]
- Maita, N.; Okada, K.; Hatakeyama, K.; Hakoshima, T. Crystal structure of the stimulatory complex of GTP cyclohydrolase I and its feedback regulatory protein GFRP. Proc. Natl. Acad. Sci. USA 2002, 99, 1212–1217. [Google Scholar] [CrossRef]
- Auerbach, G.; Herrmann, A.; Bracher, A.; Bader, G.; Gutlich, M.; Fischer, M.; Neukamm, M.; Garrido-Franco, M.; Richardson, J.; Nar, H.; et al. Zinc plays a key role in human and bacterial GTP cyclohydrolase I. Proc. Natl. Acad. Sci. USA 2000, 97, 13567–13572. [Google Scholar] [CrossRef]
- El Yacoubi, B.; Bonnett, S.; Anderson, J.N.; Swairjo, M.A.; Iwata-Reuyl, D.; de Crecy-Lagard, V. Discovery of a new prokaryotic type I GTP cyclohydrolase family. J. Biol. Chem. 2006, 281, 37586–37593. [Google Scholar] [CrossRef]
- Sankaran, B.; Bonnett, S.A.; Shah, K.; Gabriel, S.; Reddy, R.; Schimmel, P.; Rodionov, D.A.; de Crecy-Lagard, V.; Helmann, J.D.; Iwata-Reuyl, D.; et al. Zinc-independent folate biosynthesis: Genetic, biochemical, and structural investigations reveal new metal dependence for GTP cyclohydrolase IB. J. Bacteriol. 2009, 191, 6936–6949. [Google Scholar] [CrossRef]
- Maltseva, N.; Kim, Y.; Kwon, K.; Anderson, W.F.; Joachimiak, A. Crystal structure of GTP cyclohydrolase I from Yersinia pestis complexed with GTP. Available online: http://rcsb.org/pdb/explore/explore.do?structureId=4DU6/ (accessed on 26 October 2013).
- Gabelli, S.B.; Bianchet, M.A.; Xu, W.; Dunn, C.A.; Niu, Z.D.; Amzel, L.M.; Bessman, M.J. Structure and function of the E. coli dihydroneopterin triphosphate pyrophosphatase: A Nudix enzyme involved in folate biosynthesis. Structure 2007, 15, 1014–1022. [Google Scholar] [CrossRef]
- Blaszczyk, J.; Li, Y.; Gan, J.; Yan, H.; Ji, X. Structural basis for the aldolase and epimerase activities of Staphylococcus aureus dihydroneopterin aldolase. J. Mol. Biol. 2007, 368, 161–169. [Google Scholar] [CrossRef]
- Goulding, C.W.; Apostol, M.I.; Sawaya, M.R.; Phillips, M.; Parseghian, A.; Eisenberg, D. Regulation by oligomerization in a mycobacterial folate biosynthetic enzyme. J. Mol. Biol. 2005, 349, 61–72. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Yan, H. Mechanism of dihydroneopterin aldolase: Functional roles of the conserved active site glutamate and lysine residues. Biochemistry 2006, 45, 15232–15239. [Google Scholar] [CrossRef]
- Garcon, A.; Levy, C.; Derrick, J.P. Crystal structure of the bifunctional dihydroneopterin aldolase/6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase from Streptococcus pneumoniae. J. Mol. Biol. 2006, 360, 644–653. [Google Scholar] [CrossRef]
- Lopez, P.; Lacks, S.A. A bifunctional protein in the folate biosynthetic pathway of Streptococcus pneumoniae with dihydroneopterin aldolase and hydroxymethyldihydropterin pyrophosphokinase activities. J. Bacteriol. 1993, 175, 2214–2220. [Google Scholar]
- Volpe, F.; Ballantine, S.P.; Delves, C.J. Two domains with amino-acid sequence similarity are required for dihydroneopterin aldolase function in the multifunctional folic acid synthesis Fas protein of Pneumocystis carinii. Gene 1995, 160, 41–46. [Google Scholar] [CrossRef]
- Sanders, W.J.; Nienaber, V.L.; Lerner, C.G.; McCall, J.O.; Merrick, S.M.; Swanson, S.J.; Harlan, J.E.; Stoll, V.S.; Stamper, G.F.; Betz, S.F.; et al. Discovery of potent inhibitors of dihydroneopterin aldolase using CrystaLEAD high-throughput X-ray crystallographic screening and structure-directed lead optimization. J. Med. Chem. 2004, 47, 1709–1718. [Google Scholar] [CrossRef]
- Bauer, S.; Schott, A.K.; Illarionova, V.; Bacher, A.; Huber, R.; Fischer, M. Biosynthesis of tetrahydrofolate in plants: Crystal structure of 7,8-dihydroneopterin aldolase from Arabidopsis thaliana reveals a novel adolase class. J. Mol. Biol. 2004, 339, 967–979. [Google Scholar] [CrossRef]
- Pribat, A.; Jeanguenin, L.; Lara-Nunez, A.; Ziemak, M.J.; Hyde, J.E.; de Crecy-Lagard, V.; Hanson, A.D. 6-pyruvoyltetrahydropterin synthase paralogs replace the folate synthesis enzyme dihydroneopterin aldolase in diverse bacteria. J. Bacteriol. 2009, 191, 4158–4165. [Google Scholar] [CrossRef]
- Dittrich, S.; Mitchell, S.L.; Blagborough, A.M.; Wang, Q.; Wang, P.; Sims, P.F.; Hyde, J.E. An atypical orthologue of 6-pyruvoyltetrahydropterin synthase can provide the missing link in the folate biosynthesis pathway of malaria parasites. Mol. Microbiol. 2008, 67, 609–618. [Google Scholar] [CrossRef]
- Hyde, J.E.; Dittrich, S.; Wang, P.; Sims, P.F.; de Crecy-Lagard, V.; Hanson, A.D. Plasmodium falciparum: A paradigm for alternative folate biosynthesis in diverse microorganisms? Trends Parasitol. 2008, 24, 502–508. [Google Scholar] [CrossRef]
- Cicmil, N. Crystallization and preliminary X-ray crystallographic characterization of TrmFO, a folate-dependent tRNA methyltransferase from Thermotoga maritima. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 193–195. [Google Scholar]
- Derrick, J.P. The structure and mechanism of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase. Vitam. Horm. 2008, 79, 411–433. [Google Scholar]
- Shi, G.; Shaw, G.; Liang, Y.H.; Subburaman, P.; Li, Y.; Wu, Y.; Yan, H.; Ji, X. Bisubstrate analogue inhibitors of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase: New design with improved properties. Bioorg. Med. Chem. 2012, 20, 47–57. [Google Scholar] [CrossRef]
- Stammers, D.K.; Achari, A.; Somers, D.O.; Bryant, P.K.; Rosemond, J.; Scott, D.L.; Champness, J.N. A X-ray structure of the ternary complex of 7,8-dihydro-6-hydroxymethylpterinpyrophosphokinase from Escherichia coli with ATP and a substrate analogue. FEBS Lett. 1999, 456, 49–53. [Google Scholar] [CrossRef]
- Chhabra, S.; Barlow, N.; Dolezal, O.; Hattarki, M.K.; Newman, J.; Peat, T.S.; Graham, B.; Swarbrick, J.D. Exploring the chemical space around 8-mercaptoguanine as a route to new inhibitors of the folate biosynthesis enzyme HPPK. PLoS One 2013, 8, e59535. [Google Scholar]
- Chhabra, S.; Dolezal, O.; Collins, B.M.; Newman, J.; Simpson, J.S.; Macreadie, I.G.; Fernley, R.; Peat, T.S.; Swarbrick, J.D. Structure of S. aureus HPPK and the discovery of a new substrate site inhibitor. PLoS One 2012, 7, e29444. [Google Scholar]
- Garcon, A.; Bermingham, A.; Lian, L.Y.; Derrick, J.P. Kinetic and structural characterization of a product complex of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase from Escherichia coli. Biochem. J. 2004, 380, 867–873. [Google Scholar] [CrossRef]
- Xiao, B.; Shi, G.; Gao, J.; Blaszczyk, J.; Liu, Q.; Ji, X.; Yan, H. Unusual conformational changes in 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase as revealed by X-ray crystallography and NMR. J. Biol. Chem. 2001, 276, 40274–40281. [Google Scholar]
- Xiao, B.; Shi, G.; Chen, X.; Yan, H.; Ji, X. Crystal structure of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase, a potential target for the development of novel antimicrobial agents. Structure 1999, 7, 489–496. [Google Scholar] [CrossRef]
- Blaszczyk, J.; Li, Y.; Cherry, S.; Alexandratos, J.; Wu, Y.; Shaw, G.; Tropea, J.E.; Waugh, D.S.; Yan, H.; Ji, X. Structure and activity of Yersinia pestis 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase as a novel target for the development of antiplague therapeutics. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 1169–1177. [Google Scholar] [CrossRef]
- Blaszczyk, J.; Shi, G.; Li, Y.; Yan, H.; Ji, X. Reaction trajectory of pyrophosphoryl transfer catalyzed by 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase. Structure 2004, 12, 467–475. [Google Scholar] [CrossRef]
- Blaszczyk, J.; Li, Y.; Wu, Y.; Shi, G.; Ji, X.; Yan, H. Essential roles of a dynamic loop in the catalysis of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase. Biochemistry 2004, 43, 1469–1477. [Google Scholar] [CrossRef]
- Blaszczyk, J.; Shi, G.; Yan, H.; Ji, X. Catalytic center assembly of HPPK as revealed by the crystal structure of a ternary complex at 1.25 A resolution. Structure 2000, 8, 1049–1058. [Google Scholar] [CrossRef]
- Chhabra, S.; Peat, T.S.; Swarbrick, J. Synthesis and SAR study of guanine based analogues for HPPK inhibitors. Available online: http://www.rcsb.org/pdb/explore/explore.do?pdbId=4ad6/ (accessed on 27 October 2013).
- Hevener, K.E.; Yun, M.K.; Qi, J.; Kerr, I.D.; Babaoglu, K.; Hurdle, J.G.; Balakrishna, K.; White, S.W.; Lee, R.E. Structural studies of pterin-based inhibitors of dihydropteroate synthase. J. Med. Chem. 2010, 53, 166–177. [Google Scholar] [CrossRef]
- Lawrence, M.C.; Iliades, P.; Fernley, R.T.; Berglez, J.; Pilling, P.A.; Macreadie, I.G. The three-dimensional structure of the bifunctional 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase/dihydropteroate synthase of Saccharomyces cerevisiae. J. Mol. Biol. 2005, 348, 655–670. [Google Scholar] [CrossRef]
- Morgan, R.E.; Batot, G.O.; Dement, J.M.; Rao, V.A.; Eadsforth, T.C.; Hunter, W.N. Crystal structures of Burkholderia cenocepacia dihydropteroate synthase in the apo-form and complexed with the product 7,8-dihydropteroate. BMC Struct. Biol. 2011, 11, e21. [Google Scholar] [CrossRef]
- Levy, C.; Minnis, D.; Derrick, J.P. Dihydropteroate synthase from Streptococcus pneumoniae: Structure, ligand recognition and mechanism of sulfonamide resistance. Biochem. J. 2008, 412, 379–388. [Google Scholar] [CrossRef]
- Hampele, I.C.; D’Arcy, A.; Dale, G.E.; Kostrewa, D.; Nielsen, J.; Oefner, C.; Page, M.G.; Schonfeld, H.J.; Stuber, D.; Then, R.L. Structure and function of the dihydropteroate synthase from Staphylococcus aureus. J. Mol. Biol. 1997, 268, 21–30. [Google Scholar] [CrossRef]
- Yun, M.K.; Wu, Y.; Li, Z.; Zhao, Y.; Waddell, M.B.; Ferreira, A.M.; Lee, R.E.; Bashford, D.; White, S.W. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 2012, 335, 1110–1114. [Google Scholar] [CrossRef]
- Roland, S.; Ferone, R.; Harvey, R.J.; Styles, V.L.; Morrison, R.W. The characteristics and significance of sulfonamides as substrates for Escherichia coli dihydropteroate synthase. J. Biol. Chem. 1979, 254, 10337–10345. [Google Scholar]
- Lever, O.W.; Hyman, C.; Ray, P.H.; Ferone, R.; Kelsey, J.E. A galactoside derivative of a nitrosoisocytosine inhibitor of dihydropteroate synthase: Synthesis and biological evaluation. J. Heterocycl. Chem. 1986, 23, 629–631. [Google Scholar] [CrossRef]
- Valderas, M.W.; Bourne, P.C.; Barrow, W.W. Genetic basis for sulfonamide resistance in Bacillus anthracis. Microb. Drug Resist. 2007, 13, 12–21. [Google Scholar]
- Chakraborty, S.; Gruber, T.; Barry, C.E., 3rd; Boshoff, H.I.; Rhee, K.Y. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 2013, 339, 88–91. [Google Scholar] [CrossRef]
- De Beer, T.A.; Louw, A.I.; Joubert, F. Elucidation of sulfadoxine resistance with structural models of the bifunctional Plasmodium falciparum dihydropterin pyrophosphokinase-dihydropteroate synthase. Bioorg. Med. Chem. 2006, 14, 4433–4443. [Google Scholar] [CrossRef]
- Baca, A.M.; Sirawaraporn, R.; Turley, S.; Sirawaraporn, W.; Hol, W.G. Crystal structure of Mycobacterium tuberculosis 7,8-dihydropteroate synthase in complex with pterin monophosphate: New insight into the enzymatic mechanism and sulfa-drug action. J. Mol. Biol. 2000, 302, 1193–1212. [Google Scholar] [CrossRef]
- Mathieu, M.; Debousker, G.; Vincent, S.; Viviani, F.; Bamas-Jacques, N.; Mikol, V. Escherichia coli FolC structure reveals an unexpected dihydrofolate binding site providing an attractive target for anti-microbial therapy. J. Biol. Chem. 2005, 280, 18916–18922. [Google Scholar] [CrossRef]
- McDonald, D.; Atkinson, I.J.; Cossins, E.A.; Shane, B. Isolation of dihydrofolate and folylpolyglutamate synthetase activities from Neurospora. Phytochemistry 1995, 38, 327–333. [Google Scholar] [CrossRef]
- Shane, B. Pteroylpoly(gamma-glutamate) synthesis by Corynebacterium species. Studies on the mechanism of folypoly(gamma-glutamate) synthetase. J. Biol. Chem. 198, 255, 5663–5667. [Google Scholar]
- Fussenegger, M.; Meyer, T.F. Cloning and characterization of the Neisseria gonorrhoeae MS11 folC gene. Mol. Gen. Genet. 1996, 250, 277–285. [Google Scholar]
- Bognar, A.L.; Osborne, C.; Shane, B.; Singer, S.C.; Ferone, R. Folylpoly-gamma-glutamate synthetase-dihydrofolate synthetase. Cloning and high expression of the Escherichia coli folC gene and purification and properties of the gene product. J. Biol. Chem. 1985, 260, 5625–5630. [Google Scholar]
- Sun, X.; Cross, J.A.; Bognar, A.L.; Baker, E.N.; Smith, C.A. Folate-binding triggers the activation of folylpolyglutamate synthetase. J. Mol. Biol. 2001, 310, 1067–1078. [Google Scholar] [CrossRef]
- Smith, C.A.; Cross, J.A.; Bognar, A.L.; Sun, X. Mutation of Gly51 to serine in the P-loop of Lactobacillus casei folylpolyglutamate synthetase abolishes activity by altering the conformation of two adjacent loops. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 548–558. [Google Scholar]
- Sheng, Y.; Khanam, N.; Tsaksis, Y.; Shi, X.M.; Lu, Q.S.; Bognar, A.L. Mutagenesis of folylpolyglutamate synthetase indicates that dihydropteroate and tetrahydrofolate bind to the same site. Biochemistry 2008, 47, 2388–2396. [Google Scholar] [CrossRef]
- Wang, P.; Wang, Q.; Yang, Y.; Coward, J.K.; Nzila, A.; Sims, P.F.; Hyde, J.E. Characterisation of the bifunctional dihydrofolate synthase-folylpolyglutamate synthase from Plasmodium falciparum; a potential novel target for antimalarial antifolate inhibition. Mol. Biochem. Parasitol. 2010, 172, 41–51. [Google Scholar] [CrossRef]
- Kwon, Y.K.; Lu, W.; Melamud, E.; Khanam, N.; Bognar, A.; Rabinowitz, J.D. A domino effect in antifolate drug action in Escherichia coli. Nat. Chem. Biol. 2008, 4, 602–608. [Google Scholar] [CrossRef]
- Banerjee, R.V.; Shane, B.; McGuire, J.J.; Coward, J.K. Dihydrofolate synthetase and folylpolyglutamate synthetase: Direct evidence for intervention of acyl phosphate intermediates. Biochemistry 1988, 27, 9062–9070. [Google Scholar] [CrossRef]
- Bystroff, C.; Oatley, S.J.; Kraut, J. Crystal structures of Escherichia coli dihydrofolate reductase: The NADP+ holoenzyme and the folate .cntdot. NADP+ ternary complex. Substrate binding and a model for the transition state. Biochemistry 1990, 29, 3263–3277. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar]
- Cody, V.; Chan, D.; Galitsky, N.; Rak, D.; Luft, J.R.; Pangborn, W.; Queener, S.F.; Laughton, C.A.; Stevens, M.F. Structural studies on bioactive compounds. 30. Crystal structure and molecular modeling studies on the Pneumocystis carinii dihydrofolate reductase cofactor complex with TAB, a highly selective antifolate. Biochemistry 2000, 39, 3556–3564. [Google Scholar] [CrossRef]
- Liu, C.T.; Hanoian, P.; French, J.B.; Pringle, T.H.; Hammes-Schiffer, S.; Benkovic, S.J. Functional significance of evolving protein sequence in dihydrofolate reductase from bacteria to humans. Proc. Natl. Acad. Sci. USA 2013, 110, 10159–10164. [Google Scholar]
- Matherly, L.H.; Barlowe, C.K.; Phillips, V.M.; Goldman, I.D. The effects on 4-aminoantifolates on 5-formyltetrahydrofolate metabolism in L1210 cells. A biochemical basis of the selectivity of leucovorin rescue. J. Biol. Chem. 1987, 262, 710–717. [Google Scholar]
- Zervos, M.J.; Schaberg, D.R. Reversal of the in vitro susceptibility of enterococci to trimethoprim-sulfamethoxazole by folinic acid. Antimicrob. Agents Chemother. 1985, 28, 446–448. [Google Scholar] [CrossRef]
- Lewis, W.S.; Cody, V.; Galitsky, N.; Luft, J.R.; Pangborn, W.; Chunduru, S.K.; Spencer, H.T.; Appleman, J.R.; Blakley, R.L. Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. Kinetics, crystallography, and potential as selectable markers. J. Biol. Chem. 1995, 270, 5057–5064. [Google Scholar] [CrossRef]
- Buffet-Bataillon, S.; le Jeune, A.; le Gall-David, S.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Molecular mechanisms of higher MICs of antibiotics and quaternary ammonium compounds for Escherichia coli isolated from bacteraemia. J. Antimicrob. Chemother. 2012, 67, 2837–2842. [Google Scholar] [CrossRef]
- Floyd, J.L.; Smith, K.P.; Kumar, S.H.; Floyd, J.T.; Varela, M.F. LmrS is a multidrug efflux pump of the major facilitator superfamily from Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 5406–5412. [Google Scholar] [CrossRef]
- Kohler, T.; Kok, M.; Michea-Hamzehpour, M.; Plesiat, P.; Gotoh, N.; Nishino, T.; Curty, L.K.; Pechere, J.C. Multidrug efflux in intrinsic resistance to trimethoprim and sulfamethoxazole in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1996, 40, 2288–2290. [Google Scholar]
- Piddock, L.J.; Garvey, M.I.; Rahman, M.M.; Gibbons, S. Natural and synthetic compounds such as trimethoprim behave as inhibitors of efflux in Gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, 1215–1223. [Google Scholar] [CrossRef]
- Cody, V.; Galitsky, N.; Rak, D.; Luft, J.R.; Pangborn, W.; Queener, S.F. Ligand-induced conformational changes in the crystal structures of Pneumocystis carinii dihydrofolate reductase complexes with folate and NADP+. Biochemistry 1999, 38, 4303–4312. [Google Scholar] [CrossRef]
- Cody, V.; Pace, J. Structural analysis of Pneumocystis carinii and human DHFR complexes with NADPH and a series of five potent 6-[5'-(omega-carboxyalkoxy)benzyl]pyrido[2,3-d]pyrimidine derivatives. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 1–7. [Google Scholar] [CrossRef]
- Cody, V.; Pace, J.; Queener, S.F.; Adair, O.O.; Gangjee, A. Kinetic and structural analysis for potent antifolate inhibition of Pneumocystis jirovecii, Pneumocystis carinii, and human dihydrofolate reductases and their active-site variants. Antimicrob. Agents Chemother. 2013, 57, 2669–2677. [Google Scholar] [CrossRef]
- Gangjee, A.; Adair, O.; Queener, S.F. Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase inhibitors and antitumor agents: Synthesis and biological activities of 2,4-diamino-5-methyl-6-[(monosubstituted anilino) methyl]-pyrido [2,3-d] pyrimidines. J. Med. Chem. 1999, 42, 2447–2455. [Google Scholar] [CrossRef]
- Rosowsky, A.; Forsch, R.A.; Queener, S.F. Inhibition of Pneumocystis carinii, Toxoplasma gondii, and Mycobacterium avium dihydrofolate reductases by 2,4-diamino-5-[2-methoxy-5-(omega-carboxyalkyloxy)benzyl]pyrimidines: Marked improvement in potency relative to trimethoprim and species selectivity relative to piritrexim. J. Med. Chem. 2002, 45, 233–241. [Google Scholar] [CrossRef]
- Darrell, J.H.; Garrod, L.P.; Waterworth, P.M. Trimethoprim: Laboratory and clinical studies. J. Clin. Pathol. 1968, 21, 202–209. [Google Scholar] [CrossRef]
- Morgan, A.; Cofer, C.; Stevens, D.L. Iclaprim: A novel dihydrofolate reductase inhibitor for skin and soft tissue infections. Future Microbiol. 2009, 4, 131–144. [Google Scholar] [CrossRef]
- Schneider, P.; Hawser, S.; Islam, K. Iclaprim, a novel diaminopyrimidine with potent activity on trimethoprim sensitive and resistant bacteria. Bioorg. Med. Chem. Lett. 2003, 13, 4217–4221. [Google Scholar] [CrossRef]
- Oefner, C.; Bandera, M.; Haldimann, A.; Laue, H.; Schulz, H.; Mukhija, S.; Parisi, S.; Weiss, L.; Lociuro, S.; Dale, G.E. Increased hydrophobic interactions of iclaprim with Staphylococcus aureus dihydrofolate reductase are responsible for the increase in affinity and antibacterial activity. J. Antimicrob. Chemother. 2009, 63, 687–698. [Google Scholar] [CrossRef]
- Polshakov, V.I.; Birdsall, B.; Feeney, J. Characterization of rates of ring-flipping in trimethoprim in its ternary complexes with Lactobacillus casei dihydrofolate reductase and coenzyme analogues. Biochemistry 1999, 38, 15962–15969. [Google Scholar] [CrossRef]
- Maskell, J.P.; Sefton, A.M.; Hall, L.M. Multiple mutations modulate the function of dihydrofolate reductase in trimethoprim-resistant Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2001, 45, 1104–1108. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Perdikouli, M.; Beeck, A.; Verhoef, J.; Fluit, A.C. Resistance to trimethoprim-sulfamethoxazole and modifications in genes coding for dihydrofolate reductase and dihydropteroate synthase in European Streptococcus pneumoniae isolates. J. Antimicrob. Chemother. 2001, 48, 935–936. [Google Scholar] [CrossRef]
- Skold, O. Resistance to trimethoprim and sulfonamides. Vet. Res. 2001, 32, 261–273. [Google Scholar] [CrossRef]
- Barrow, E.W.; Bourne, P.C.; Barrow, W.W. Functional cloning of Bacillus anthracis DHFR and confirmation of natural resistance to trimethoprim. Antimicrob. Agents Chemother. 2004, 48, 4643–4649. [Google Scholar] [CrossRef]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006, 119, S3–S10. [Google Scholar] [CrossRef]
- Beierlein, J.M.; Karri, N.G.; Anderson, A.C. Targeted mutations of Bacillus anthracis dihydrofolate reductase condense complex structure-activity relationships. J. Med. Chem. 2010, 53, 7327–7336. [Google Scholar] [CrossRef]
- Bourne, C.R.; Barrow, E.W.; Bunce, R.A.; Bourne, P.C.; Berlin, K.D.; Barrow, W.W. Inhibition of antibiotic-resistant Staphylococcus aureus by the broad-spectrum dihydrofolate reductase inhibitor RAB1. Antimicrob. Agents Chemother. 2010, 54, 3825–3833. [Google Scholar] [CrossRef]
- Frey, K.M.; Lombardo, M.N.; Wright, D.L.; Anderson, A.C. Towards the understanding of resistance mechanisms in clinically isolated trimethoprim-resistant, methicillin-resistant Staphylococcus aureus dihydrofolate reductase. J. Struct. Biol. 2010, 170, 93–97. [Google Scholar] [CrossRef]
- Bourne, C.R.; Wakeham, N.; Nammalwar, B.; Tseitin, V.; Bourne, P.C.; Barrow, E.W.; Mylvaganam, S.; Ramnarayan, K.; Bunce, R.A.; Berlin, K.D.; et al. Structure-activity relationship for enantiomers of potent inhibitors of B. anthracis dihydrofolate reductase. Biochim. Biophys. Acta 2013, 1834, 46–52. [Google Scholar] [CrossRef]
- Bowden, K.; Harris, N.V.; Watson, C.A. Structure-activity relationships of dihydrofolate reductase inhibitors. J. Chemother. 1993, 5, 377–388. [Google Scholar]
- Frey, K.M.; Liu, J.; Lombardo, M.N.; Bolstad, D.B.; Wright, D.L.; Anderson, A.C. Crystal structures of wild-type and mutant methicillin-resistant Staphylococcus aureus dihydrofolate reductase reveal an alternate conformation of NADPH that may be linked to trimethoprim resistance. J. Mol. Biol. 2009, 387, 1298–1308. [Google Scholar] [CrossRef]
- Popov, V.M.; Chan, D.C.; Fillingham, Y.A.; Yee, W.A.; Wright, D.L.; Anderson, A.C. Analysis of compleses of inhibitors with Cryptosporidium hominis DHFR leads to a new trimethoprim derivative. Bioorg. Med. Chem. Lett. 2006, 16, 4366–4370. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Barton, H.A.; Wright, G.D. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 2012, 7, e34953. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Bourne, C.R. Utility of the Biosynthetic Folate Pathway for Targets in Antimicrobial Discovery. Antibiotics 2014, 3, 1-28. https://doi.org/10.3390/antibiotics3010001
AMA Style
Bourne CR. Utility of the Biosynthetic Folate Pathway for Targets in Antimicrobial Discovery. Antibiotics. 2014; 3(1):1-28. https://doi.org/10.3390/antibiotics3010001
Chicago/Turabian StyleBourne, Christina R. 2014. "Utility of the Biosynthetic Folate Pathway for Targets in Antimicrobial Discovery" Antibiotics 3, no. 1: 1-28. https://doi.org/10.3390/antibiotics3010001