Bioactive Polymeric Composites for Tooth Mineral Regeneration: Physicochemical and Cellular Aspects

Abstract
:1. Introduction
1.1. Hard Tissue Regenerating Materials Based on Calcium Phosphates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (acronym) | Formula | Ca/P | pKsp# | pH stability range## |
|---|---|---|---|---|
| Dicalcium phosphate dihydrate (DCPD) | CaHPO4·2H2O | 1.00 | 6.6 | 2.0–6.0 |
| Octacalcium phosphate (OCP) | Ca8(HPO4)2(PO4)4·5H2O | 1.33 | 96.6 | 5.5–7.0 |
| Amorphous calcium phosphate (ACP) | Ca3(PO4)2·nH2O* | 1.50 @ | Nd | ∼5.0–12.0** |
| α-tricalcium phosphate (α-TCP) | Ca3(PO4)2 | 1.50 | 25.5 | N/a |
| β-tricalcium phosphate (β-TCP) | Ca3(PO4)2 | 1.50 | 28.9 | N/a |
| Hydroxyapatite (HAP) | Ca10(PO4)6(OH)2 | 1.67 | 116.8 | 9.5–12.0 |
| Fluoroapatite (FAP) | Ca10(PO4)6F2 | 1.67 | 120.0 | 7.0–12.0 |
1.2. ACP-based Dental Materials
2. Experimental Section
| Physicochemical evaluation | |
|---|---|
| Atomic emission spectroscopy (AES) | Compositional analysis of ACP fillers (Ca/PO4 ratio of the solid); kinetics of Ca and PO4 ions release from composites |
| Dilatometry | Volumetric changes of composite specimens as a consequence of polymerization shrinkage (PS) |
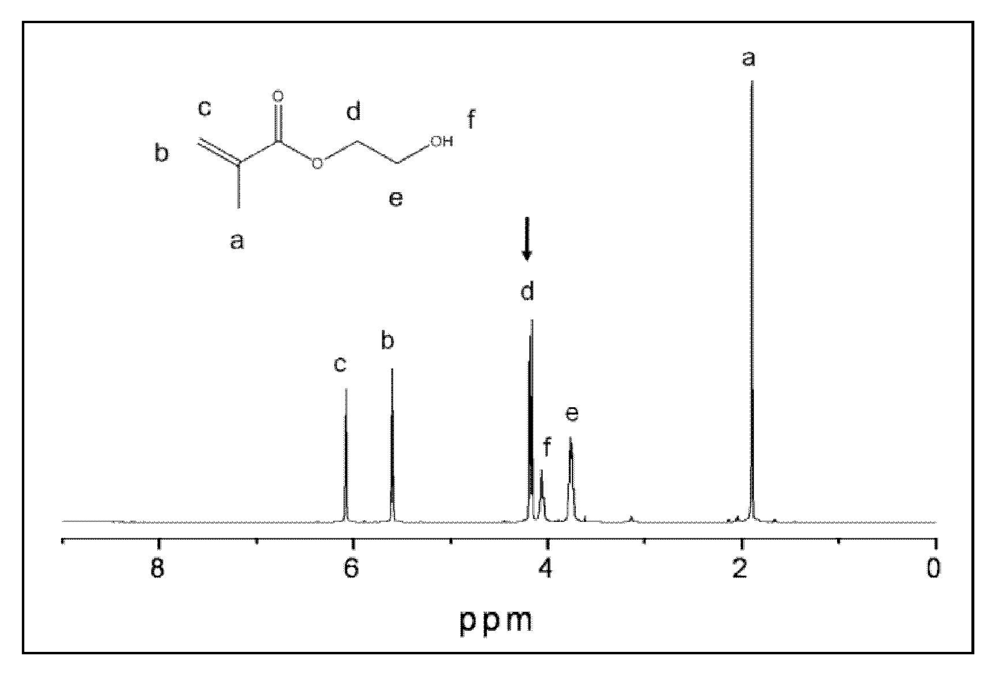
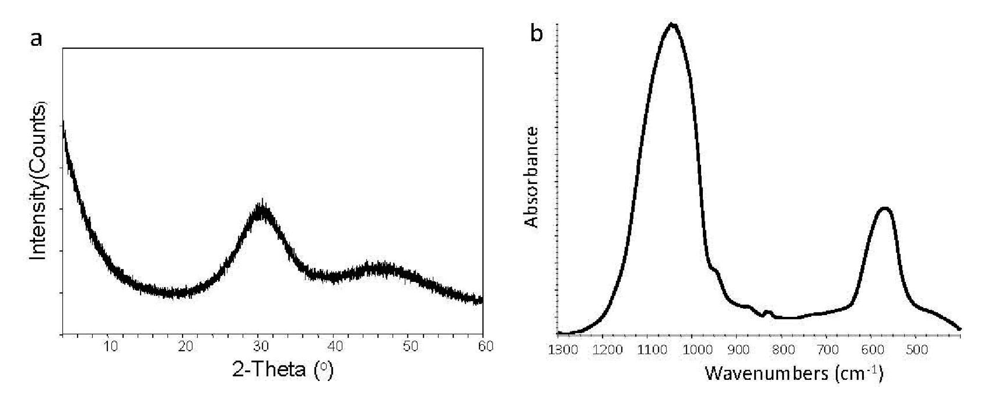
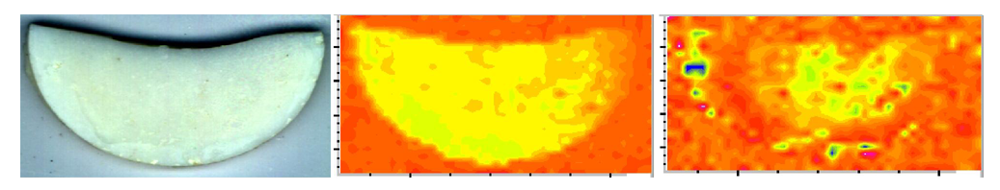
| Fourier-transform infrared (FTIR) spectroscopy and microspectroscopy (FTIR-m) | Validation of ACP structure; distribution of the resin and ACP filler on composite's surface; degree of vinyl conversion (DVC) of copolymers and composites |
| Gravimetry | The overall mass changes resulting from water sorption, filler dissolution and leachability of the unreacted species; water uptake and hygroscopic expansion (HE) of copolymers and composites upon exposure to relative humidity (RH) or aqueous immersion |
| Mechanical tests | Biaxial flexure strength (BFS), shear bond strength (SBS) of copolymer and composite specimens in dry and wet state |
| Nuclear magnetic resonance (1H NMR) spectroscopy | Identification and quantification of leachables in the extracts of copolymers and composites |
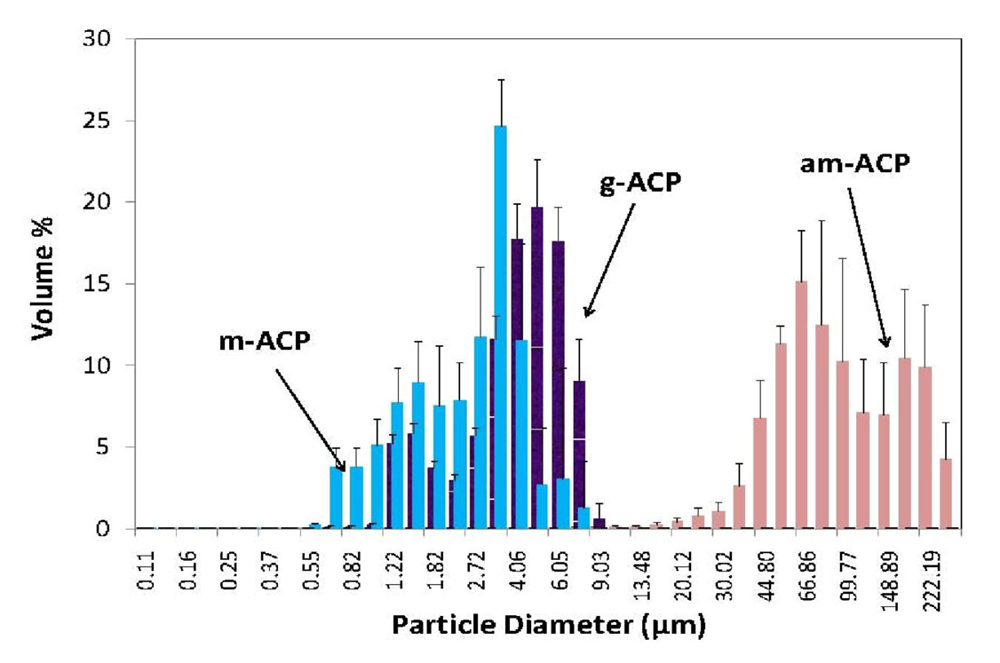
| Particle size distribution (PSD) analysis | Histograms of the volume and number PSD; size range and median diameter (dm) of ACP filler |
| Scanning electron microscopy (SEM) | Morphology and topology of ACP filler, surface characteristics of copolymers and composites before and after aqueous immersion |
| Tensometry | Determination of the stresses developing within the composites due to shrinkage upon polymerization (PSS) |
| Thermogravimetric analysis (TGA) | Temperature-dependent mass changes of the fillers and composites |
| X-ray diffraction (XRD) | Long-range (non)crystalline order of the fillers and their stability upon aqueous immersion; intra-composite ACP to HAP transformation |
| Cellular responses | |
| Colorimetry | Viability of cells exposed to the extracts from copolymer and/or composite specimens |
| Phase contrast microscopy | Effects of the copolymer and composite extracts on cell morphology |
2.1. Synthesis, Modification and Characterization of ACP Fillers
2.2. Formulation and Characterization of Experimental Resins
| Component | Acronym/Comm. name |
|---|---|
| Base monomers | |
| 2,2-Bis(p-2′-hydroxy-3′-methcryloxypropoxy)phenyl-propane | Bis-GMA |
| Ethoxylated bisphenol A dimethacrylate | EBPADMA |
| Urethane dimethacrylate | UDMA |
| Diluent monomers | |
| 2-hydroxyethyl methacrylate | HEMA |
| Hexamethylene dimethacrylate | HmDMA |
| Poly(ethylene glycol) extended urethane dimethacrylate | PEG-U |
| Triethylene glycol dimethacrylate | TEGDMA |
| Adhesive (multifunctional) monomers | |
| Methacryloyloxyethyl phthalate | MEP |
| Zirconyl dimethacrylate | ZrDMA |
| Pyromellitic glycerol dimethacrylate | PMGDMA |
| Components of polymerization initiating systems | |
| Benzoyl peroxide | BPO |
| Camphorquinone | CQ |
| Diphenyl(2,4,6-trimethylbenzoyl) phosphine oxide & 2-hydroxy-2-methyl-1-phenyl-1-propanone | 4265 Darocur |
| 2,2′-Dihydroxyethyl-p-toluidine | DHEPT |
| Ethyl-4-N,N-dimethylamino benzoate | 4EDMAB |
| Bis(2,6-dimethoxybenzoyl)-2,4,4-trimethylpentyl phosphine oxide & 1-hydroxycyclohexyl phenyl ketone | 1850 Irgacure |
| 2-benzyl-2-(dimethylamino)-1-(4-(4-morphollinyl)phenyl)-1-butanone | 369 Irgacure |
2.3. Fabrication and Physicochemical Evaluation of Experimental ACP Composites

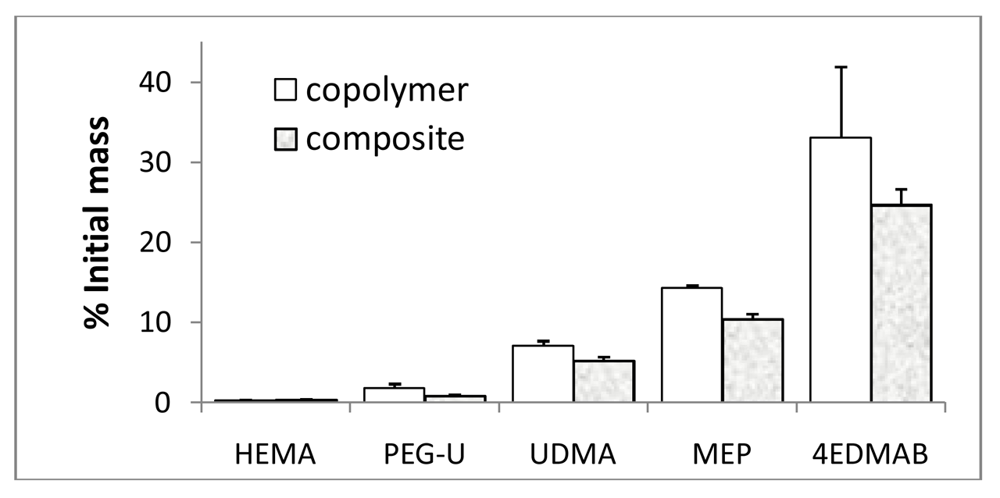
2.4. Leachability of Unreacted Monomers from Copolymers and ACP Composites
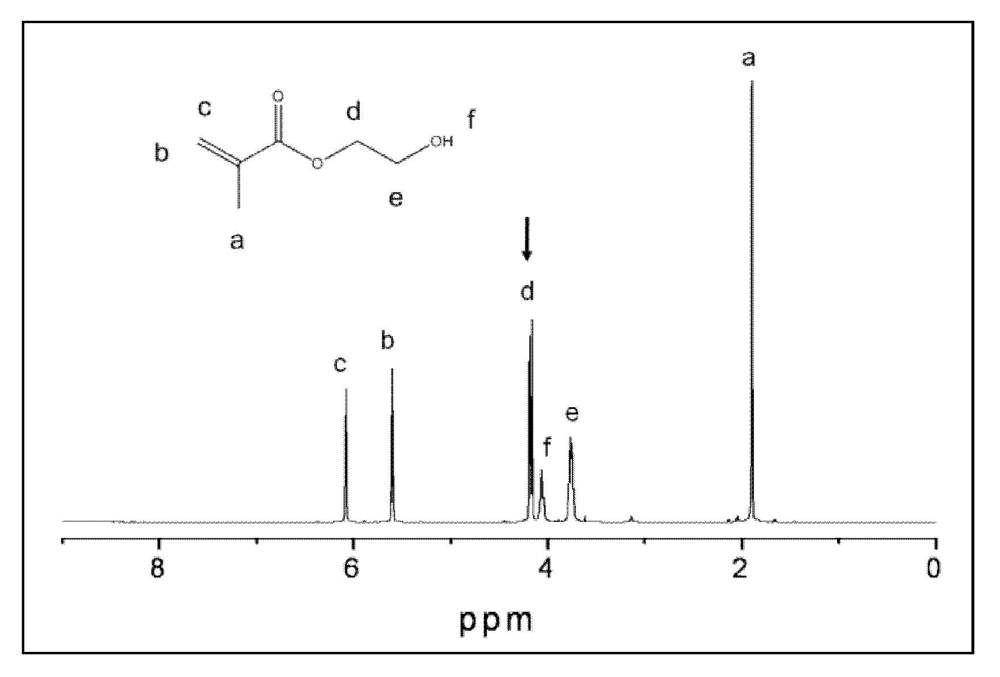
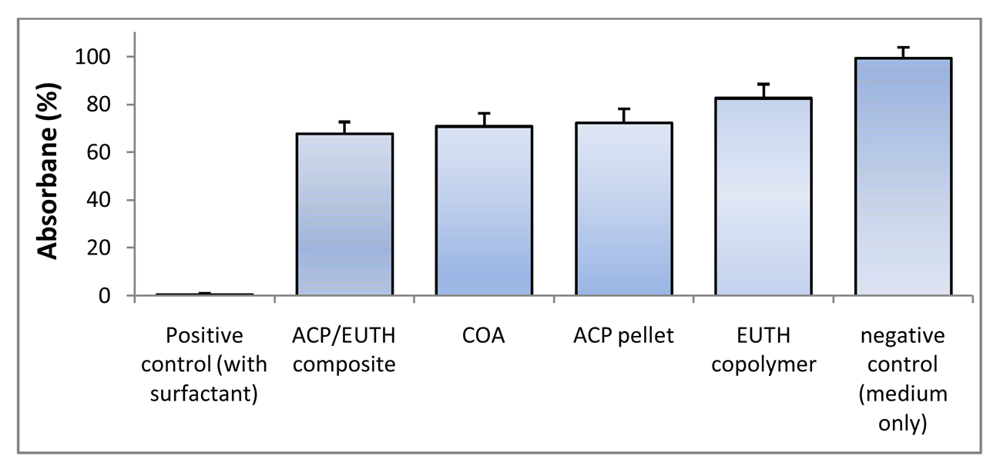
2.5. I n Vitro Cytotoxicity of Copolymers and ACP Composites
2.6. Statistical Methodology
3. Results and Discussion
3.1. Effects of Precipitating Conditions and Treatments on Properties of ACP Fillers

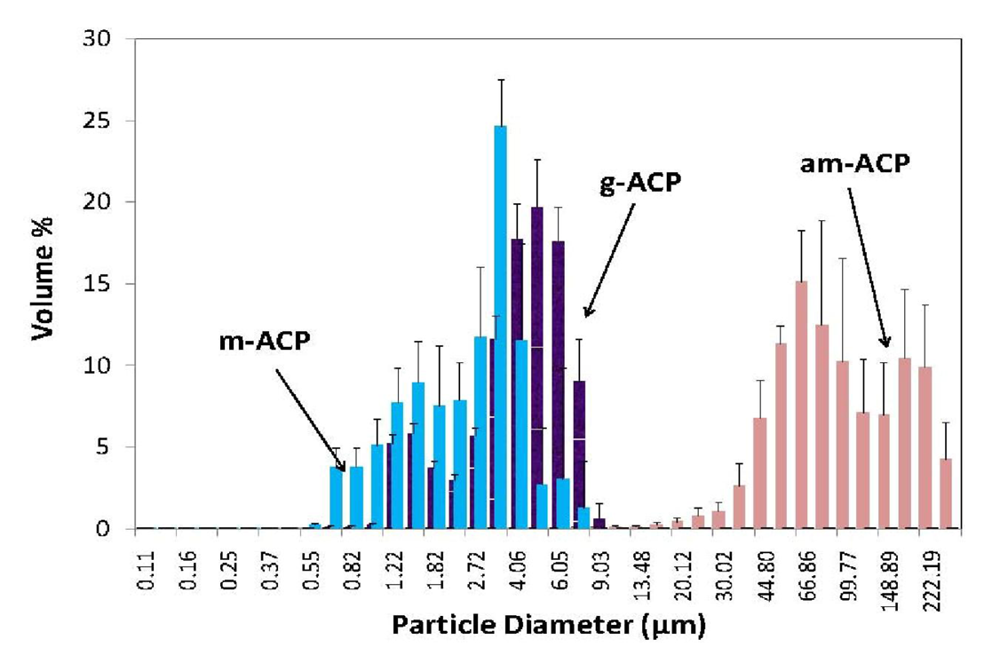
| Parameter | am-ACP | g-ACP | m-ACP |
|---|---|---|---|
| dm (μm) | 80.0 ± 4.7 | 4.5 ± 0.8 | 3.3 ± 0.5 |
| BFS (MPa) | 42.2 ± 6.7 | 50.0 ± 8.0 | 56.4 ± 7.7 |
| WSmax (mass%) | 3.1 ± 0.4 | 2.5 ± 0.5 | 1.7 ± 0.2 |
| ΔG0 (kJ/mol) | −(5.7 ± 0.2) | −(4.9 ± 0.6) | −(5.1 ± 0.3) |
3.2. Structure/Composition/Property Relationships in Experimental Resins and Their ACP Composites
| Base monomer | DVC (%) | WSmax (mass %) | PS (vol %) | BFS (MPa) | ΔG0 (kJ/mol) | ||
|---|---|---|---|---|---|---|---|
| Dry | Wet | ||||||
| Bis-GMA | copolymer | 88.6 ± 2.7 | 3.4 ± 0.3 | nd | 136 ± 35 | 131 ± 34 | n/a |
| composite | 80.8 ± 4.6 | 2.8 ± 0.2 | 6.4 ± 1.5 | 61 ± 12 | 53 ± 11 | −[5.96 ± 0.12] | |
| EBPADMA | copolymer | 90.8 ± 1.6 | 2.0 ± 0.3 | nd | 116 ± 22 | 117 ± 34 | n/a |
| composite | 81.2 ± 3.6 | 2.6 ± 0.2 | 7.4 ± 1.1 | 59 ± 9 | 53 ± 9 | −[7.23 ± 0.23] | |
| UDMA | copolymer | 87.9 ± 1.7 | 2.6 ± 0.3 | nd | 167 ± 33 | 125 ± 35 | n/a |
| composite | 86.7 ± 2.0 | 2.8 ± 0.1 | 6.7 ± 0.9 | 61 ± 9 | 46 ± 12 | −[5.35 ± 0.17] | |
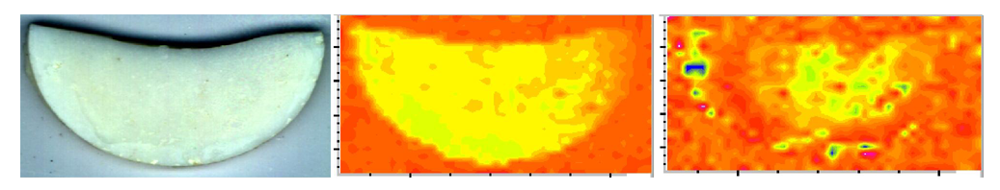
| ETHM resin | DVC (%) | WSmax (mass %) | PS (vol %) | BFS (MPa) | ΔG0 (kJ/mol) | |
|---|---|---|---|---|---|---|
| Series 1 | am-ACP | 84.8 ± 6.5 | 3.5 ± 0.5 | 6.9 ± 0.6 | 44.5 ± 8.2 | −[4.65 ± 0.31] |
| Series 2 | am-ACP | 72.2 ± 3.8 | 2.5 ± 0.3 | nd | 36.1 ± 6.7 | −[4.56 ± 0.23] |
| m-ACP | 76.7 ± 3.9 | 1.8 ± 0.2 | nd | 56.5 ± 9.4 | −[4.18 ± 0.39] | |
| Property | LC-UPHM | DC-UPHM | ||||
|---|---|---|---|---|---|---|
| copolymer | composite | copolymer | composite | |||
| am-ACP | g-ACP | am-ACP | g-ACP | |||
| DVC (%) | 95.7 ± 2.2 | 86.4 ± 1.9 | 88.4 ± 2.4 | 79.3 ± 3.4 | 76.0 ± 4.4 | 85.4 ± 3.2 |
| PS (vol%) | n/d | 7.1 ± 0.3 | 6.9 ± 0.1 | n/d | n/d | n/d |
| PSS (MPa) | n/d | 4.8 ± 0.2 | 4.1 ± 0.2 | n/d | 3.7 ± 0.3 | 3.6 ± 0.2 |
| WSmax (mass%) RH | 3.2 ± 0.3 | 3.2 ± 0.2 | 3.2 ± 0.1 | 2.5 ± 0.5 | 2.9 ± 0.1 | 2.6 ± 0.2 |
| immersion | 6.7 ± 0.6 | 8.7 ± 0.4 | 9.4 ± 0.4 | 6.2 ± 0.5 | 7.4 ± 0.5 | 8.6 ± 0.4 |
| HE (vol%) | 5.4 ± 2.4 | 12.5 ± 1.8 | 11.8 ± 1.3 | 6.7 ± 1.7 | 13.0 ± 1.1 | 13.6 ± 1.7 |
| BFS (MPa) | 137.1 ± 24.9 | 39.4 ± 3.3 | 44.4 ± 4.4 | 124.3 ± 3.4 | 49.4 ± 9.4 | 47.3 ± 8.9 |
| ΔG0 (kJ/mol) | n/a | −[7.37 ± 0.33] | −[7.14 ± 0.46] | n/a | −[7.44 ± 0.39] | −[6.96 ± 0.32] |

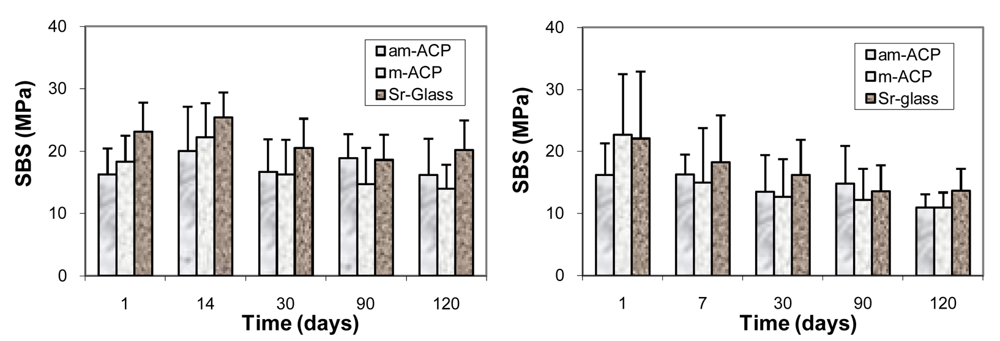
3.3. Leachability of Unreacted Monomers from Copolymers and ACP Composites
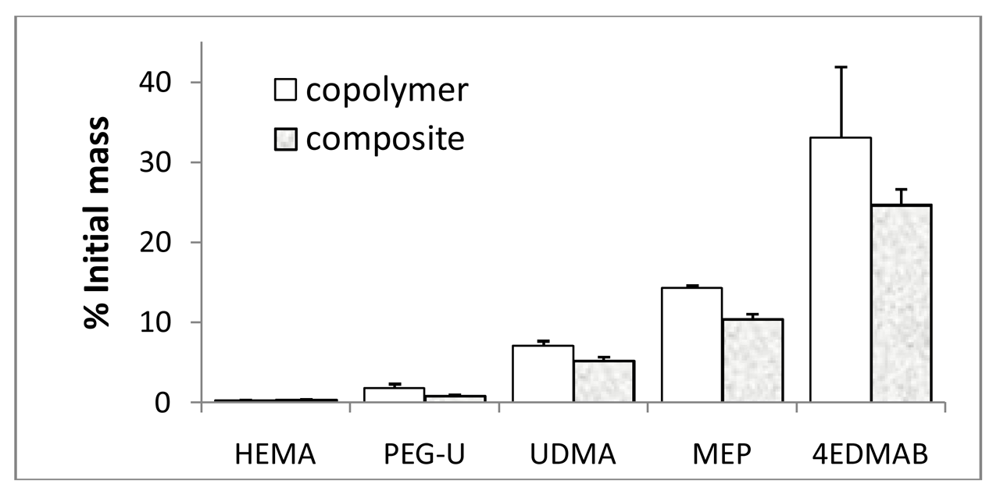
3.4. In Vitro Cytotoxicity of Copolymers and ACP Composites



4. Conclusions
Disclaimer
Acknowledgments
Appendix 1. List of acronyms used throughout the manuscript
| ACP | amorphous calcium phosphate |
| ADAF | American Dental Association Foundation |
| AES | atomic emission spectroscopy |
| Ag-ACP | silver-modified ACP |
| Al-ACP | aluminum-modified ACP |
| ALP | alkaline phosphatase |
| am-ACP | as made ACP |
| ANOVA | analysis of variance |
| APTMS | 3-aminopropyltrimethoxysilane |
| APTMS-ACP | APTMS silanized ACP |
| ASTM | American Society for Testing and Materials |
| BFS | biaxial flexural strength |
| BHT | butylated hydroxyl toluene |
| Bis-GMA | 2,2-bis[p-(2-hydroxy-3-methacryloxypropoxy)phenyl]propane |
| BPO | benzyl peroxide |
| BTHZ | Bis-GMA/TEGDMA/HEMA/ZrDMA resin |
| CC | chemical cure |
| C factor | cavity configuration factor |
| CaP | calcium phosphate |
| CES | commercial endodontic sealer |
| COA | commercial orthodontic adhesive |
| CQ | camphorquinone |
| 4265 Darocur | commercial polymerization initiating system |
| DC | dual cure |
| DCPD | dicalcium phosphate dihydrate |
| DHEPT | 2,2′-dihydroxyethyl-p-toluidine |
| dm | median particle diameter |
| DMSO | dimethylsulfoxide |
| DVC | degree of vinyl conversion |
| EBPADMA | ethoxylated bisphenol A dimethacrylate |
| 4EDMAB | ethyl-4-N,N-dimethylamino benzoate |
| EDTA | ethylenediamine tetraacetic acid |
| ETHM | EBPADMA/TEGDMA/HEMA/MEP resin |
| EUTH | EBPADMA/UDMA/TEGDMA/HEMA resin |
| FAP | fluoroapatite |
| Fe2+-ACP | iron (II)-modified ACP |
| Fe3+-ACP | iron (III)-modified ACP |
| FTIR | Fourier transform infrared spectroscopy |
| FTIR-m | FTIR micro-spectroscopy |
| ΔGo | Gibbs free energy |
| g-ACP | ground ACP |
| HAP | hydroxyapatite |
| HE | hygroscopic expansion |
| HEMA | 2-hydroxyethyl methacrylate |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethane sulfonic acid |
| HmDMA | hexamethylene dimethacrylate |
| IAP | ion activity product |
| 1850 Irgacure | commercial polymerization initiating system |
| 369 Irgacure | commercial polymerization initiating system |
| Ksp | thermodynamic solubility product |
| LC | light cure |
| m-ACP | milled ACP |
| MEP | methacryloyloxyethyl phthalate |
| MPTMS | methacryloxypropyltrimethoxysilane |
| MPTMS-ACP | MPTMS silanized ACP |
| MTT | dehydrogenase activity assay |
| n | number of specimens (or repetitive experiments) |
| NIDCR | National Institute of Dental and Craniofacial Research |
| NIR | near infrared spectroscopy |
| NIST | National Institute of Standards and Technology |
| NMR | nuclear magnetic resonance |
| OCP | octacalcium phosphate pentahydrate |
| PBS | phosphate buffered saline solution |
| PEG-U | poly(ethylene glycol) extended urethane dimethacrylate |
| PEO | poly(ethylene oxide) |
| PEO-ACP | PEO modified ACP |
| PRC | Paffenbarger Research Center |
| PS | polymerization shrinkage |
| PSD | particle size distribution |
| PSS | polymerization shrinkage stress |
| R | ideal gas constant |
| RH | relative humidity |
| SBS | shear bond strength |
| SEM | scanning electron microscopy |
| SD | standard deviation |
| Si-ACP | silicon-modified ACP |
| T | absolute temperature |
| TCP | tricalcium phosphate |
| TEGDMA | triethylene glycol dimethacrylate |
| TGA | thermogravimetric analysis |
| TRITON | alkyl aryl polyether alcohol (nonionic surfactant) |
| TWEEN | poly(oxyethylene) sorbitan monolaureate (nonionic surfactant) |
| UPHM | UDMA/PEG-U/HEMA/MEP resin |
| WS | water sorption |
| Wst1 | mitochondrial dehydrogenase activity assay |
| XRD | X-ray diffraction |
| Zn-ACP | zinc-modified ACP |
| ZONYL FSN | non-ionic fluoro-surfactant |
| ZONYL FSP | anionic fluoro-surfactant |
| Zr-ACP | zirconia-modified ACP |
| ZrDMA | zirconyl dimethacrylate |
References
- Dorozhkin, S.V. Amorphous calcium (ortho)phosphates. Acta Biomater. 2010, 6, 4457–4475. [Google Scholar]
- Dorozhkin, S.V. Calcium orthophosphates as bioceramics: State of the art. J. Funct. Biomater. 2010, 1, 22–107. [Google Scholar]
- Wang, L.; Nancollas, G.H. Pathways to biomineralization and biodemineralization of calcium phosphates: The thermodynamic and kinetic controls. Dalton Trans. 2009, 15, 2665–2672. [Google Scholar]
- Putlyaev, V.I.; Safronova, T.V. A new generation of calcium phosphate biomaterials: The role of phase and chemical compositions. Glass Ceram. 2006, 63, 99–102. [Google Scholar]
- Vallet-Regi, M.; Gonzales-Calbert, J.M. Calcium phosphates as substitution of bone tissues. Prog. Solid State Chem. 2004, 32, 1–31. [Google Scholar]
- Hench, L.L.; Xynos, I.D.; Polak, J.M. Bioactive glasses for in situ tissue regeneration. J. Biomater. Sci. Ploym. Ed. 2004, 15, 543–562. [Google Scholar]
- Barrere, F.; van Blitterswijk, C.A.; de Groot, K. Bone regeneration: Molecular and cellular interactions with calcium phosphate ceramics. Int. J. Nanomed. 2006, 1, 317–332. [Google Scholar]
- Kartsogiannis, V.; Ng, K.W. Cell lines and primary cell cultures in the study of bone cell biology. Mol. Cell. Endocrinol. 2004, 228, 79–102. [Google Scholar]
- Annaz, B.; Hing, K.A.; Kayser, M.; Buckland, T.; di Silvio, L. An ultrastructural study of cellular response to variation in porosity in phase-pure hydroxyapatite. J. Microsc. 2004, 216, 97–109. [Google Scholar]
- Devlin, H.; Sloan, P. Early bone healing events in the human extraction socket. Int. J. Oral Maxillofac. Surg. 2002, 31, 641–645. [Google Scholar]
- Radin, S.; Ducheyne, P.; Berthold, P.; Decker, S. Effect of serum proteins and osteoblasts on the surface transformation of a calcium phosphate coating: A physicochemical and ultrastructural study. J. Biolmed. Mater. Res. 1998, 39, 234–243. [Google Scholar]
- De Bruijn, J.D.; Flach, J.S.; Leenders, H.; van den Brink, J.; van Blitterswijk, C.A. Degradation and interface characteristics of plasma-sprayed hydroxyapatite coatings with different crystallinities. Bioceramics 1992, 5, 291–298. [Google Scholar]
- Midy, V.; Dard, M.; Hollande, E. Evaluation of the effect of three calcium phosphate powders on osteoblast cells. J. Mater. Sci. Mater. Med. 2001, 12, 259–265. [Google Scholar]
- Siebers, M.C.; Walboomers, X.F.; Leeuwenburgh, S.C.G.; Wolke, J.C.G.; Jansen, J.A. Electrostatic spray deposition (ESD) of calcium phosphate coatings, an in vitro study with osteoblast-like cells. Biomaterials 2004, 25, 2019–2027. [Google Scholar]
- Wang, C.; Duan, Y.; Markovic, B.; Barbara, J.; Howlett, C.R.; Zhang, X.; Zreiqat, H. Phenotypic expression of bone-related genes in osteoblasts grown on calcium phosphate ceramics with different phase compositions. Biomaterials 2004, 25, 2507–2514. [Google Scholar]
- Arinzeh, T.L.; Tran, T.; Mcalary, J.; Daculsi, G. A comparative study of biphasic calcium phosphate ceramics for human mesenchymal stem-cell induced bone formation. Biomaterials 2005, 26, 3631–3638. [Google Scholar]
- Adams, C.S.; Mansfield, K.; Perlot, R.L.; Shapiro, I.M. Matrix regulation of skeletal cell apoptosis—Role of calcium and phosphate ions. J. Biol. Chem. 2001, 276, 20316–20322. [Google Scholar]
- Dvorak, M.M.; Siddiqua, A.; Ward, D.T.; Carter, H.; Dallas, S.H.; Nemeth, E.F.; Riccardi, D. Physiological changes in extracellular calcium concentration directly control osteoblast function in the absence of claciotropic hormones. Proc. Natl. Acad. Sci. USA 2004, 101, 5140–5145. [Google Scholar]
- Redey, S.A.; Nardin, M.; Bernache-Assolant, D.; Delannoy, L.S.; Marie, P.J. Behavior of human osteoblastic cells on stoichiometric hydroxyapatite and type A carbonate apatite: Role of surface energy. J. Biolmed. Mater. Res. 2000, 50, 353–364. [Google Scholar]
- Lu, X.; Leng, Y. Quantitative analysis of osteoblast behavior on microgrooved hydroxyapatite and titanium substrata. J. Biolmed. Mater. Res. 2003, 66, 677–687. [Google Scholar]
- Chou, Y.F.; Dunn, J.C.Y.; Wu, B.M. In vitro response of MC3T3-E1 preosteoblast within three-dimensional apatite-coated PLGA scaffolds. J. Biolmed. Mater. Res. 2005, 75B, 81–90. [Google Scholar]
- De Bruijn, J.D.; Bovell, Y.P.; Davies, J.E.; van Blitterswijk, C.A. Osteoclastic resorption of calcium phosphates is potentiated in postosteogenic culture conditions. J. Biolmed. Mater. Res. 1994, 28, 105–112. [Google Scholar]
- Chou, Y.F.; Huang, W.; Dunn, J.C.Y.; Miller, T.A.; Wu, B.M. The effect of biomimetic apatite structure on osteoblast viability, proliferation and gene expression. Biomaterials 2005, 26, 285–295. [Google Scholar]
- Heyman, D.; Pradal, G.; Benahmed, M. Cellular mechanisms of calcium phosphate ceramics degradation. Histol. Histopathol. 1999, 14, 871–877. [Google Scholar]
- Lu, J.; Descamps, M.; Dejon, J.; Koubi, G.; Hardonin, P.; Lemaitre, J.; Proust, J.P. The biodegradation mechanism of calcium phosphate biomaterials in bone. J. Biolmed. Mater. Res. 2002, 63, 408–412. [Google Scholar]
- Zerbo, I.R.; Bronckers, A.L.J.J.; de Lange, G. Localization of osteogenic and osteoclastic cells in porous [beta]-tricalcium phosphate particles used for human maxillary sinus floor elevation. Biomaterials 2005, 26, 1445–1451. [Google Scholar]
- Haders, D.J.; Kazanecki, C.C.; Denhardt, D.T.; Riman, R.E. Crystallographically engineered, hydrothermally crystallized hydroxyapatite films: An in vitro study of bioactivity. J. Mater. Sci. Mater. Med. 2010, 21, 1531–1542. [Google Scholar]
- Reynolds, E.C.; Black, C.L.; Cai, F.; Cross, K.J.; Eakins, D.; Huq, N.L.; Morgan, M.V.; Nowicki, A.; Perich, J.W.; Riley, P.F.; et al. Advances in enamel remineralization: Casein phosphopeptide-amorphous calcium phosphate. J. Clin. Dent. 1999, 10, 86–88. [Google Scholar]
- Ten Cate, J.M. Current concepts on the theories of the mechanism of action of fluoride. Acta Odontol. Scand. 1999, 57, 325–329. [Google Scholar]
- Kashet, S. Historical review of remineralization research. J. Clin. Dent. 1999, 10, 56–64. [Google Scholar]
- Featherstone, J.D. Fluoride, remineralization and root caries. Am. J. Dent. 1994, 7, 271–274. [Google Scholar]
- Chow, L.C.; Vogel, G.L. Enhancing remineralization. Oper. Dent. 2001, 6, 27–38. [Google Scholar]
- Reynolds, E.C.; Cai, F.; Shen, P.; Walker, G.D. Retention in plaque and remineralization of enamel lesions by various forms of calcium in a mouthrinse or sugar-free chewing gum. J. Dental Res. 2003, 82, 206–211. [Google Scholar]
- Reynolds, E.C.; Cai, F.; Cohrane, N.J.; Shen, P.; Walker, G.D.; Morgan, M.V.; Reynolds, C. Fluoride and casein phosphopeptide-amorphous calcium phosphate. J. Dental Res. 2008, 87, 344–348. [Google Scholar]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D. Amorphous calcium phosphate-based bioactive polymeric composites for mineralized tissue regeneration. J. Res. Natl. Inst. Stand. Technol. 2003, 108, 167–182. [Google Scholar]
- Skrtic, D.; Antonucci, J.M. Design, characterization and evaluation of biomimetic polymeric dental composites with remineralization potential. In Encyclopedia of Polymer Composites: Properties, Performance and Applications; Lechov, M., Prandzheva, S., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2010; pp. 281–318. [Google Scholar]
- Langhorst, S.E.; O'Donnell, J.N.R.; Skrtic, D. In vitro remineralization effectiveness of polymeric ACP composites: Quantitative micro-radiographic study. Dental Mater. 2009, 25, 884–891. [Google Scholar]
- Tung, M.S.; Eichmiller, F.C. Dental applications of amorphous calcium phosphates. J. Clin. Dent. 1999, 10, 1–6. [Google Scholar]
- Santerre, J.P.; Shajii, L.; Leung, B.W. Relation of dental composite formulations to their degradation and the release of hydrolyzed polymeric-resin-derived products. Crit. Rev. Oral Biol. Med. 2001, 12, 136–151. [Google Scholar]
- Pelka, M.; Distle, R.W.; Petshelt, A. Elution parameters and HPLC-detection of single components from resin composite. Clin. Oral Investig. 1999, 3, 194–200. [Google Scholar]
- Spahl, W.; Budzikiewicz, H.; Geurtsen, W. Determination of leachable components from four commercial dental composites by gas and liquid chromatography/mass spectrometry. J. Dent. 1998, 26, 137–145. [Google Scholar]
- Antonucci, J.M.; Skrtic, D. Physicochemical and mechanical evaluation of cation-modified ACP acrylic resin composites. Polym. Prepr. 2006, 47, 113–114. [Google Scholar]
- Antonucci, J.M.; Liu, D.W.; Skrtic, D. Amorphous calcium phosphate based composites: Effect of surfactants and poly(ethylene) oxide on filler and composite properties. J. Dispers. Sci. Technol. 2007, 28, 819–824. [Google Scholar]
- Antonucci, J.M.; Skrtic, D. Bioactive and biocompatible polymeric composites based on amorphous calcium phosphate. In Integrated Biomaterials for Medical Applications, Vol. 1: Biomaterials—Protocols and Techniques; Ramalingam, M., Tiwari, A., Eds.; VBRI Press: Saidabad, India, 2011; in press. [Google Scholar]
- O'Donnell, J.N.R.; Skrtic, D. Degree of vinyl conversion, polymerization shrinkage and stress development in experimental endodontic composites. J. Biomim. Biomater. Tissue Eng. 2009, 4, 1–12. [Google Scholar]
- Lee, S.Y.; Regnault, W.F.; Antonucci, J.M.; Skrtic, D. Effect of particle size of an amorphous calcium phosphate filler on the mechanical strength and ion release of polymeric composites. J. Biomed. Mater. Res. 2007, 80B, 11–17. [Google Scholar]
- Stansbury, J.W.; Dickens, S.H. Network formation and compositional drift during photo-initiated copolymerization of dimethacrylate monomers. Polymer 2001, 42, 6363–6369. [Google Scholar]
- ASTM Standard F394-78: Standard Test Method for Biaxial Strength (Modulus of Rupture) of Ceramic Substrates; ASTM International: West Conshochoken, PA, USA, 1996.
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D.; Eidelman, N. Dental composites based on hybrid and surface-modified amorphous calcium phosphates—A FTIR microspectroscopic study. Biomaterials 2004, 25, 1141–1150. [Google Scholar]
- Lu, H.; Stansbury, J.W.; Dickens, S.H.; Eichmiller, F.C.; Bowman, C.N. Probing the origins and control of shrinkage stress in dental resin-composites: I. Shrinkage stress characterization technique. J. Mater. Sci. Mater. Med. 2004, 15, 1097–1103. [Google Scholar]
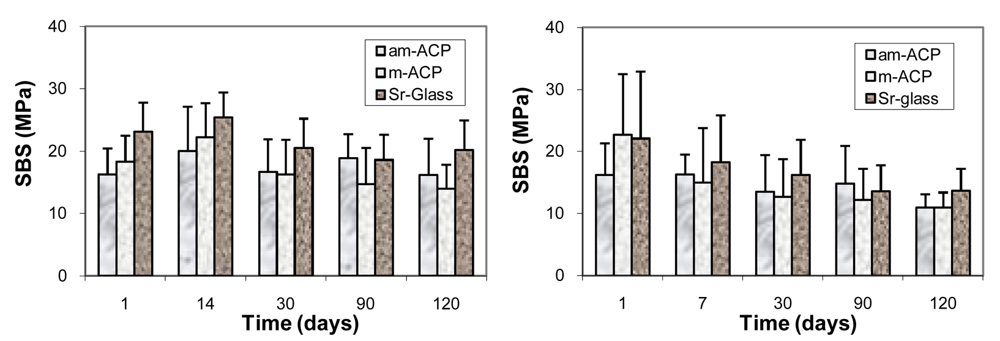
- Schumacher, G.E.; Antonucci, J.M.; O'Donnell, J.N.R.; Skrtic, D. The use of amorphous calcium phosphate composites as bioactive materials and their effect on the strength of the composite/adhesive/dentin bond. J. Am. Dental Assoc. 2007, 138, 1476–1484. [Google Scholar]
- O'Donnell, J.N.R.; Schumacher, G.E.; Antonucci, J.M.; Skrtic, D. Adhesion of amorphous calcium phosphate composites bonded to dentin: A study in failure modality. J. Biolmed. Mater. Res. 2009, 90B, 238–249. [Google Scholar]
- Simon, C.G., Jr.; Antonucci, J.M.; Liu, D.W.; Skrtic, D. In vitro cytotoxicity of amorphous calcium phosphate composites. J. Bioact. Compat. Polym. 2005, 20, 279–295. [Google Scholar]
- Ishiyama, M.; Shiga, M.; Sasamoto, K.; Mizoguchi, H.; He, P.G. A new sulfonated tetrazolium salt that produces a highly water-soluble formazan dye. Chem. Pharm. Bull. 1993, 41, 1118–1122. [Google Scholar]
- Amjad, Z. Inhibition of the amorphous calcium phosphate phase transformation reaction by polymeric and non-polymeric inhibitors. Phosphorus Res. Bull. 2004, 7, 45–54. [Google Scholar]
- Ofir, P.B.Y.; Govrin-Lipman, R.; Garti, N.; Furedi-Milhofer, H. The influence of polyelectrolytes on the formation and phase transformation of amorphous calcium phosphate. Cryst. Growth Des. 2004, 4, 177–183. [Google Scholar]
- Eanes, E.D. Amorphous calcium phosphate: Thermodynamic and kinetic considerations. In Calcium Phosphates in Biological and Industrial Systems; Amjad, Z., Ed.; Kluwer Academic Publ: Boston, MA, USA, 1998; pp. 21–39. [Google Scholar]
- Skrtic, D.; Antonucci, J.M.; Eanes, E.D.; Brunworth, R.T. Silica- and zirconia-hybridized amorphous calcium phosphate. Effect on transformation to hydroxyapatite. J. Biolmed. Mater. Res. 2002, 59, 597–604. [Google Scholar]
- Venhoven, B.A.M.; de Gee, A.J.; Davidson, C.L. Polymerization contraction and conversion of light-curing Bis-GMA based methacrylate resins. Biomaterials 1993, 14, 871–875. [Google Scholar]
- Labella, R.; Lambrecths, P.; van Meerbeck, B.; Vanherle, G. Polymerization shrinkage and elasticity of flowable composites and filled adhesives. Dental Mater. 1999, 15, 128–137. [Google Scholar]
- Guggenbarger, R.; Weinmann, W. Exploring beyond methacrylates. Am. J. Dent. 2000, 13, 82D–84D. [Google Scholar]
- Tilbrook, D.A.; Clarke, R.L.; Howle, N.E.; Braden, M. Photocurable epoxy-polyol matrices for use in dental composites. Biomaterials 2000, 21, 1743–1753. [Google Scholar]
- Arima, T.; Hamada, T.; Mccabe, J.F. The effects of cross-linking agents on some properties of HEMA-based resins. J. Dental Res. 1995, 74, 1597–1601. [Google Scholar]
- Garcia-Fiero, J.L.; Aleman, J.V. Sorption of water by epoxide prepolymers. Macromolecules 1982, 15, 1145–1149. [Google Scholar]
- Skrtic, D.; Antonucci, J.M. Dental composites based on amorphous calcium phosphate—Resin composition/physicochemical properties study. J. Biomater. Appl. 2007, 21, 375–393. [Google Scholar]
- Antonucci, J.M.; Regnault, W.F.; Skrtic, D. Polymerization shrinkage and polymerization stress development in amorphous calcium phosphate/urethane dimethacrylate polymeric composites. J. Compos. Mater. 2010, 44, 355–367. [Google Scholar]
- Momoi, Y.; McCabe, J.F. Hygroscopic expansion of resin based composites during 6 months water storage. Br. Dental J. 1994, 176, 91–96. [Google Scholar]
- Huang, C.; Tay, F.R.; Cheung, G.S.P.; Kei, L.H.; Wei, S.H.Y.; Pashley, D.H. Hygroscopic expansion of a compomer and a composite on artificial gap reduction. J. Dent. 2002, 30, 11–19. [Google Scholar]
- Ferracane, J.L. Developing a more complete understanding of stresses produced in dental composites during polymerization. Dental Mater. 2005, 21, 36–42. [Google Scholar]
- Braga, R.R.; Ferracane, J.L. Contraction stress related to degree of conversion and reaction kinetics. J. Dental Res. 2002, 81, 114–118. [Google Scholar]
- Calheiros, F.C.; Braga, R.R.; Kawano, Y.; Ballester, R.Y. Relationship between contraction stress and degree of conversion in restorative composites. Dental Mater. 2004, 20, 939–946. [Google Scholar]
- Stansbury, J.W.; Trujillo-Lemon, M.; Lu, H.; Ding, X.; Lin, Y.; Ge, J. Conversion-dependent shrinkage stress and strain in dental resins and composites. Dental Mater. 2005, 21, 56–67. [Google Scholar]
- Kleverlaan, C.J.; Feilzer, A.J. Polymerization shrinkage and contraction stress of dental resin composites. Dental Mater. 2005, 21, 1150–1157. [Google Scholar]
- Choi, K.K.; Ruy, G.J.; Choi, S.M.; Lee, M.J.; Park, S.J.; Ferracane, J.L. Effects of cavity configuration on composite restoration. Oper. Dent. 2004, 29, 462–469. [Google Scholar]
- Uno, S.; Tanaka, T.; Inoue, S.; Sano, S. The influence of configuration factors on cavity adaptation in compomer restorations. Dental Mater. 1999, 18, 19–31. [Google Scholar]
- Feilzer, A.J.; de Gee, A.J.; Davidson, C.L. Quantitative determination of stress reduction by flow in composite restorations. Dental Mater. 1990, 6, 167–171. [Google Scholar]
- Alster, D.; Feilzer, A.J.; de Gee, A.J.; Davidson, C.L. Polymerization contraction stress in thin resin composite layers as a function of layer thickness. Dental Mater. 1997, 13, 146–150. [Google Scholar]
- Choi, K.K.; Condon, J.R.; Ferracane, J.L. The effects of adhesive thickness on polymerization contraction stress of composite. J. Dental Res. 2000, 79, 812–817. [Google Scholar]
- Watts, D.C.; Satterthwaite, J.D. Axial shrinkage stress depends upon both c-factor and composite mass. J. Dental Res. 2008, 24, 1–8. [Google Scholar]
- Munksgaard, E.C.; Hansen, E.K.; Kato, H. Wall-to-wall polymerization contraction of composite resins versus filler content. Scand. J. Dent. 1987, 95, 526–531. [Google Scholar]
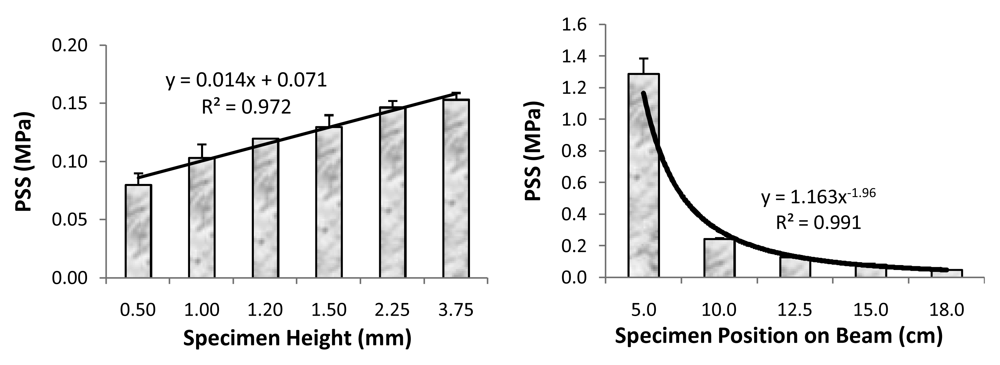
- Antonucci, J.M.; Giuseppetti, A.A.; O'Donnell, J.N.R.; Schumacher, G.E.; Skrtic, D. Polymerization stress development in dental composites: Effect of cavity design factor. Materials 2009, 2, 169–180. [Google Scholar]
- Antonucci, J.M.; O'Donnell, J.N.R.; Schumacher, G.E.; Skrtic, D. Amorphous calcium phosphate composites and their effect on the composite/adhesive/dentin bond. J. Adhes. Sci. Technol. 2009, 23, 1133–1147. [Google Scholar]
- Davis, C.H.; O'Donnell, J.N.R.; Skrtic, D. Determination of leachable components from an experimental ACP endodontic sealer by 1H NMR. Dental Mater. 2011, in press. [Google Scholar]
- Durner, J.; Walther, U.I.; Zaspel, J.; Hickel, R.; Reichl, F.X. Metabolism of TEGDMA and HEMA in human cells. Biomaterials 2010, 31, 818–823. [Google Scholar]
- Floyd, C.J.E.; Dickens, S.H. Network structure of Bis-GMA- and UDMA-based resin systems. Dental Mater. 2006, 22, 1143–1149. [Google Scholar]
- Chatterjee, K.; Lin-Gibson, S.; Wallace, W.E.; Parekh, S.H.; Lee, Y.J.; Cicerone, M.T.; Young, M.F.; Simon, C.G. The effect of 3D hydrogel scaffold modulus on osteoblast differentiation and mineralization revealed by combinatorial screening. Biomaterials 2010, 31, 5051–5062. [Google Scholar]
- Dalby, M.J.; Gadegaard, N.; Tare, R.; Andar, A.; Riehle, M.O.; Herzyk, P.; Wilkinson, C.D.W.; Oreffo, R.O.C. The control of human mesenchymal cell differentiation using nanoscale symmetry and disorder. Nat. Mater. 2007, 6, 997–1003. [Google Scholar]
- Franz, A.; Kőnig, F.; Lucas, T.; Watts, D.C.; Schedle, A. Cytotoxic effects of dental bonding substances as a function of degree of conversion. Dental Mater. 2009, 25, 232–239. [Google Scholar]
- O'Donnell, J.; Sun, J.; Skrtic, D. In vitro cytotoxicity of the experimental ACP endodontic sealer. Int. Assn. Dent. Res. 2011, in press. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Skrtic, D.; Antonucci, J.M. Bioactive Polymeric Composites for Tooth Mineral Regeneration: Physicochemical and Cellular Aspects. J. Funct. Biomater. 2011, 2, 271-307. https://doi.org/10.3390/jfb2030271
Skrtic D, Antonucci JM. Bioactive Polymeric Composites for Tooth Mineral Regeneration: Physicochemical and Cellular Aspects. Journal of Functional Biomaterials. 2011; 2(3):271-307. https://doi.org/10.3390/jfb2030271
Chicago/Turabian StyleSkrtic, Drago, and Joseph M. Antonucci. 2011. "Bioactive Polymeric Composites for Tooth Mineral Regeneration: Physicochemical and Cellular Aspects" Journal of Functional Biomaterials 2, no. 3: 271-307. https://doi.org/10.3390/jfb2030271
APA StyleSkrtic, D., & Antonucci, J. M. (2011). Bioactive Polymeric Composites for Tooth Mineral Regeneration: Physicochemical and Cellular Aspects. Journal of Functional Biomaterials, 2(3), 271-307. https://doi.org/10.3390/jfb2030271