Astrocyte Regulation of CNS Inflammation and Remyelination

{kind=link}
Abstract
:1. Introductions
2. Astrocytic Regulation of Adaptive Immune Responses
3. Astrocytic Regulation of Innate Immune Responses
4. Regulation of Astrocytic Phenotype Influences Oligodendrocyte Differentiation
5. Phenotypic Plasticity of Astrocytes as Regulated by Inflammation
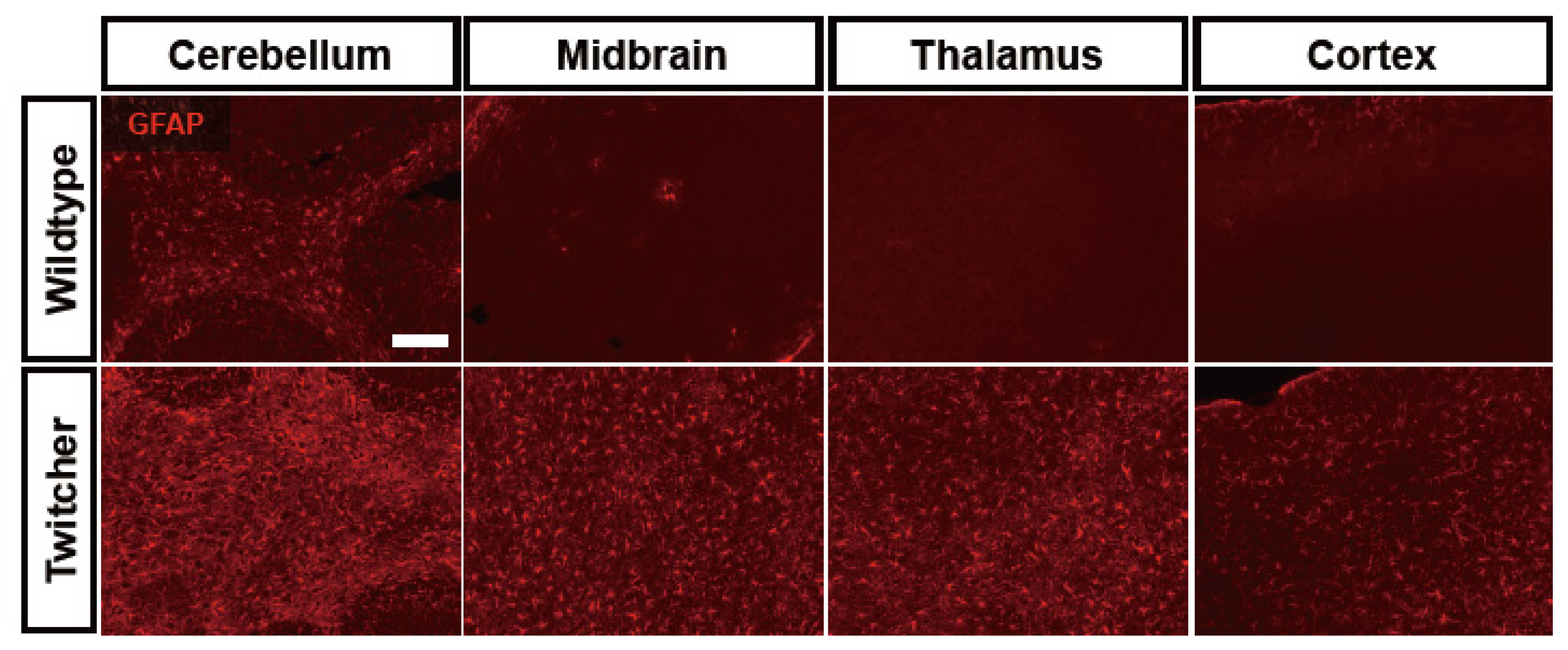
6. Astrocyte Dysfunction in Leukodystrophies
7. Conclusions
8. Therapeutic Potential of Astrocytes
Conflict of Interest
References
- De Jager, P.L.; Hafler, D.A. New therapeutic approaches for multiple sclerosis. Annu. Rev. Med. 2007, 58, 417–432. [Google Scholar] [CrossRef]
- Runia, T.F.; van Pelt-Gravesteijn, E.D.; Hintzen, R.Q. Recent gains in clinical multiple sclerosis research. CNS Neurol. Disord. Drug Targets 2012, 11, 497–505. [Google Scholar] [CrossRef]
- Shirani, A.; Zhao, Y.; Karim, M.E.; Evans, C.; Kingwell, E.; van der Kop, M.L.; Oger, J.; Gustafson, P.; Petkau, J.; Tremlett, H. Association between use of interferon beta and progression of disability in patients with relapsing-remitting multiple sclerosis. JAMA 2012, 308, 247–256. [Google Scholar] [CrossRef]
- Stys, P.K.; Zamponi, G.W.; van Minnen, J.; Geurts, J.J. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012, 13, 507–514. [Google Scholar]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Tsunoda, I.; Fujinami, R.S. Inside-Out versus Outside-In models for virus induced demyelination: Axonal damage triggering demyelination. Springer Semin. Immunopathol. 2002, 24, 105–125. [Google Scholar]
- Vogel, F.S. Demyelinization induced in living rabbits by means of a lipolytic enzyme preparation. J. Exp. Med. 1951, 93, 297–304. [Google Scholar]
- Oberheim, N.A.; Goldman, S.A.; Nedergaard, M. Heterogeneity of astrocytic form and function. Methods Mol. Biol. 2012, 814, 23–45. [Google Scholar]
- Chiu, F.C.; Norton, W.T.; Fields, K.L. The cytoskeleton of primary astrocytes in culture contains actin, glial fibrillary acidic protein, and the fibroblast-type filament protein, vimentin. J. Neurochem. 1981, 37, 147–155. [Google Scholar] [CrossRef]
- Von Herrath, M.G.; Fujinami, R.S.; Whitton, J.L. Microorganisms and autoimmunity: Making the barren field fertile? Nat. Rev. Microbiol. 2003, 1, 151–157. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef]
- Voskuhl, R.R.; Peterson, R.S.; Song, B.; Ao, Y.; Morales, L.B.; Tiwari-Woodruff, S.; Sofroniew, M.V. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J. Neurosci. 2009, 29, 11511–11522. [Google Scholar] [CrossRef]
- Toft-Hansen, H.; Fuchtbauer, L.; Owens, T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia 2011, 59, 166–176. [Google Scholar] [CrossRef]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Nair, A.; Frederick, T.J.; Miller, S.D. Astrocytes in multiple sclerosis: A product of their environment. Cell. Mol. Life Sci. 2008, 65, 2702–2720. [Google Scholar] [CrossRef]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Prochiantz, A.; Mallat, M. Astrocyte diversity. Ann. N. Y. Acad. Sci. 1988, 540, 52–63. [Google Scholar] [CrossRef]
- Cornet, A.; Bettelli, E.; Oukka, M.; Cambouris, C.; Avellana-Adalid, V.; Kosmatopoulos, K.; Liblau, R.S. Role of astrocytes in antigen presentation and naive T-cell activation. J. Neuroimmunol. 2000, 106, 69–77. [Google Scholar] [CrossRef]
- Soos, J.M.; Ashley, T.A.; Morrow, J.; Patarroyo, J.C.; Szente, B.E.; Zamvil, S.S. Differential expression of B7 co-stimulatory molecules by astrocytes correlates with T cell activation and cytokine production. Int. Immunol. 1999, 11, 1169–1179. [Google Scholar] [CrossRef]
- Tan, L.J.; Vanderlugt, C.L.; McRae, B.L.; Miller, S.D. Regulation of the effector stages of experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance induction. III. A role for anergy/deletion. Autoimmunity 1998, 27, 13–28. [Google Scholar]
- Kort, J.J.; Kawamura, K.; Fugger, L.; Weissert, R.; Forsthuber, T.G. Efficient presentation of myelin oligodendrocyte glycoprotein peptides but not protein by astrocytes from HLA-DR2 and HLA-DR4 transgenic mice. J. Neuroimmunol. 2006, 173, 23–34. [Google Scholar] [CrossRef]
- Girvin, A.M.; Gordon, K.B.; Welsh, C.J.; Clipstone, N.A.; Miller, S.D. Differential abilities of central nervous system resident endothelial cells and astrocytes to serve as inducible antigen-presenting cells. Blood 2002, 99, 3692–3701. [Google Scholar] [CrossRef]
- De Keyser, J.; Laureys, G.; Demol, F.; Wilczak, N.; Mostert, J.; Clinckers, R. Astrocytes as potential targets to suppress inflammatory demyelinating lesions in multiple sclerosis. Neurochem. Int. 2010, 57, 446–450. [Google Scholar] [CrossRef]
- Stuve, O.; Youssef, S.; Slavin, A.J.; King, C.L.; Patarroyo, J.C.; Hirschberg, D.L.; Brickey, W.J.; Soos, J.M.; Piskurich, J.F.; Chapman, H.A.; Zamvil, S.S. The role of the MHC class II transactivator in class II expression and antigen presentation by astrocytes and in susceptibility to central nervous system autoimmune disease. J. Immunol. 2002, 169, 6720–6732. [Google Scholar]
- De Keyser, J.; Wilczak, N.; Leta, R.; Streetland, C. Astrocytes in multiple sclerosis lack beta-2 adrenergic receptors. Neurology 1999, 53, 1628–1633. [Google Scholar] [CrossRef]
- John, G.R.; Chen, L.; Rivieccio, M.A.; Melendez-Vasquez, C.V.; Hartley, A.; Brosnan, C.F. Interleukin-1beta induces a reactive astroglial phenotype via deactivation of the Rho GTPase-Rock axis. J. Neurosci. 2004, 24, 2837–2845. [Google Scholar] [CrossRef]
- Nikcevich, K.M.; Gordon, K.B.; Tan, L.; Hurst, S.D.; Kroepfl, J.F.; Gardinier, M.; Barrett, T.A.; Miller, S.D. IFN-gamma-activated primary murine astrocytes express B7 costimulatory molecules and prime naive antigen-specific T cells. J. Immunol. 1997, 158, 614–621. [Google Scholar]
- Zeinstra, E.M.; Wilczak, N.; Wilschut, J.C.; Glazenburg, L.; Chesik, D.; Kroese, F.G.; de Keyser, J. 5HT4 agonists inhibit interferon-gamma-induced MHC class II and B7 costimulatory molecules expression on cultured astrocytes. J. Neuroimmunol. 2006, 179, 191–195. [Google Scholar]
- Constantinescu, C.S.; Tani, M.; Ransohoff, R.M.; Wysocka, M.; Hilliard, B.; Fujioka, T.; Murphy, S.; Tighe, P.J.; Das Sarma, J.; Trinchieri, G.; Rostami, A. Astrocytes as antigen-presenting cells: Expression of IL-12/IL-23. J. Neurochem. 2005, 95, 331–340. [Google Scholar] [CrossRef]
- Chastain, E.M.; Duncan, D.S.; Rodgers, J.M.; Miller, S.D. The role of antigen presenting cells in multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 265–274. [Google Scholar]
- Hassan-Zahraee, M.; Ladiwala, U.; Lavoie, P.M.; McCrea, E.; Sekaly, R.P.; Owens, T.; Antel, J.P. Superantigen presenting capacity of human astrocytes. J. Neuroimmunol. 2000, 102, 131–136. [Google Scholar]
- Wang, X.; Haroon, F.; Karray, S.; Martina, D.; Schluter, D. Astrocytic Fas ligand expression is required to induce T-cell apoptosis and recovery from experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2013, 43, 115–124. [Google Scholar] [CrossRef]
- Aubagnac, S.; Brahic, M.; Bureau, J.F. Bone marrow chimeras reveal non-H-2 hematopoietic control of susceptibility to Theiler’s virus persistent infection. J. Virol. 2002, 76, 5807–5812. [Google Scholar] [CrossRef]
- Carpentier, P.A.; Getts, M.T.; Miller, S.D. Pro-inflammatory functions of astrocytes correlate with viral clearance and strain-dependent protection from TMEV-induced demyelinating disease. Virology 2008, 375, 24–36. [Google Scholar]
- Hertzenberg, D.; Lehmann-Horn, K.; Kinzel, S.; Husterer, V.; Cravens, P.D.; Kieseier, B.C.; Hemmer, B.; Bruck, W.; Zamvil, S.S.; Stuve, O.; Weber, M.S. Developmental maturation of innate immune cell function correlates with susceptibility to central nervous system autoimmunity. Eur J. Immunol 2013. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, O.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar]
- Lyons, A.; Downer, E.J.; Crotty, S.; Nolan, Y.M.; Mills, K.H.; Lynch, M.A. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: A role for IL-4. J. Neurosci. 2007, 27, 8309–8313. [Google Scholar] [CrossRef]
- Chew, L.J.; Fusar-Poli, P.; Schmitz, T. Oligodendroglial alterations and the role of microglia in white matter injury: Relevance to schizophrenia. Dev. Neurosci. 2013, 35, 102–129. [Google Scholar] [CrossRef]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef]
- Yeo, Y.A.; Martinez Gomez, J.M.; Croxford, J.L.; Gasser, S.; Ling, E.A.; Schwarz, H. CD137 ligand activated microglia induces oligodendrocyte apoptosis via reactive oxygen species. J. Neuroinflammation 2012, 9, 173. [Google Scholar]
- Brand-Schieber, E.; Werner, P.; Iacobas, D.A.; Iacobas, S.; Beelitz, M.; Lowery, S.L.; Spray, D.C.; Scemes, E. Connexin43, the major gap junction protein of astrocytes, is down-regulated in inflamed white matter in an animal model of multiple sclerosis. J. Neurosci. Res. 2005, 80, 798–808. [Google Scholar] [CrossRef]
- Lutz, S.E.; Zhao, Y.; Gulinello, M.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 2009, 29, 7743–7752. [Google Scholar] [CrossRef]
- Paulson, H.L.; Garbern, J.Y.; Hoban, T.F.; Krajewski, K.M.; Lewis, R.A.; Fischbeck, K.H.; Grossman, R.I.; Lenkinski, R.; Kamholz, J.A.; Shy, M.E. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann. Neurol. 2002, 52, 429–434. [Google Scholar] [CrossRef]
- Moore, C.S.; Milner, R.; Nishiyama, A.; Frausto, R.F.; Serwanski, D.R.; Pagarigan, R.R.; Whitton, J.L.; Miller, R.H.; Crocker, S.J. Astrocytic tissue inhibitor of metalloproteinase-1 (TIMP-1) promotes oligodendrocyte differentiation and enhances CNS myelination. J. Neurosci. 2011, 31, 6247–6254. [Google Scholar] [CrossRef]
- Nash, B.; Thomson, C.E.; Linington, C.; Arthur, A.T.; McClure, J.D.; McBride, M.W.; Barnett, S.C. Functional duality of astrocytes in myelination. J. Neurosci. 2011, 31, 13028–13038. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Honda, S.; Sasaki, Y.; Ohsawa, K.; Imai, Y.; Nakamura, Y.; Inoue, K.; Kohsaka, S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J. Neurosci. 2001, 21, 1975–1982. [Google Scholar]
- Shigemoto-Mogami, Y.; Koizumi, S.; Tsuda, M.; Ohsawa, K.; Kohsaka, S.; Inoue, K. Mechanisms underlying extracellular ATP-evoked interleukin-6 release in mouse microglial cell line, MG-5. J. Neurochem. 2001, 78, 1339–1349. [Google Scholar]
- Inoue, K.; Nakajima, K.; Morimoto, T.; Kikuchi, Y.; Koizumi, S.; Illes, P.; Kohsaka, S. ATP stimulation of Ca2+-dependent plasminogen release from cultured microglia. Br. J. Pharmacol. 1998, 123, 1304–1310. [Google Scholar] [CrossRef]
- Hide, I.; Tanaka, M.; Inoue, A.; Nakajima, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J. Neurochem. 2000, 75, 965–972. [Google Scholar]
- Ovanesov, M.V.; Ayhan, Y.; Wolbert, C.; Moldovan, K.; Sauder, C.; Pletnikov, M.V. Astrocytes play a key role in activation of microglia by persistent Borna disease virus infection. J. Neuroinflammation 2008, 5, 50. [Google Scholar] [CrossRef]
- Tanuma, N.; Sakuma, H.; Sasaki, A.; Matsumoto, Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006, 112, 195–204. [Google Scholar] [CrossRef]
- Min, K.J.; Yang, M.S.; Kim, S.U.; Jou, I.; Joe, E.H. Astrocytes induce hemeoxygenase-1 expression in microglia: A feasible mechanism for preventing excessive brain inflammation. J. Neurosci. 2006, 26, 1880–1887. [Google Scholar] [CrossRef]
- Muller, M.; Carter, S.L.; Hofer, M.J.; Manders, P.; Getts, D.R.; Getts, M.T.; Dreykluft, A.; Lu, B.; Gerard, C.; King, N.J.; Campbell, I.L. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J. Immunol. 2007, 179, 2774–2786. [Google Scholar]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar]
- Gutowski, N.J.; Newcombe, J.; Cuzner, M.L. Tenascin-R and C in multiple sclerosis lesions: Relevance to extracellular matrix remodelling. Neuropathol. Appl. Neurobiol. 1999, 25, 207–214. [Google Scholar]
- Stoffels, J.M.; de Jonge, J.C.; Stancic, M.; Nomden, A.; van Strien, M.E.; Ma, D.; Siskova, Z.; Maier, O.; Ffrench-Constant, C.; Franklin, R.J.; et al. Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain 2013, 136, 116–131. [Google Scholar]
- Moore, C.S.; Abdullah, S.L.; Brown, A.; Arulpragasam, A.; Crocker, S.J. How factors secreted from astrocytes impact myelin repair. J. Neurosci. Res. 2011, 89, 13–21. [Google Scholar] [CrossRef]
- Back, S.A.; Tuohy, T.M.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.L.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar]
- Sosunov, A.A.; Guilfoyle, E.; Wu, X.; McKhann, G.M., II; Goldman, J.E. Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in alexander disease. J. Neurosci. 2013, 33, 7439–7450. [Google Scholar]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Eng, L.F.; Ghirnikar, R.S. GFAP and astrogliosis. Brain Pathol. 1994, 4, 229–237. [Google Scholar] [CrossRef]
- Norenberg, M.D. Astrocyte responses to CNS injury. J. Neuropathol. Exp. Neurol. 1994, 53, 213–220. [Google Scholar] [CrossRef]
- Gomez-Pinilla, F.; Vu, L.; Cotman, C.W. Regulation of astrocyte proliferation by FGF-2 and heparan sulfate in vivo. J. Neurosci. 1995, 15, 2021–2029. [Google Scholar]
- McKeon, R.J.; Jurynec, M.J.; Buck, C.R. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J. Neurosci. 1999, 19, 10778–10788. [Google Scholar]
- Leadbeater, W.E.; Gonzalez, A.M.; Logaras, N.; Berry, M.; Turnbull, J.E.; Logan, A. Intracellular trafficking in neurones and glia of fibroblast growth factor-2, fibroblast growth factor receptor 1 and heparan sulphate proteoglycans in the injured adult rat cerebral cortex. J. Neurochem. 2006, 96, 1189–1200. [Google Scholar] [CrossRef]
- Lee, S.C.; Liu, W.; Dickson, D.W.; Brosnan, C.F.; Berman, J.W. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J. Immunol. 1993, 150, 2659–2667. [Google Scholar]
- Rouach, N.; Calvo, C.F.; Glowinski, J.; Giaume, C. Brain macrophages inhibit gap junctional communication and downregulate connexin 43 expression in cultured astrocytes. Eur. J. Neurosci. 2002, 15, 403–407. [Google Scholar] [CrossRef]
- Rouach, N.; Avignone, E.; Meme, W.; Koulakoff, A.; Venance, L.; Blomstrand, F.; Giaume, C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol. Cell. 2002, 94, 457–475. [Google Scholar] [CrossRef]
- Crocker, S.J.; Pagenstecher, A.; Campbell, I.L. The TIMPs tango with MMPs and more in the central nervous system. J. Neurosci. Res. 2004, 75, 1–11. [Google Scholar] [CrossRef]
- Crocker, S.J.; Milner, R.; Pham-Mitchell, N.; Campbell, I.L. Cell and agonist-specific regulation of genes for matrix metalloproteinases and their tissue inhibitors by primary glial cells. J. Neurochem. 2006, 98, 812–823. [Google Scholar] [CrossRef]
- Welser-Alves, J.V.; Crocker, S.J.; Milner, R. A dual role for microglia in promoting tissue inhibitor of metalloproteinase (TIMP) expression in glial cells in response to neuroinflammatory stimuli. J. Neuroinflammation 2011, 8, 61. [Google Scholar] [CrossRef]
- Fields, J.; Cisneros, I.E.; Borgmann, K.; Ghorpade, A. Extracellular regulated kinase 1/2 signaling is a critical regulator of interleukin-1beta-mediated astrocyte tissue inhibitor of metalloproteinase-1 expression. PLoS One 2013, 8, e56891. [Google Scholar]
- Pagenstecher, A.; Stalder, A.K.; Kincaid, C.L.; Shapiro, S.D.; Campbell, I.L. Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in the mouse central nervous system in normal and inflammatory states. Am. J. Pathol. 1998, 152, 729–741. [Google Scholar]
- Crocker, S.J.; Whitmire, J.K.; Frausto, R.F.; Chertboonmuang, P.; Soloway, P.D.; Whitton, J.L.; Campbell, I.L. Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am. J. Pathol. 2006, 169, 2104–2116. [Google Scholar] [CrossRef]
- Suryadevara, R.; Holter, S.; Borgmann, K.; Persidsky, R.; Labenz-Zink, C.; Persidsky, Y.; Gendelman, H.E.; Wu, L.; Ghorpade, A. Regulation of tissue inhibitor of metalloproteinase-1 by astrocytes: Links to HIV-1 dementia. Glia 2003, 44, 47–56. [Google Scholar] [CrossRef]
- Ichiyama, T.; Kajimoto, M.; Suenaga, N.; Maeba, S.; Matsubara, T.; Furukawa, S. Serum levels of matrix metalloproteinase-9 and its tissue inhibitor (TIMP-1) in acute disseminated encephalomyelitis. J. Neuroimmunol. 2006, 172, 182–186. [Google Scholar] [CrossRef]
- Avolio, C.; Ruggieri, M.; Giuliani, F.; Liuzzi, G.M.; Leante, R.; Riccio, P.; Livrea, P.; Trojano, M. Serum MMP-2 and MMP-9 are elevated in different multiple sclerosis subtypes. J. Neuroimmunol. 2003, 136, 46–53. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Sofroniew, M.V.; Messing, A.; deLanerolle, N.C.; Rempe, D.; Rodriguez, J.J.; Nedergaard, M. Neurological diseases as primary gliopathies: A reassessment of neurocentrism. ASN Neuro 2012, 4. [Google Scholar] [CrossRef]
- Sharma, R.; Fischer, M.T.; Bauer, J.; Felts, P.A.; Smith, K.J.; Misu, T.; Fujihara, K.; Bradl, M.; Lassmann, H. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. 2010, 120, 223–236. [Google Scholar] [CrossRef]
- Crocker, S.J. Gliodystrophy: Astroglial Heterogeneity in Neurodegenerative Disease; American Society of Neurochemistry: Baltimore, MD, USA, 2012. [Google Scholar]
- Kondo, Y.; Wenger, D.A.; Gallo, V.; Duncan, I.D. Galactocerebrosidase-deficient oligodendrocytes maintain stable central myelin by exogenous replacement of the missing enzyme in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 18670–18675. [Google Scholar] [CrossRef]
- Ijichi, K.; Brown, G.D.; Moore, C.S.; Lee, J.P.; Winokur, P.N.; Pagarigan, R.; Snyder, E.Y.; Bongarzone, E.R.; Crocker, S.J. MMP-3 mediates psychosine-induced globoid cell formation: Implications for leukodystrophy pathology. Glia 2013, 61, 765–777. [Google Scholar] [CrossRef]
- Mohri, I.; Taniike, M.; Taniguchi, H.; Kanekiyo, T.; Aritake, K.; Inui, T.; Fukumoto, N.; Eguchi, N.; Kushi, A.; Sasai, H.; et al. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J. Neurosci. 2006, 26, 4383–4393. [Google Scholar] [CrossRef]
- Hanefeld, F.; Holzbach, U.; Kruse, B.; Wilichowski, E.; Christen, H.J.; Frahm, J. Diffuse white matter disease in three children: an encephalopathy with unique features on magnetic resonance imaging and proton magnetic resonance spectroscopy. Neuropediatrics 1993, 24, 244–248. [Google Scholar] [CrossRef]
- Schiffmann, R.; Moller, J.R.; Trapp, B.D.; Shih, H.H.; Farrer, R.G.; Katz, D.A.; Alger, J.R.; Parker, C.C.; Hauer, P.E.; Kaneski, C.R.; et al. Childhood ataxia with diffuse central nervous system hypomyelination. Ann. Neurol. 1994, 35, 331–340. [Google Scholar] [CrossRef]
- Van Der Knaap, M.S.; Kamphorst, W.; Barth, P.G.; Kraaijeveld, C.L.; Gut, E.; Valk, J. Phenotypic variation in leukoencephalopathy with vanishing white matter. Neurology 1998, 51, 540–547. [Google Scholar] [CrossRef]
- Van der Knaap, M.S.; Leegwater, P.A.; Konst, A.A.; Visser, A.; Naidu, S.; Oudejans, C.B.; Schutgens, R.B.; Pronk, J.C. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann. Neurol. 2002, 51, 264–270. [Google Scholar] [CrossRef]
- Leegwater, P.A.; Vermeulen, G.; Konst, A.A.; Naidu, S.; Mulders, J.; Visser, A.; Kersbergen, P.; Mobach, D.; Fonds, D.; van Berkel, C.G.; et al. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat. Genet. 2001, 29, 383–388. [Google Scholar] [CrossRef]
- Dietrich, J.; Lacagnina, M.; Gass, D.; Richfield, E.; Mayer-Proschel, M.; Noble, M.; Torres, C.; Proschel, C. EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nat. Med. 2005, 11, 277–283. [Google Scholar]
- Van Der Knaap, M.S.; Pronk, J.C.; Scheper, G.C. Vanishing white matter disease. Lancet Neurol. 2006, 5, 413–423. [Google Scholar] [CrossRef]
- Campbell, I.L.; Hofer, M.J.; Pagenstecher, A. Transgenic models for cytokine-induced neurological disease. Biochim. Biophys. Acta 2010, 1802, 903–917. [Google Scholar] [CrossRef]
- Zimmermann, J.; Krauthausen, M.; Hofer, M.J.; Heneka, M.T.; Campbell, I.L.; Muller, M. CNS-targeted production of IL-17A induces glial activation, microvascular pathology and enhances the neuroinflammatory response to systemic endotoxemia. PLoS One 2013, 8, e57307. [Google Scholar]
- Pagenstecher, A.; Lassmann, S.; Carson, M.J.; Kincaid, C.L.; Stalder, A.K.; Campbell, I.L. Astrocyte-targeted expression of IL-12 induces active cellular immune responses in the central nervous system and modulates experimental allergic encephalomyelitis. J. Immunol. 2000, 164, 4481–4492. [Google Scholar]
- Boztug, K.; Carson, M.J.; Pham-Mitchell, N.; Asensio, V.C.; DeMartino, J.; Campbell, I.L. Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J. Immunol. 2002, 169, 1505–1515. [Google Scholar]
- Quintana, A.; Muller, M.; Frausto, R.F.; Ramos, R.; Getts, D.R.; Sanz, E.; Hofer, M.J.; Krauthausen, M.; King, N.J.; Hidalgo, J.; Campbell, I.L. Site-specific production of IL-6 in the central nervous system retargets and enhances the inflammatory response in experimental autoimmune encephalomyelitis. J. Immunol. 2009, 183, 2079–2088. [Google Scholar] [CrossRef]
- Freude, S.; Hausmann, J.; Hofer, M.; Pham-Mitchell, N.; Campbell, I.L.; Staeheli, P.; Pagenstecher, A. Borna disease virus accelerates inflammation and disease associated with transgenic expression of interleukin-12 in the central nervous system. J. Virol. 2002, 76, 12223–12232. [Google Scholar] [CrossRef]
- Greco, T.M.; Seeholzer, S.H.; Mak, A.; Spruce, L.; Ischiropoulos, H. Quantitative mass spectrometry-based proteomics reveals the dynamic range of primary mouse astrocyte protein secretion. J. Proteome Res. 2010, 9, 2764–2774. [Google Scholar] [CrossRef]
- Jha, M.K.; Seo, M.; Kim, J.H.; Kim, B.G.; Cho, J.Y.; Suk, K. The secretome signature of reactive glial cells and its pathological implications. Biochim. Biophys. Acta 2012. [Google Scholar] [CrossRef]
- Davies, S.J.; Shih, C.H.; Noble, M.; Mayer-Proschel, M.; Davies, J.E.; Proschel, C. Transplantation of specific human astrocytes promotes functional recovery after spinal cord injury. PLoS One 2011, 6, e17328. [Google Scholar]
- Noble, M.; Davies, J.E.; Mayer-Proschel, M.; Proschel, C.; Davies, S.J. Precursor cell biology and the development of astrocyte transplantation therapies: Lessons from spinal cord injury. Neurotherapeutics 2011, 8, 677–693. [Google Scholar] [CrossRef]
- Han, X.; Chen, M.; Wang, F.; Windrem, M.; Wang, S.; Shanz, S.; Xu, Q.; Oberheim, N.A.; Bekar, L.; Betstadt, S.; et al. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 2013, 12, 342–353. [Google Scholar] [CrossRef]
- Papadeas, S.T.; Kraig, S.E.; O’Banion, C.; Lepore, A.C.; Maragakis, N.J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17803–17808. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Claycomb, K.I.; Johnson, K.M.; Winokur, P.N.; Sacino, A.V.; Crocker, S.J. Astrocyte Regulation of CNS Inflammation and Remyelination. Brain Sci. 2013, 3, 1109-1127. https://doi.org/10.3390/brainsci3031109
Claycomb KI, Johnson KM, Winokur PN, Sacino AV, Crocker SJ. Astrocyte Regulation of CNS Inflammation and Remyelination. Brain Sciences. 2013; 3(3):1109-1127. https://doi.org/10.3390/brainsci3031109
Chicago/Turabian StyleClaycomb, Kumiko I., Kasey M. Johnson, Paige N. Winokur, Anthony V. Sacino, and Stephen J. Crocker. 2013. "Astrocyte Regulation of CNS Inflammation and Remyelination" Brain Sciences 3, no. 3: 1109-1127. https://doi.org/10.3390/brainsci3031109