Genetic Deletion of Prostacyclin IP Receptor Exacerbates Transient Global Cerebral Ischemia in Aging Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. No Change in Cerebral Blood Flow after Global Cerebral Ischemia in Young and Aged WT and IP KO Mice
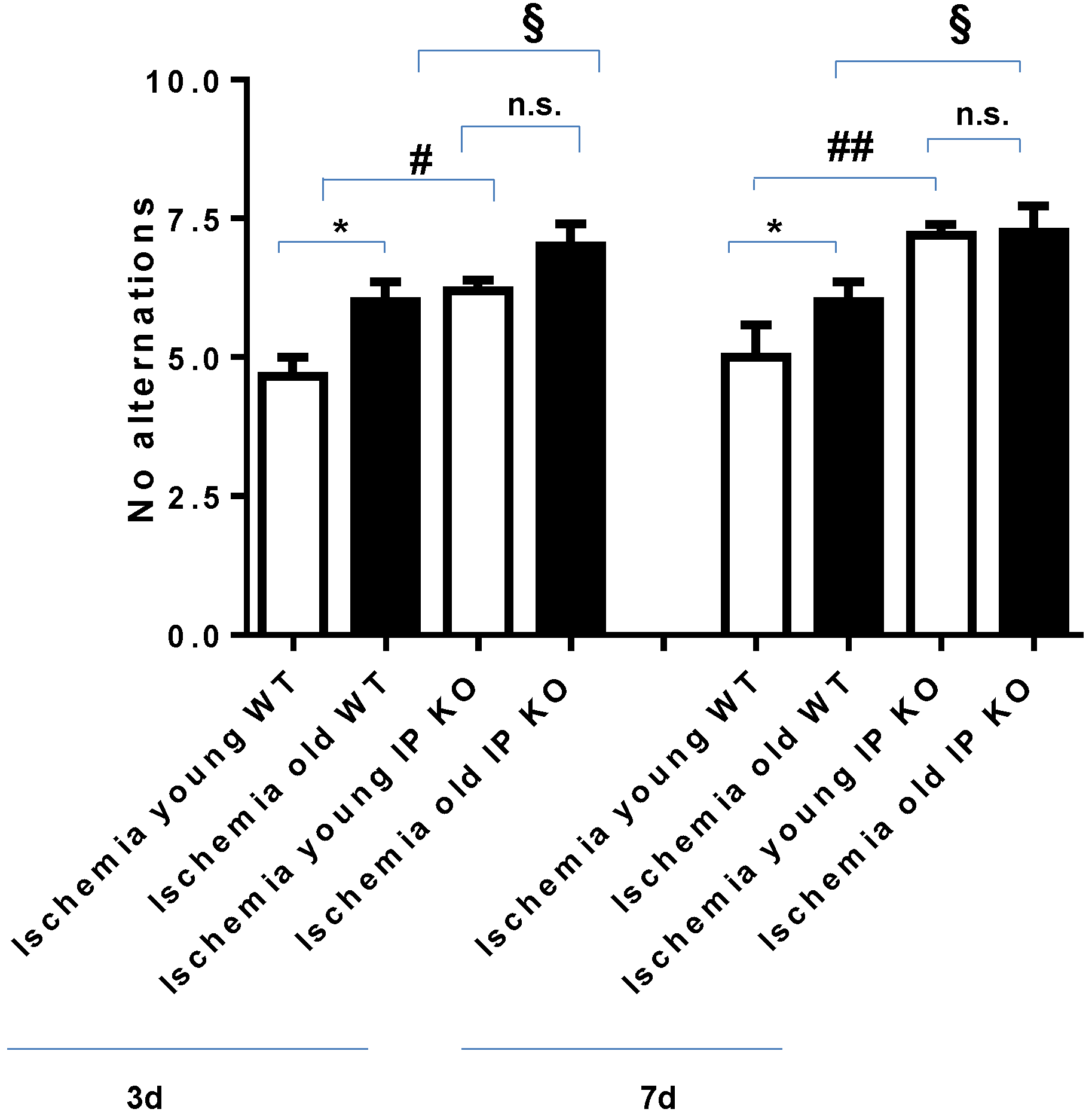
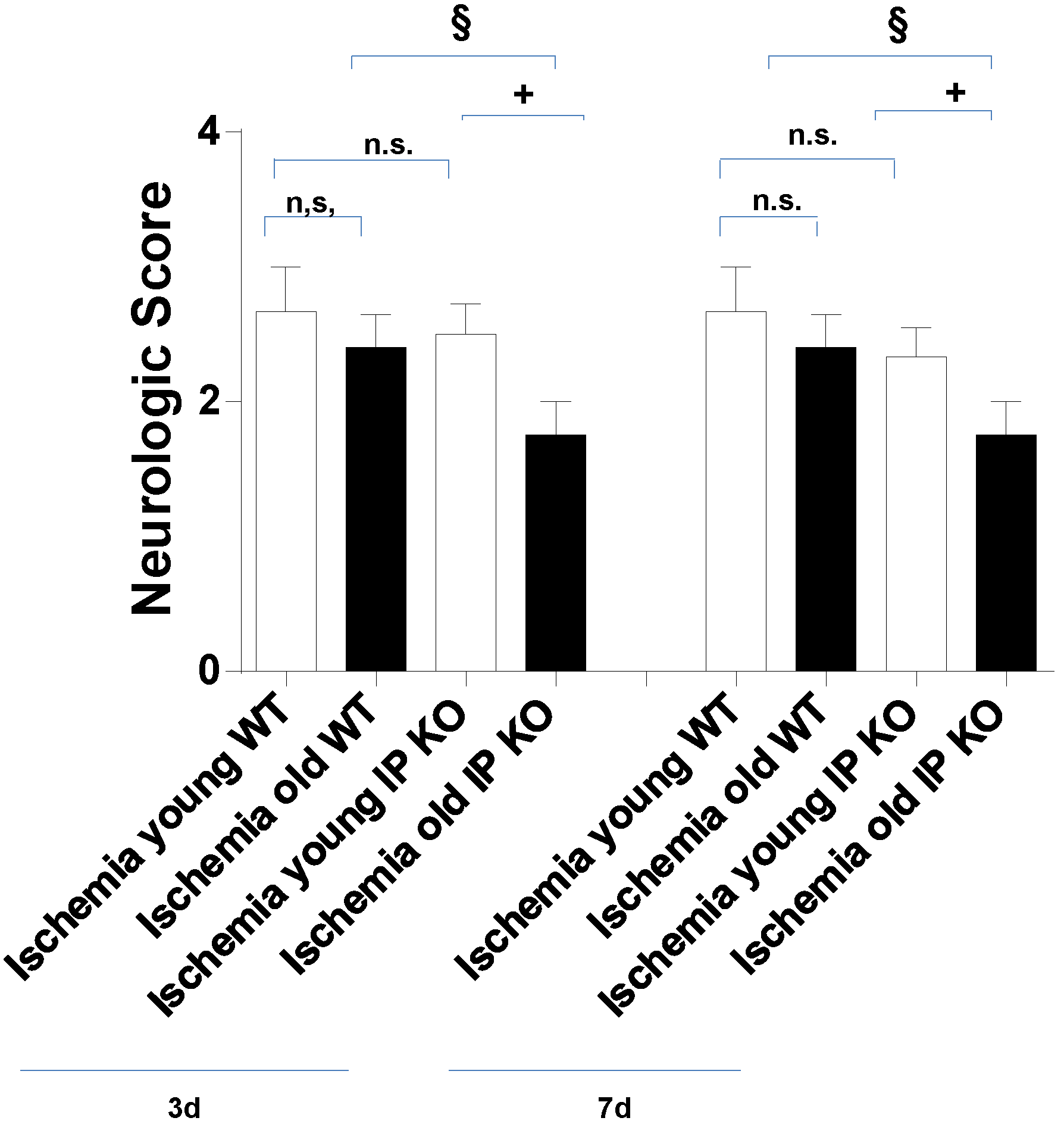
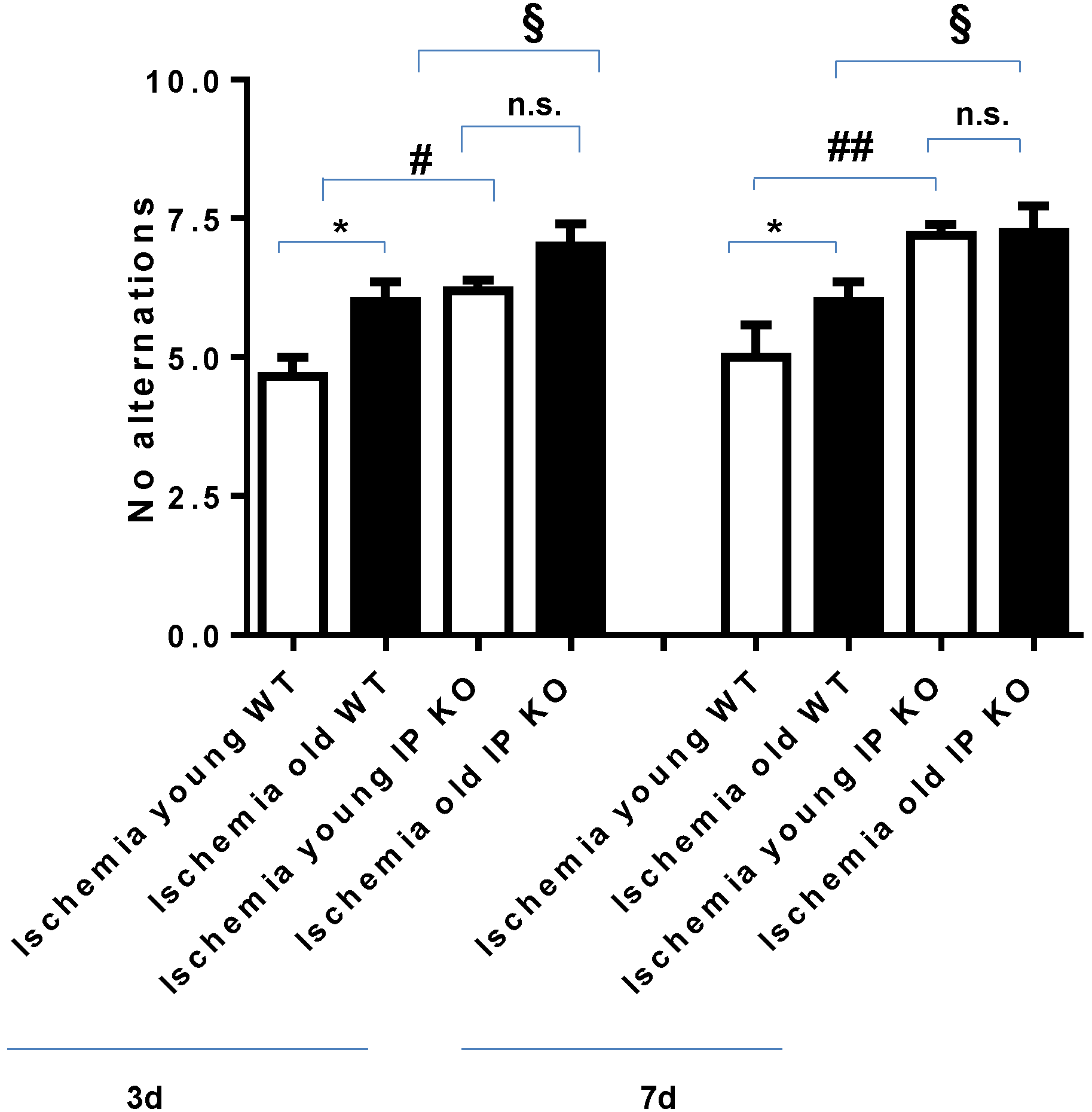
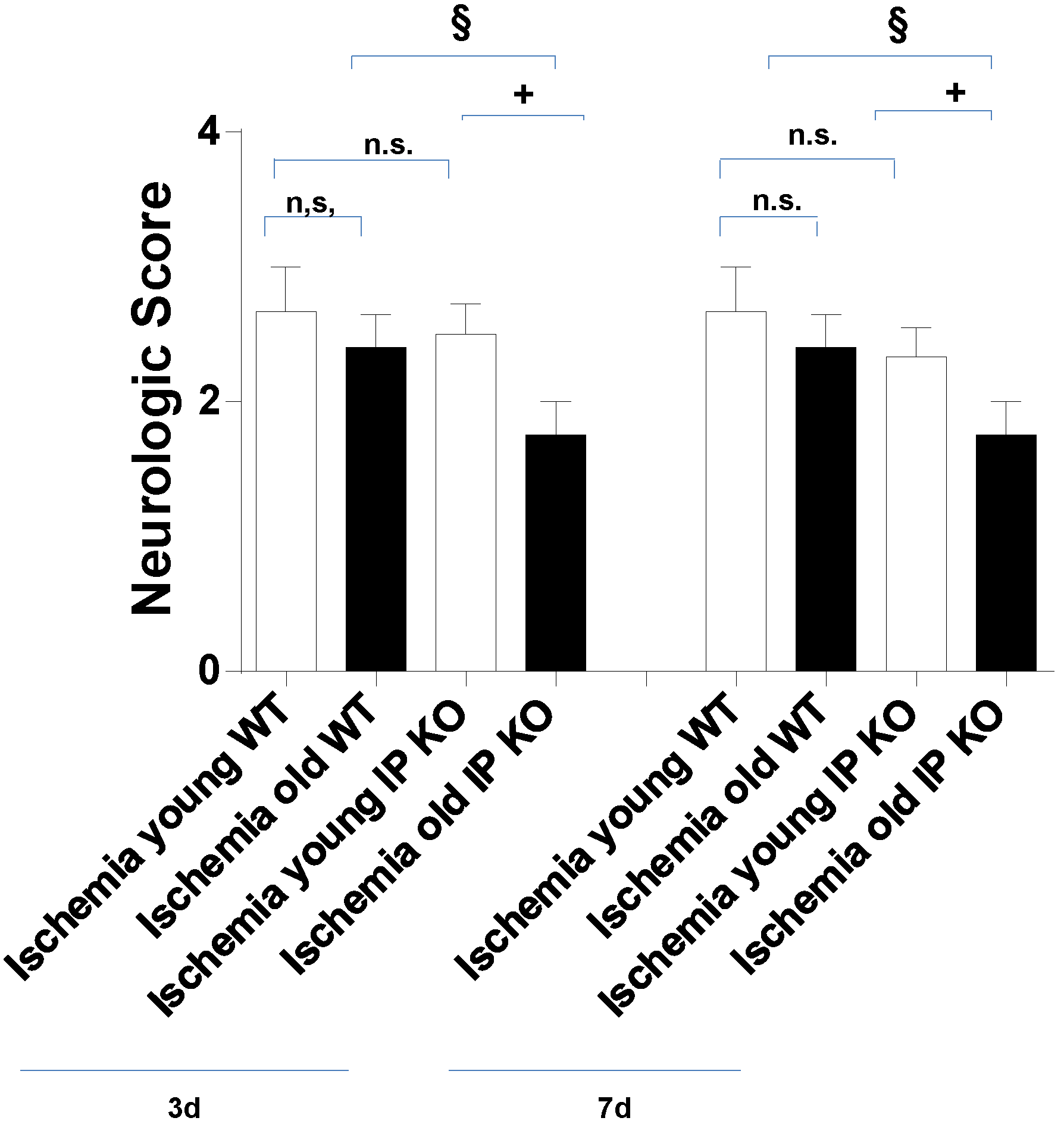
2.2. Cognitive Deficiency and Motor Dysfunction Induced by Global Cerebral Ischemia Were Enhanced in Young and Aged IP KO Mice
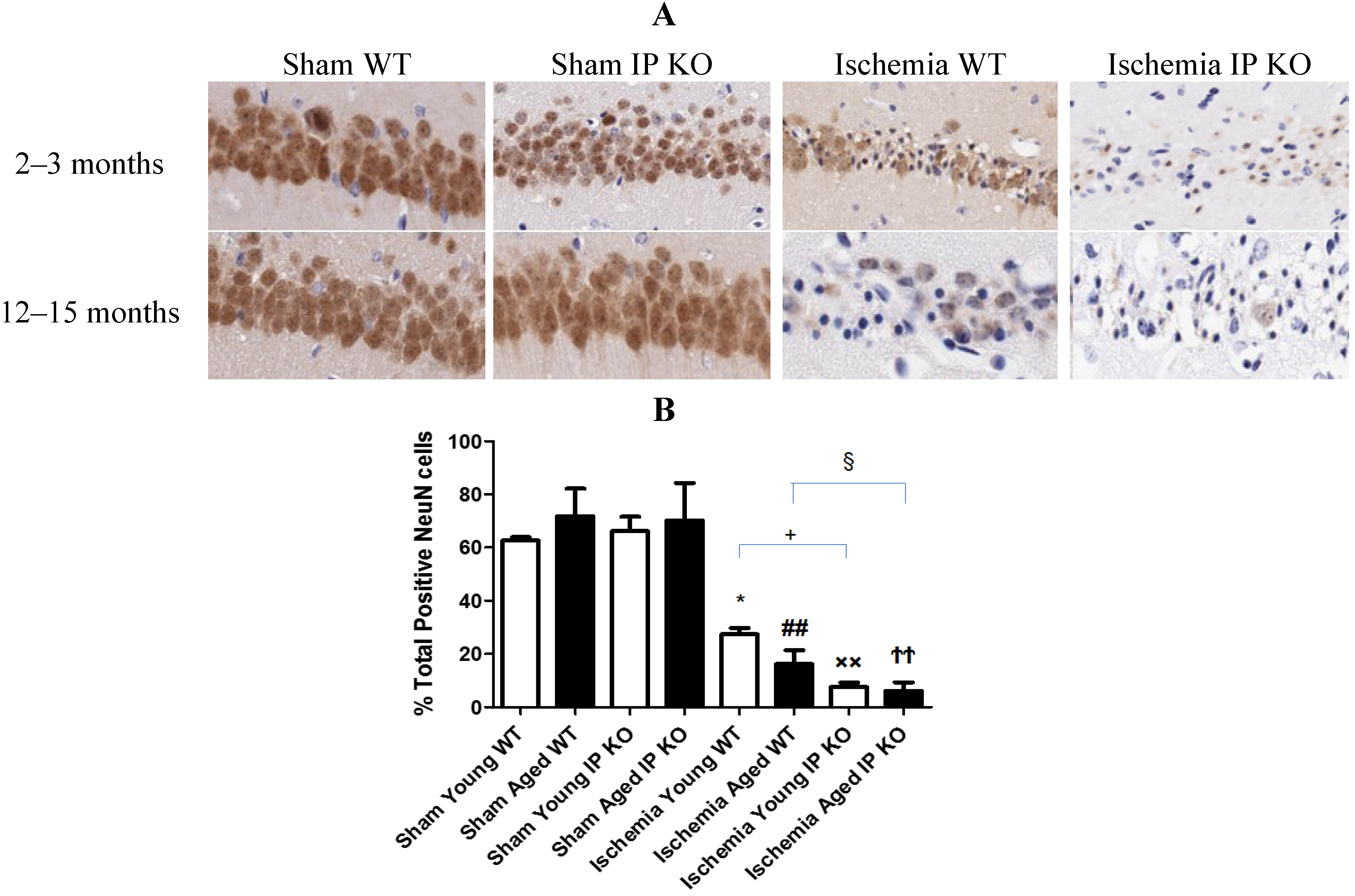
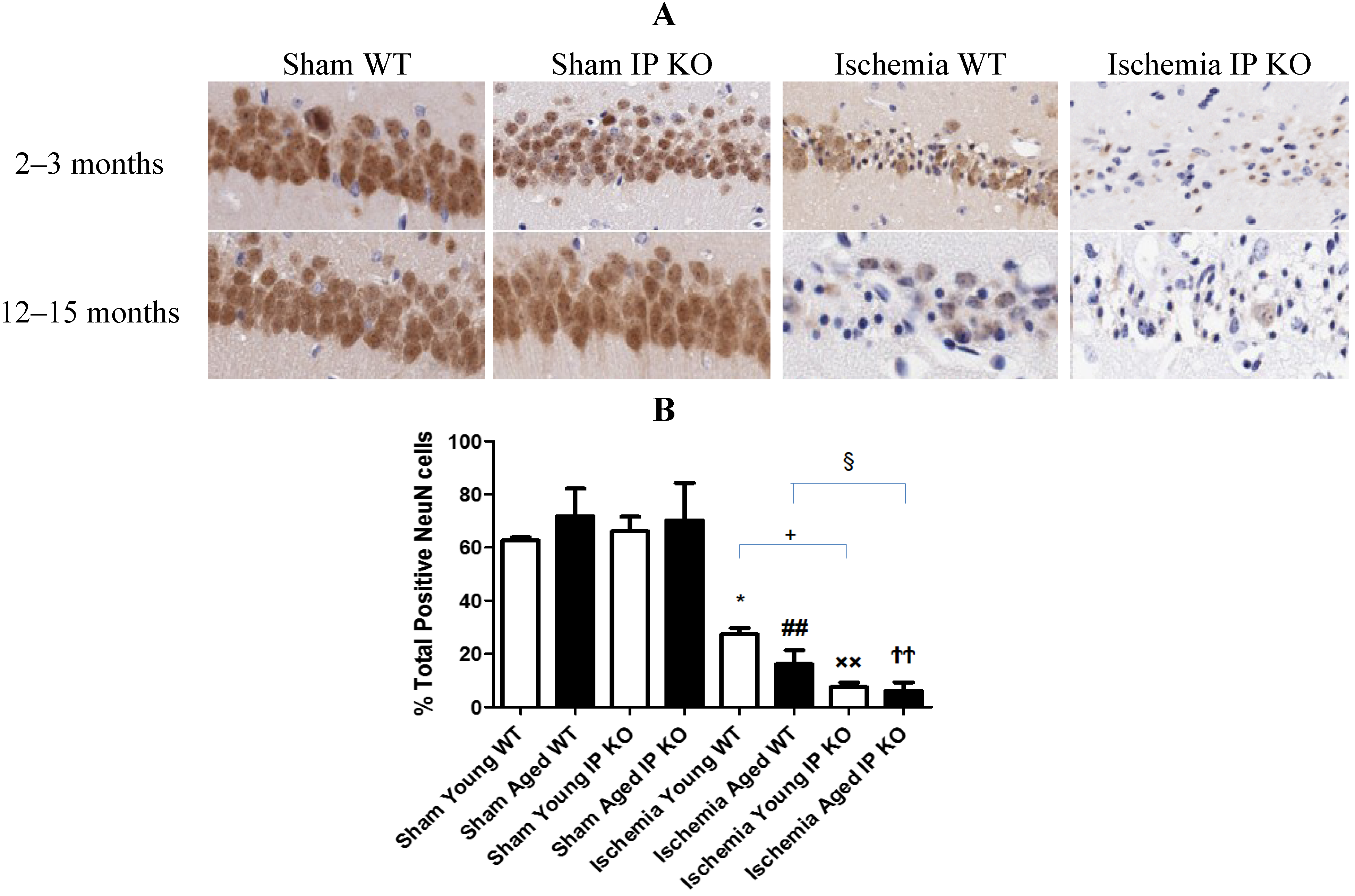
2.3. Genetic Deletion of IP Receptor Enhanced Hippocampal CA1 Neuronal Cell Loss in Young and Aged IP KO Mice
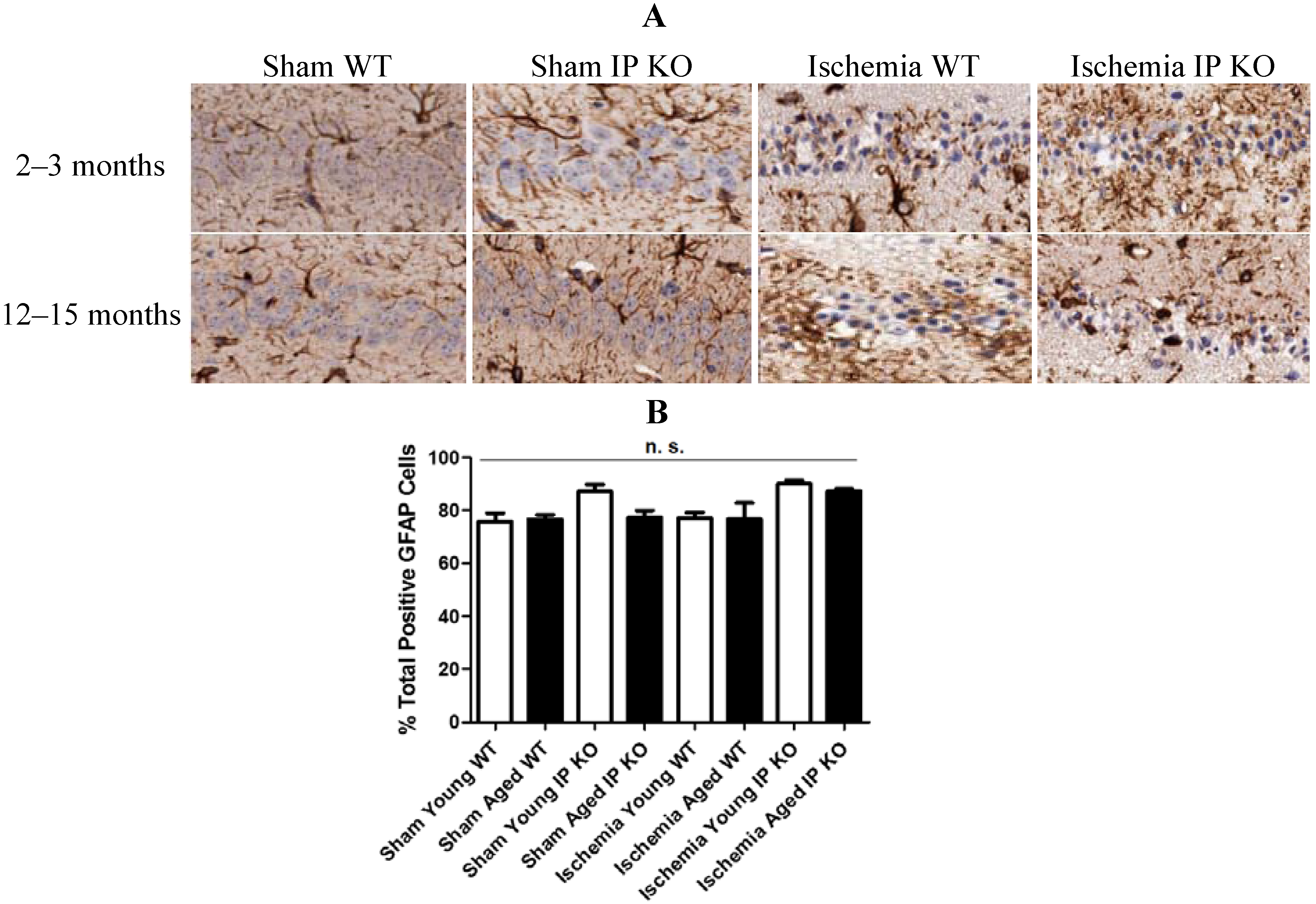
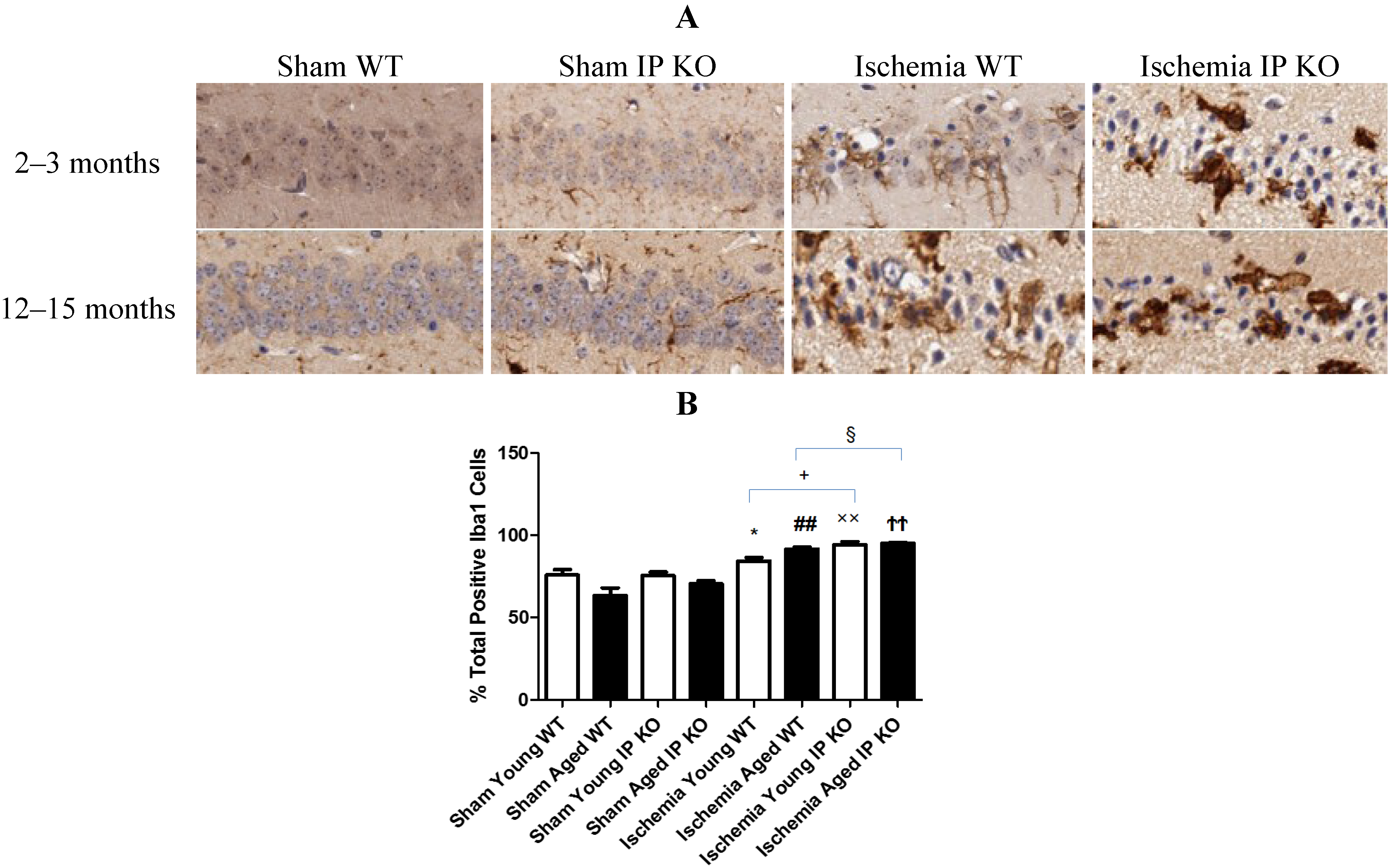
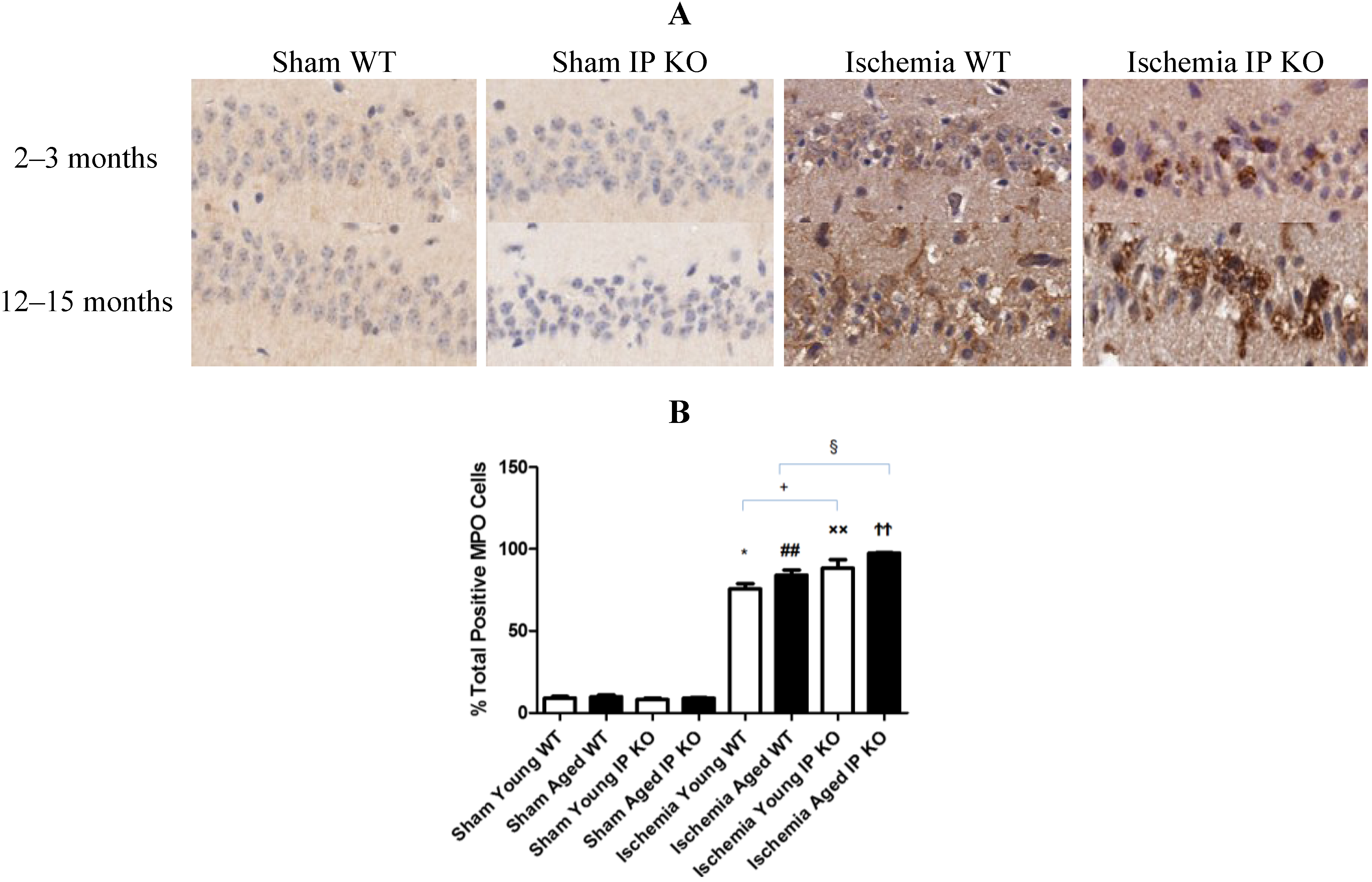
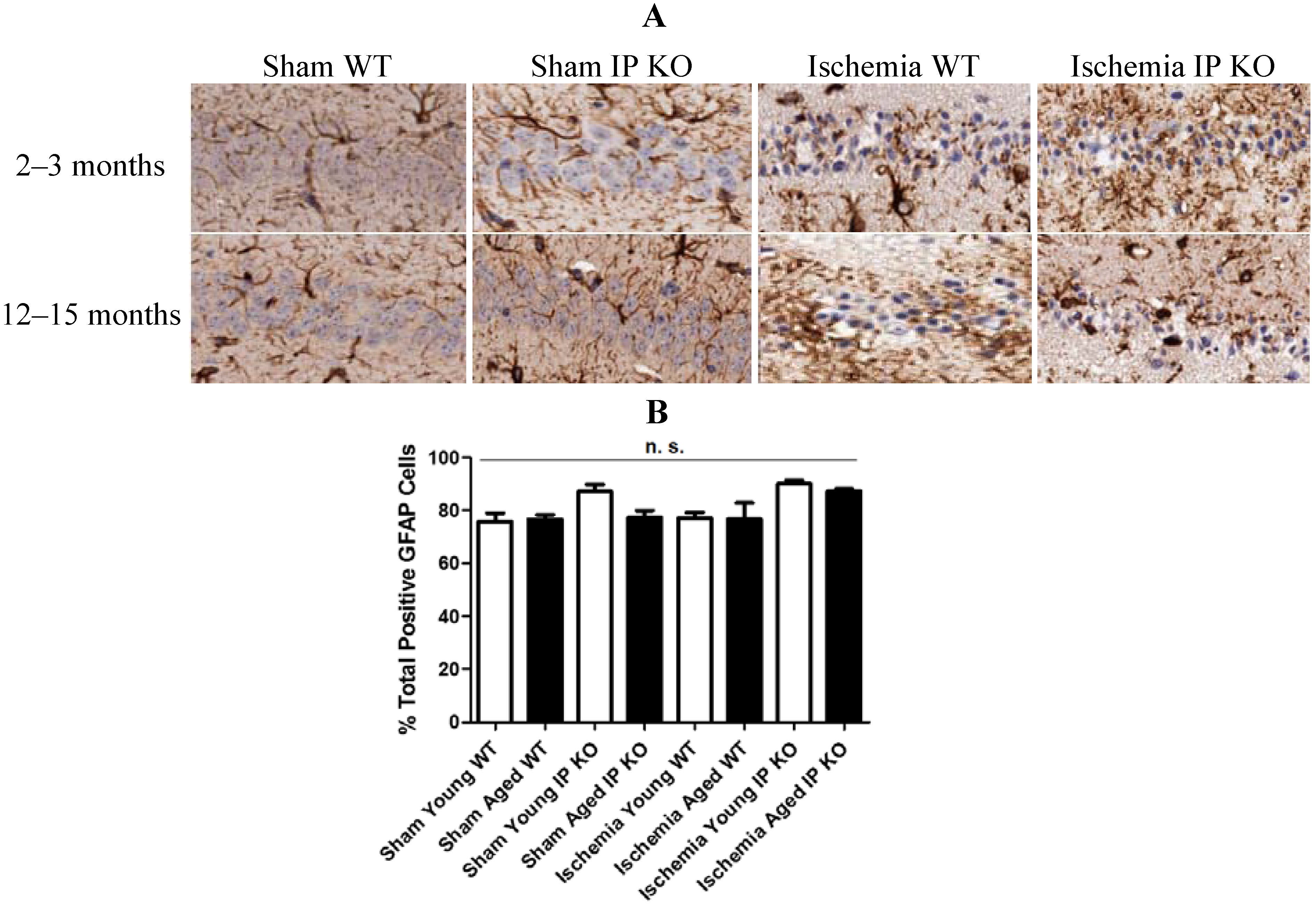
2.4. Genetic Deletion of IP Receptor Exacerbated Microglia Activation and Neutrophil Infiltration in Young and Aged IP KO Mice


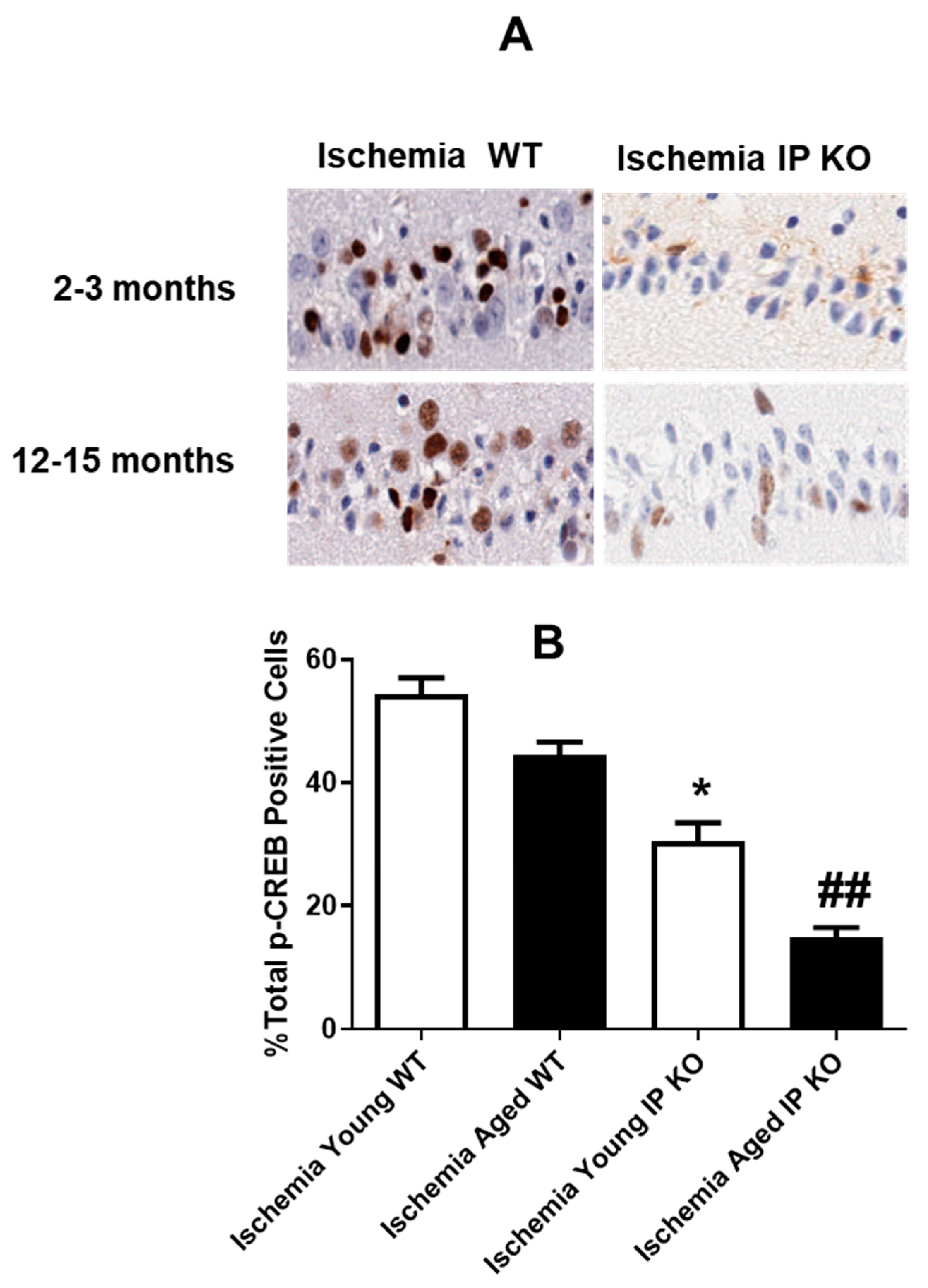
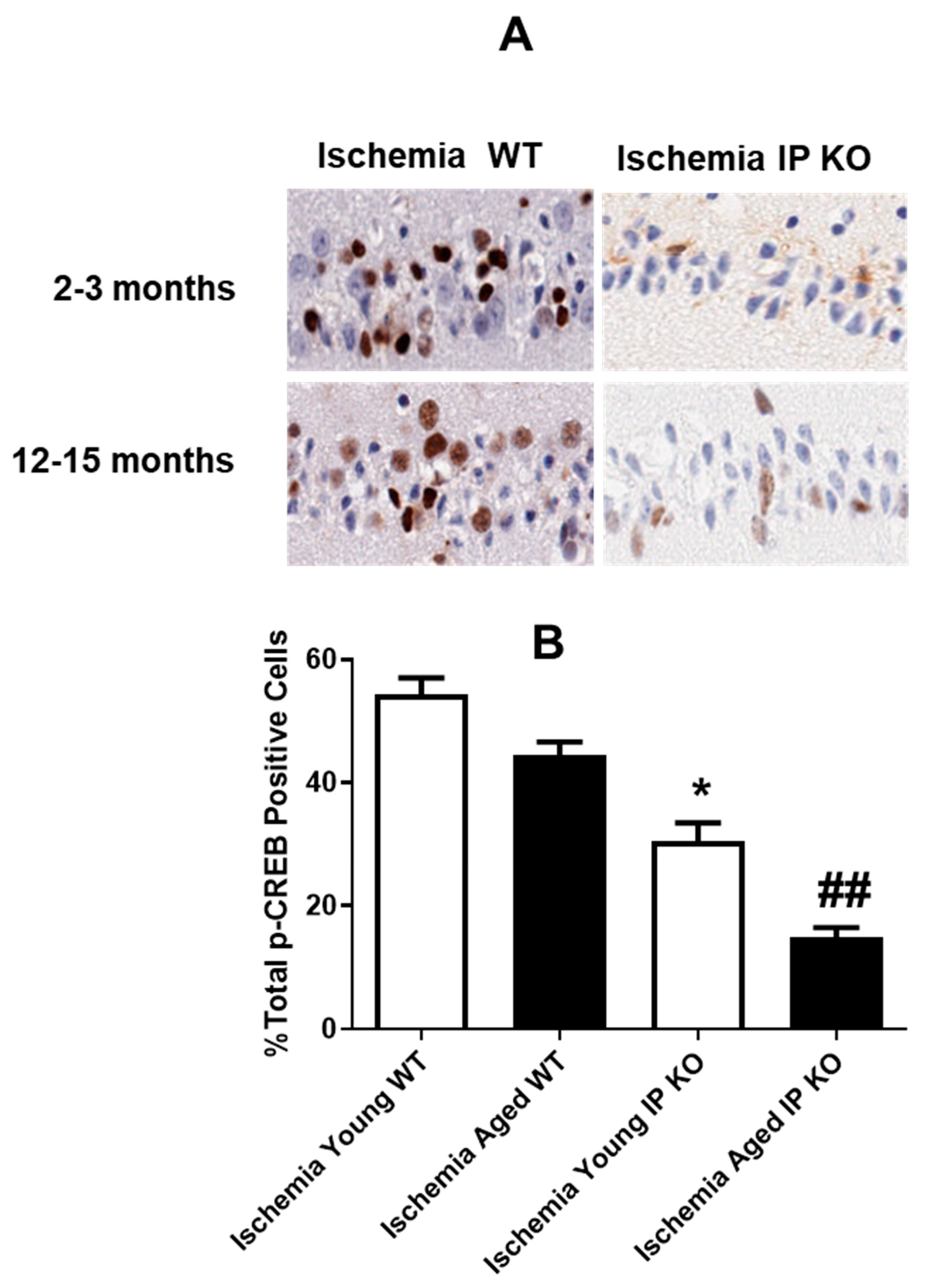
2.5. PGI2 IP Receptor Regulates CREB Phosphorylation after Global Cerebral Ischemia
3. Discussion
4. Experimental Section
4.1. Experimental Animals
4.2. Experimental Groups
4.3. Induction of Global Cerebral Ischemia: BCCAO
4.4. T-Maze Spontaneous Alternation
4.5. Open Field Test
4.6. Immunohistochemistry and Quantification
4.7. Statistical Analysis
5. Conclusions
Acknowledgements
Conflict of Interest
References
- Peskine, A.; Picq, C.; Pradat-Diehl, P. Cerebral anoxia and disability. Brain Inj. 2004, 18, 1243–1254. [Google Scholar] [CrossRef]
- Yager, J.; Andersen, A.E. Clinical practice. Anorexia nervosa. N. Engl. J. Med. 2005, 353, 1481–1488. [Google Scholar] [CrossRef]
- Ritter, L.; Funk, J.; Schenkel, L.; Tipton, A.; Downey, K.; Wilson, J.; Coull, B.; McDonagh, P. Inflammatory and hemodynamic changes in the cerebral microcirculation of aged rats after global cerebral ischemia and reperfusion. Microcirculation 2008, 15, 297–310. [Google Scholar] [CrossRef]
- Neumar, R.W. Molecular mechanisms of ischemic neuronal injury. Ann. Emerg. Med. 2000, 36, 483–506. [Google Scholar]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef]
- Woodward, D.F.; Jones, R.L.; Narumiya, S. International union of basic and clinical pharmacology. LXXXIII: Classification of prostanoid receptors, updating 15 years of progress. Pharmacol. Rev. 2011, 63, 471–538. [Google Scholar]
- Kobayashi, T.; Narumiya, S. Prostanoids in health and disease; lessons from receptor-knockout mice. Adv. Exp. Med. Biol. 2002, 507, 593–597. [Google Scholar] [CrossRef]
- Dorris, S.L.; Peebles, R.S., Jr. PGI2 as a regulator of inflammatory diseases. Mediators Inflamm. 2012, 2012. 926968. [Google Scholar]
- Anwaar, I.; Gottsater, A.; Ohlsson, K.; Mattiasson, I.; Lindgarde, F. Increasing levels of leukocyte-derived inflammatory mediators in plasma and camp in platelets during follow-up after acute cerebral ischemia. Cerebrovasc. Dis. 1998, 8, 310–317. [Google Scholar] [CrossRef]
- Masuda, Y.; Yasuba, M.; Zushi, K.; Ochi, Y.; Kadokawa, T.; Okegawa, T. Effect of OP-2507, a stable prostacyclin analogue on cerebral ischemia induced by unilateral ligation of common carotid artery in gerbils. Arch. Int. Pharmacodyn. Ther. 1988, 294, 125–136. [Google Scholar]
- Gryglewski, R.J.; Nowak, S.; Kostka-Trabka, E.; Kusmiderski, J.; Dembinska-Kiec, A.; Bieron, K.; Basista, M.; Blaszczyk, B. Treatment of ischaemic stroke with prostacyclin. Stroke 1983, 14, 197–202. [Google Scholar] [CrossRef]
- Lundblad, C.; Grande, P.O.; Bentzer, P. Increased cortical cell loss and prolonged hemodynamic depression after traumatic brain injury in mice lacking the IP receptor for prostacyclin. J. Cereb. Blood Flow Metab. 2008, 28, 367–376. [Google Scholar] [CrossRef]
- Saleem, S.; Shah, Z.A.; Maruyama, T.; Narumiya, S.; Dore, S. Neuroprotective properties of prostaglandin I2 IP receptor in focal cerebral ischemia. Neuroscience 2010, 170, 317–323. [Google Scholar] [CrossRef]
- Chan, E.C.; Dusting, G.J.; Guo, N.; Peshavariya, H.M.; Taylor, C.J.; Dilley, R.; Narumiya, S.; Jiang, F. Prostacyclin receptor suppresses cardiac fibrosis: Role of CREB phosphorylation. J. Mol. Cell. Cardiol. 2010, 49, 176–185. [Google Scholar] [CrossRef]
- Moncada, D.; Viola, H. Phosphorylation state of creb in the rat hippocampus: A molecular switch between spatial novelty and spatial familiarity? Neurobiol. Learn. Mem. 2006, 86, 9–18. [Google Scholar] [CrossRef]
- Guzowski, J.F.; McGaugh, J.L. Antisense oligodeoxynucleotide-mediated disruption of hippocampal camp response element binding protein levels impairs consolidation of memory for water maze training. Proc. Natl. Acad. Sci. USA 1997, 94, 2693–2698. [Google Scholar] [CrossRef]
- Harada, N.; Taoka, Y.; Okajima, K. Role of prostacyclin in the development of compression trauma-induced spinal cord injury in rats. J. Neurotrauma 2006, 23, 1739–1749. [Google Scholar] [CrossRef]
- Sato, Y.; Kitani, K.; Kanai, S.; Nokubo, M.; Ohta, M. Differences in tolerance to hypoxia/anoxia in mice of different ages. Res. Commun. Chem. Pathol. Pharmacol. 1991, 73, 209–220. [Google Scholar]
- Hoth, K.F.; Poppas, A.; Moser, D.J.; Paul, R.H.; Cohen, R.A. Cardiac dysfunction and cognition in older adults with heart failure. Cogn. Behav. Neurol. 2008, 21, 65–72. [Google Scholar] [CrossRef]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets 2012, 14, 20–35. [Google Scholar] [CrossRef]
- Danton, G.H.; Dietrich, W.D. Inflammatory mechanisms after ischemia and stroke. J. Neuropathol. Exp. Neurol. 2003, 62, 127–136. [Google Scholar]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Xiao, F. Bench to bedside: Brain edema and cerebral resuscitation: The present and future. Acad. Emerg. Med. 2002, 9, 933–946. [Google Scholar] [CrossRef]
- Breckwoldt, M.O.; Chen, J.W.; Stangenberg, L.; Aikawa, E.; Rodriguez, E.; Qiu, S.; Moskowitz, M.A.; Weissleder, R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Natl. Acad. Sci. USA 2008, 105, 18584–18589. [Google Scholar] [CrossRef]
- Jang, S.; Dilger, R.N.; Johnson, R.W. Luteolin inhibits microglia and alters hippocampal-dependent spatial working memory in aged mice. J. Nutr. 2010, 140, 1892–1898. [Google Scholar] [CrossRef]
- McGettigan, P.; Henry, D. Current problems with non-specific cox inhibitors. Curr. Pharm. Des. 2000, 6, 1693–1724. [Google Scholar] [CrossRef]
- Garcia Rodriguez, L.A.; Egan, K.; FitzGerald, G.A. Traditional nonsteroidal anti-inflammatory drugs and postmenopausal hormone therapy: A drug-drug interaction? PLoS Med. 2007, 4, e157. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- Montminy, M.R.; Gonzalez, G.A.; Yamamoto, K.K. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990, 13, 184–188. [Google Scholar] [CrossRef]
- Gonzalez, G.A.; Montminy, M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989, 59, 675–680. [Google Scholar] [CrossRef]
- Yiu, A.P.; Rashid, A.J.; Josselyn, S.A. Increasing CREB function in the CA1 region of dorsal hippocampus rescues the spatial memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2011, 36, 2169–2186. [Google Scholar] [CrossRef]
- Mizuno, M.; Yamada, K.; Maekawa, N.; Saito, K.; Seishima, M.; Nabeshima, T. CREB phosphorylation as a molecular marker of memory processing in the hippocampus for spatial learning. Behav. Brain Res. 2002, 133, 135–141. [Google Scholar] [CrossRef]
- Zhou, Y.; Won, J.; Karlsson, M.G.; Zhou, M.; Rogerson, T.; Balaji, J.; Neve, R.; Poirazi, P.; Silva, A.J. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat. Neurosci. 2009, 12, 1438–1443. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Shioda, N.; Han, F.; Moriguchi, S.; Nakajima, A.; Yokosuka, A.; Mimaki, Y.; Sashida, Y.; Yamakuni, T.; Ohizumi, Y.; et al. Nobiletin improves brain ischemia-induced learning and memory deficits through stimulation of camkii and CREB phosphorylation. Brain Res. 2009, 1295, 218–229. [Google Scholar]
- Chen, Y.; Huang, X.; Zhang, Y.W.; Rockenstein, E.; Bu, G.; Golde, T.E.; Masliah, E.; Xu, H. Alzheimer’s beta-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of beta-amyloid. J. Neurosci. 2012, 32, 11390–11395. [Google Scholar] [CrossRef]
- Kim, D.H.; Jeon, S.J.; Son, K.H.; Jung, J.W.; Lee, S.; Yoon, B.H.; Choi, J.W.; Cheong, J.H.; Ko, K.H.; Ryu, J.H. Effect of the flavonoid, oroxylin a, on transient cerebral hypoperfusion-induced memory impairment in mice. Pharmacol. Biochem. Behav. 2006, 85, 658–668. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tahara, Y.; Matsumoto, M.; Iguchi, M.; Sano, H.; Murayama, T.; Arai, H.; Oida, H.; Yurugi-Kobayashi, T.; Yamashita, J.K.; et al. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J. Clin. Invest. 2004, 114, 784–794. [Google Scholar]
- Olsson, T.; Wieloch, T.; Smith, M.L. Brain damage in a mouse model of global cerebral ischemia. Effect of NMDA receptor blockade. Brain Res. 2003, 982, 260–269. [Google Scholar] [CrossRef]
- Matchett, G.A.; Calinisan, J.B.; Matchett, G.C.; Martin, R.D.; Zhang, J.H. The effect of granulocyte-colony stimulating factor in global cerebral ischemia in rats. Brain Res. 2007, 1136, 200–207. [Google Scholar]
- Krajewska, M.; You, Z.; Rong, J.; Kress, C.; Huang, X.; Yang, J.; Kyoda, T.; Leyva, R.; Banares, S.; Hu, Y.; et al. Neuronal deletion of caspase 8 protects against brain injury in mouse models of controlled cortical impact and kainic acid-induced excitotoxicity. PLoS One 2011, 6, e24341. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shakil, H.; Saleem, S. Genetic Deletion of Prostacyclin IP Receptor Exacerbates Transient Global Cerebral Ischemia in Aging Mice. Brain Sci. 2013, 3, 1095-1108. https://doi.org/10.3390/brainsci3031095
Shakil H, Saleem S. Genetic Deletion of Prostacyclin IP Receptor Exacerbates Transient Global Cerebral Ischemia in Aging Mice. Brain Sciences. 2013; 3(3):1095-1108. https://doi.org/10.3390/brainsci3031095
Chicago/Turabian StyleShakil, Hania, and Sofiyan Saleem. 2013. "Genetic Deletion of Prostacyclin IP Receptor Exacerbates Transient Global Cerebral Ischemia in Aging Mice" Brain Sciences 3, no. 3: 1095-1108. https://doi.org/10.3390/brainsci3031095