Effects of Ethanol Exposure during Distinct Periods of Brain Development on Hippocampal Synaptic Plasticity


 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Effects of Ethanol Exposure on Body Weight and Litter Size

{kind=link}
{kind=link}
{kind=link}
| % weight gain during pregnancy | Number of pups per litter | |
|---|---|---|
| Ad libitum | 61.6 ± 11.3 | 13.7 ± 2.3 |
| Ethanol 1st | 74.4 ± 3.8 | 12.8 ± 1.3 |
| Pair-fed 1st | 66.3 ± 9.3 | 18 ± 1.7 * |
| Ethanol 2nd | 51.1 ± 3.2 | 15.3 ± 0.5 |
| Pair-fed 2nd | 63.1 ± 4.3 | 13.0 ± 0.6 |
| Ethanol 3rd | 65.4 ± 4.3 | 14.0 ± 0.8 |
| Pair-fed 3rd | 69.7 ± 5.2 | 15.6 ± 0.2 |
| Ad libitum | Ethanol-exposed | Pair-fed | |||||
|---|---|---|---|---|---|---|---|
| Weight (g) | 1st | 2nd | 3rd | 1st | 2nd | 3rd | |
| PND2 | 8.0 ± 0.3 | 8.3 ± 0.3 | 7.5 ± 0.2 | 8.1 ± 0.2 | 6.9 ± 0.4 * | 7.8 ± 0.2 | 7.7 ± 0.2 |
| PND5 | 15.0 ± 0.5 | 14.6 ± 0.4 | 14.5 ± 0.4 | 14.9 ± 0.4 | 13.6 ± 0.4 | 14.4 ± 0.4 | 14.6 ± 0.4 |
| PND10 | 23.6 ± 0.8 | 23.3 ± 0.6 | 23.2 ± 0.7 | 24.0 ± 0.7 | 22.2 ± 0.7 | 23.0 ± 0.7 | 24.8 ± 0.6 |
| PND14 | 33.3 ± 1.1 | 32.8 ± 0.9 | 33.4 ± 0.9 | 34.2 ± 1.0 | 32.1 ± 0.9 | 32.0 ± 0.9 | 35.8 ± 0.9 |
| PND18 | 43.7 ± 1.4 | 41.8 ± 1.2 | 43.2 ± 1.2 | 45.1 ± 1.5 | 41.2 ± 1.2 # | 41.0 ± 1.2 # | 47.2 ± 1.3 |
| PND22 | 60.4 ± 1.9 | 60.4 ± 1.7 | 59.8 ± 1.7 | 63.3 ± 1.9 | 54.7±1.7 $$ | 59.1±1.7 $$ | 67.3 ± 1.7 |
| Weight (g) | Male | Female | |
|---|---|---|---|
| Ad libitum | -- | 413.1 ± 6.2 | 271.5 ± 14.3 |
| Ethanol-exposed | 1st | 417.5 ± 5.2 | 281.1 ± 11.4 |
| 2nd | 400.0 ± 7.0 | 267.0 ± 6.1 | |
| 3rd | 408.4 ± 4.7 | 257.1 ± 5.4 | |
| Pair-fed | 1st | 413.0 ± 1.6 | 263.1 ± 3.0 |
| 2nd | 413.2 ± 3.4 | 269.7 ± 4.0 | |
| 3rd | 421.6 ± 4.1 | 272.1 ± 5.7 |
2.2. Intoxication Levels
2.3. Effects of Ethanol Exposure on Basal Synaptic Transmission and Post-Tetanic Potentiation
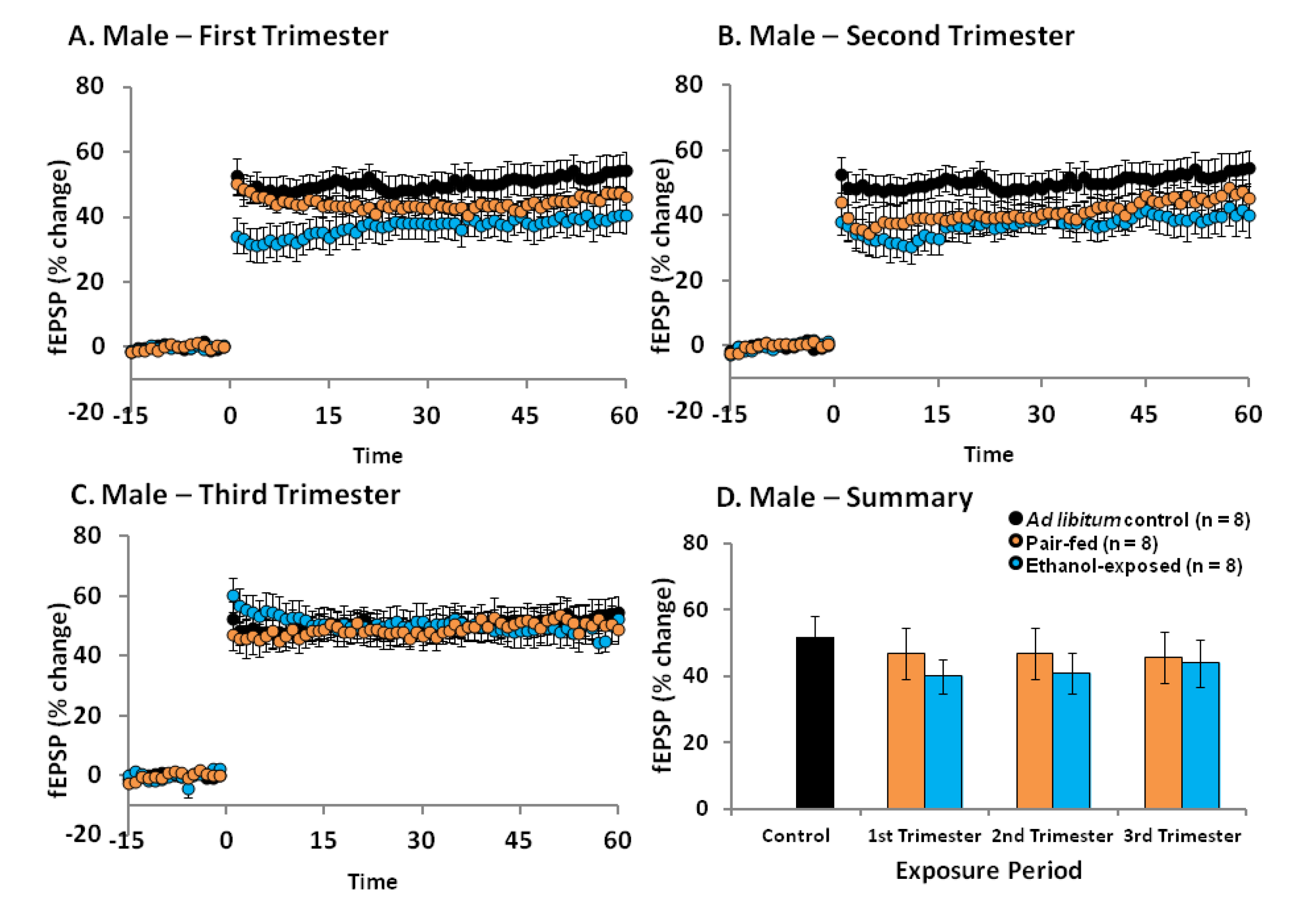
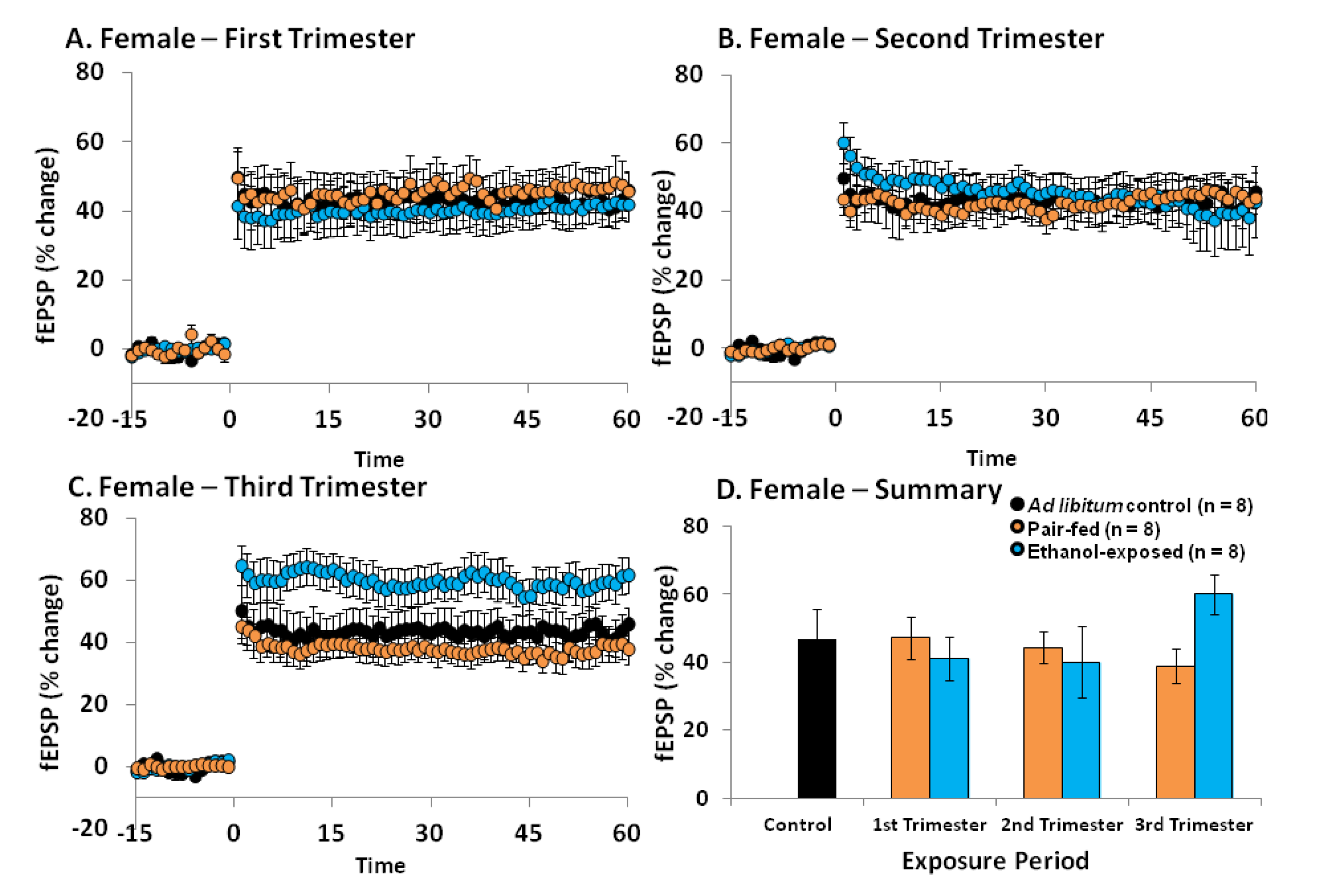
2.4. Effects of Ethanol Exposure on Young Adult DG LTP
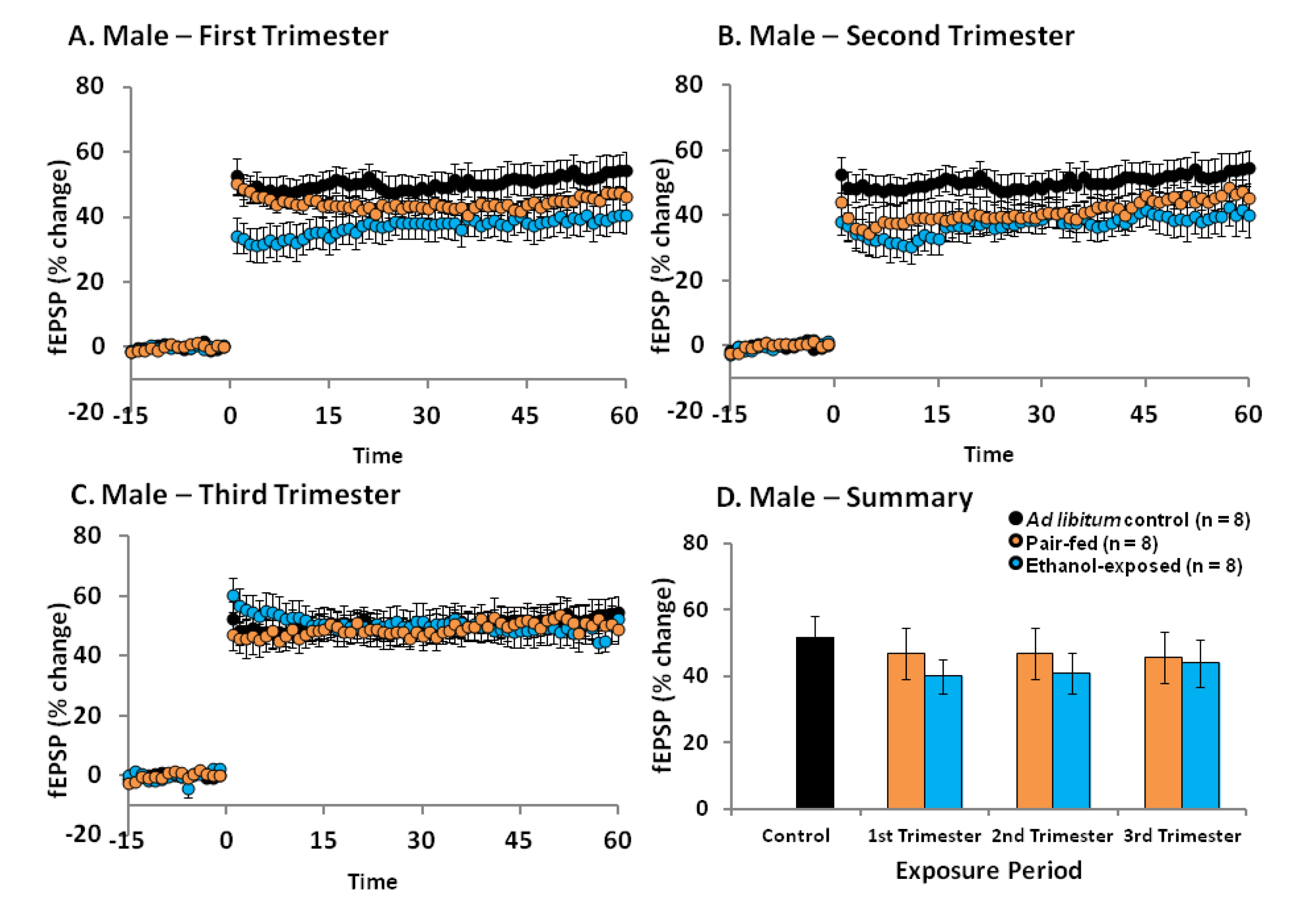

2.5. Discussion
3. Experimental Section
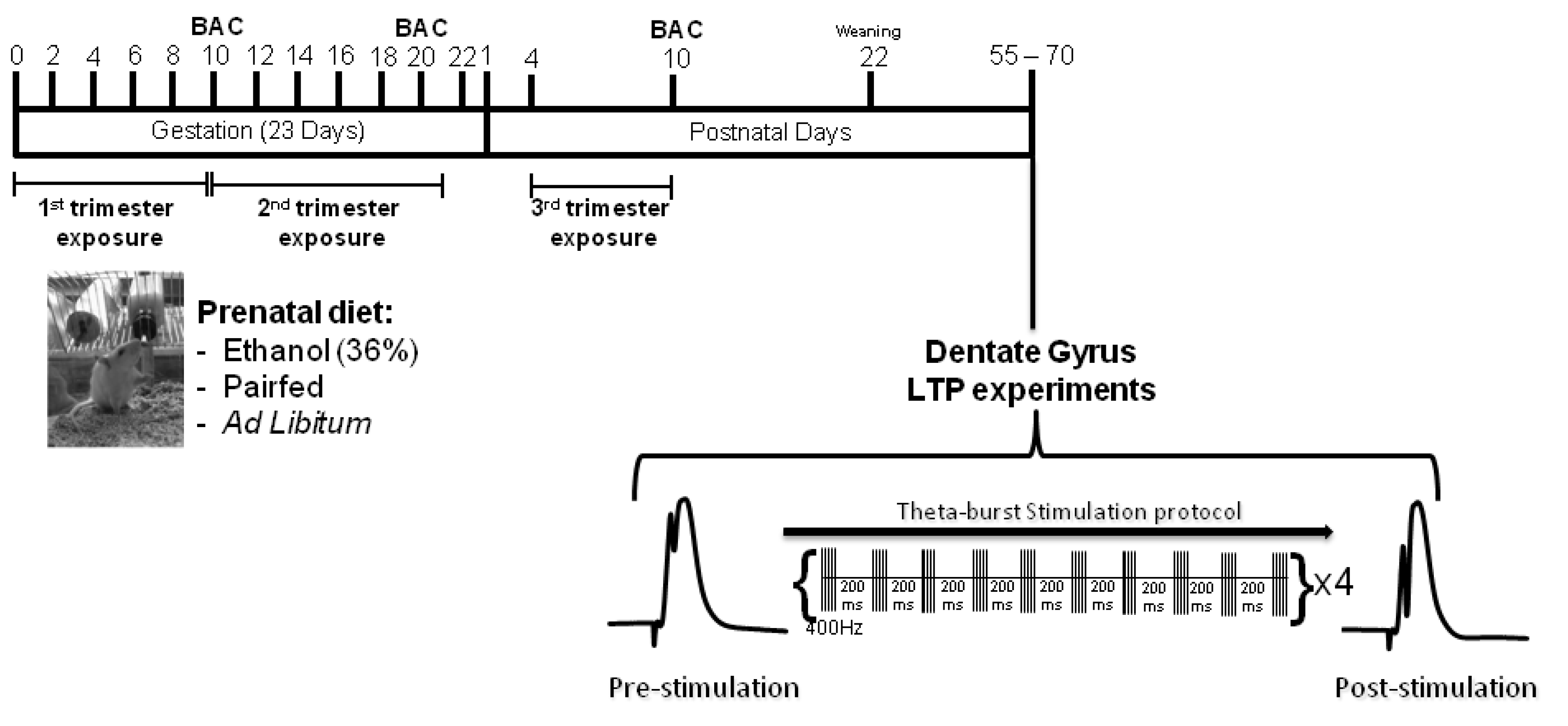
3.1. Animals and Breeding

3.2. Prenatal Diets
3.3. Blood Alcohol Concentration Assay
3.4. Litters and Weaning
3.5. In Vivo Electrophysiology
3.6. Statistical Analyses
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Sokol, R.J.; Delaney-Black, V.; Nordstrom, B. Fetal alcohol spectrum disorder. JAMA 2003, 290, 2996–2999. [Google Scholar] [CrossRef]
- Jones, K.L. The fetal alcohol syndrome. Addict. Dis. 1975, 2, 79–88. [Google Scholar]
- May, P.A.; Gossage, J.P.; Kalberg, W.O.; Robinson, L.K.; Buckley, D.; Manning, M.; Hoyme, H.E. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev. Disabil. Res. Rev. 2009, 15, 176–192. [Google Scholar] [CrossRef]
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 302, 999–1001. [Google Scholar] [CrossRef]
- Bliss, T.V.; Lomo, T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 1973, 232, 331–356. [Google Scholar]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef]
- West, J.R.; Hodges, C.A.; Black, A.C., Jr. Prenatal exposure to ethanol alters the organization of hippocampal mossy fibers in rats. Science 1981, 211, 957–959. [Google Scholar]
- Gabriel, K.I.; Johnston, S.; Weinberg, J. Prenatal ethanol exposure and spatial navigation: Effects of postnatal handling and aging. Dev. Psychobiol. 2002, 40, 345–357. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Peterson, S.D. Sex differences in vulnerability to developmental spatial learning deficits induced by limited binge alcohol exposure in neonatal rats. Neurobiol. Learn. Mem. 1995, 64, 265–275. [Google Scholar] [CrossRef]
- Kelly, S.J.; Goodlett, C.R.; Hulsether, S.A.; West, J.R. Impaired spatial navigation in adult female but not adult male rats exposed to alcohol during the brain growth spurt. Behav. Brain Res. 1988, 27, 247–257. [Google Scholar] [CrossRef]
- Thomas, J.D.; Wasserman, E.A.; West, J.R.; Goodlett, C.R. Behavioral deficits induced by bingelike exposure to alcohol in neonatal rats: importance of developmental timing and number of episodes. Dev. Psychobiol. 1996, 29, 433–452. [Google Scholar] [CrossRef]
- Christie, B.R.; Swann, S.E.; Fox, C.J.; Froc, D.; Lieblich, S.E.; Redila, V.; Webber, A. Voluntary exercise rescues deficits in spatial memory and long-term potentiation in prenatal ethanol-exposed male rats. Eur. J. Neurosci. 2005, 21, 1719–1726. [Google Scholar] [CrossRef]
- Richardson, D.P.; Byrnes, M.L.; Brien, J.F.; Reynolds, J.N.; Dringenberg, H.C. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur. J. Neurosci. 2002, 16, 1593–1598. [Google Scholar] [CrossRef]
- Blanchard, B.A.; Riley, E.P.; Hannigan, J.H. Deficits on a spatial navigation task following prenatal exposure to ethanol. Neurotoxicol. Teratol. 1987, 9, 253–258. [Google Scholar] [CrossRef]
- Popovic, M.; Caballero-Bleda, M.; Guerri, C. Adult rat’s offspring of alcoholic mothers are impaired on spatial learning and object recognition in the Can test. Behav. Brain Res. 2006, 174, 101–111. [Google Scholar] [CrossRef]
- Wozniak, D.F.; Hartman, R.E.; Boyle, M.P.; Vogt, S.K.; Brooks, A.R.; Tenkova, T.; Young, C.; Olney, J.W.; Muglia, L.J. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis. 2004, 17, 403–414. [Google Scholar] [CrossRef]
- Malisza, K.L.; Allman, A.A.; Shiloff, D.; Jakobson, L.; Longstaffe, S.; Chudley, A.E. Evaluation of spatial working memory function in children and adults with fetal alcohol spectrum disorders: A functional magnetic resonance imaging study. Pediatr. Res. 2005, 58, 1150–1157. [Google Scholar] [CrossRef]
- Sowell, E.R.; Lu, L.H.; O’Hare, E.D.; McCourt, S.T.; Mattson, S.N.; O’Connor, M.J.; Bookheimer, S.Y. Functional magnetic resonance imaging of verbal learning in children with heavy prenatal alcohol exposure. Neuroreport 2007, 18, 635–639. [Google Scholar] [CrossRef]
- Willoughby, K.A.; Sheard, E.D.; Nash, K.; Rovet, J. Effects of prenatal alcohol exposure on hippocampal volume, verbal learning, and verbal and spatial recall in late childhood. J. Int. Neuropsychol. Soc. 2008, 14, 1022–1033. [Google Scholar] [CrossRef]
- Mattson, S.N.; Riley, E.P. Implicit and explicit memory functioning in children with heavy prenatal alcohol exposure. J. Int. Neuropsychol. Soc. 1999, 5, 462–471. [Google Scholar]
- Titterness, A.K.; Christie, B.R. Prenatal ethanol exposure enhances NMDAR-dependent long-term potentiation in the adolescent female dentate gyrus. Hippocampus 2012, 22, 69–81. [Google Scholar] [CrossRef]
- Sickmann, H.; Patten, A.R.; Morch, K.; Sawchuk, S.; Zhang, C.; Parton, R.; Svlavik, L.; Christie, B.R. Gender-differences in hippocampal long-term potentiation in adult rats following prenatal ethanol exposure. Hippocampus 2013. submitted for publication. [Google Scholar]
- Sutherland, R.J.; McDonald, R.J.; Savage, D.D. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus 1997, 7, 232–238. [Google Scholar] [CrossRef]
- Brady, M.L.; Diaz, M.R.; Iuso, A.; Everett, J.C.; Valenzuela, C.F.; Caldwell, K.K. Moderate Prenatal Alcohol Exposure Reduces Plasticity and Alters NMDA Receptor Subunit Composition in the Dentate Gyrus. J. Neurosci. 2013, 33, 1062–1067. [Google Scholar] [CrossRef]
- Varaschin, R.K.; Akers, K.G.; Rosenberg, M.J.; Hamilton, D.A.; Savage, D.D. Effects of the cognition-enhancing agent ABT-239 on fetal ethanol-induced deficits in dentate gyrus synaptic plasticity. J. Pharmacol. Exp. Ther. 2010, 334, 191–198. [Google Scholar] [CrossRef]
- Patten, A.; Sickmann, H.M.; Dyer, R.; Innis, S.M.; Christie, B.R. Omega-3 Fatty Acids Reverse the Long-Term Deficits in Hippocampal Synaptic Plasticity Caused by Prenatal Ethanol Exposure. Neurosci. Lett. 2013. accepted for publication. [Google Scholar]
- Brodie, C.; Vernadakis, A. Critical periods to ethanol exposure during early neuroembryogenesis in the chick embryo: Cholinergic neurons. Brain Res. Dev. Brain Res. 1990, 56, 223–228. [Google Scholar] [CrossRef]
- Cartwright, M.M.; Smith, S.M. Stage-dependent effects of ethanol on cranial neural crest cell development: Partial basis for the phenotypic variations observed in fetal alcohol syndrome. Alcohol. Clin. Exp. Res. 1995, 19, 1454–1462. [Google Scholar] [CrossRef]
- West, J.R. Fetal alcohol-induced brain damage and the problem of determining temporal vulnerability: A review. Alcohol Drug Res. 1987, 7, 423–441. [Google Scholar]
- Davis, W.L.; Crawford, L.A.; Cooper, O.J.; Farmer, G.R.; Thomas, D.L.; Freeman, B.L. Ethanol induces the generation of reactive free radicals by neural crest cells in vitro. J. Craniofac. Genet. Dev. Biol. 1990, 10, 277–293. [Google Scholar]
- Brocardo, P.S.; Boehme, F.; Patten, A.; Cox, A.; Gil-Mohapel, J.; Christie, B.R. Anxiety- and depression-like behaviors are accompanied by an increase in oxidative stress in a rat model of fetal alcohol spectrum disorders: Protective effects of voluntary physical exercise. Neuropharmacology 2012, 62, 1607–1618. [Google Scholar]
- Patten, A.R.; Brocardo, P.S.; Christie, B.R. Omega-3 supplementation can restore glutathione levels and prevent oxidative damage caused by prenatal ethanol exposure. J. Nutr. Biochem. 2012, 24, 760–769. [Google Scholar]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Feldman, H.S.; Jones, K.L.; Lindsay, S.; Slymen, D.; Klonoff-Cohen, H.; Kao, K.; Rao, S.; Chambers, C. Prenatal alcohol exposure patterns and alcohol-related birth defects and growth deficiencies: A prospective study. Alcohol. Clin. Exp. Res. 2012, 36, 670–676. [Google Scholar]
- Nava-Ocampo, A.A.; Velazquez-Armenta, Y.; Brien, J.F.; Koren, G. Elimination kinetics of ethanol in pregnant women. Reprod. Toxicol. 2004, 18, 613–617. [Google Scholar]
- Boehme, F.; Gil-Mohapel, J.; Cox, A.; Patten, A.; Giles, E.; Brocardo, P.S.; Christie, B.R. Voluntary exercise induces adult hippocampal neurogenesis and BDNF expression in a rodent model of fetal alcohol spectrum disorders. Eur. J. Neurosci. 2011, 33, 1799–1811. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.; Boehme, F.; Patten, A.; Cox, A.; Kainer, L.; Giles, E.; Brocardo, P.S.; Christie, B.R. Altered adult hippocampal neuronal maturation in a rat model of fetal alcohol syndrome. Brain Res. 2011, 1384, 29–41. [Google Scholar]
- Gil-Mohapel, J.; Boehme, F.; Kainer, L.; Christie, B.R. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: Insights from different rodent models. Brain Res. Rev. 2010, 64, 283–303. [Google Scholar] [CrossRef]
- Tan, S.E.; Berman, R.F.; Abel, E.L.; Zajac, C.S. Prenatal alcohol exposure alters hippocampal slice electrophysiology. Alcohol 1990, 7, 507–511. [Google Scholar]
- Byrnes, M.L.; Richardson, D.P.; Brien, J.F.; Reynolds, J.N.; Dringenberg, H.C. Spatial acquisition in the Morris water maze and hippocampal long-term potentiation in the adult guinea pig following brain growth spurt—prenatal ethanol exposure. Neurotoxicol. Teratol. 2004, 26, 543–551. [Google Scholar] [CrossRef]
- Bellinger, F.P.; Bedi, K.S.; Wilson, P.; Wilce, P.A. Ethanol exposure during the third trimester equivalent results in long-lasting decreased synaptic efficacy but not plasticity in the CA1 region of the rat hippocampus. Synapse 1999, 31, 51–58. [Google Scholar] [CrossRef]
- Cronise, K.; Marino, M.D.; Tran, T.D.; Kelly, S.J. Critical periods for the effects of alcohol exposure on learning in rats. Behav. Neurosci. 2001, 115, 138–145. [Google Scholar] [CrossRef]
- Ethen, M.K.; Ramadhani, T.A.; Scheuerle, A.E.; Canfield, M.A.; Wyszynski, D.F.; Druschel, C.M.; Romitti, P.A. Alcohol consumption by women before and during pregnancy. Matern. Child. Health J. 2009, 13, 274–285. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. 3), 511–533. [Google Scholar]
- Dobbing, J.; Sands, J. Quantitative growth and development of human brain. Arch. Dis. Child. 1973, 48, 757–767. [Google Scholar] [CrossRef]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef]
- Lan, N.; Yamashita, F.; Halpert, A.G.; Sliwowska, J.H.; Viau, V.; Weinberg, J. Effects of prenatal ethanol exposure on hypothalamic-pituitary-adrenal function across the estrous cycle. Alcohol. Clin. Exp. Res. 2009, 33, 1075–1088. [Google Scholar]
- He, F.Q. Prenatal Ethanol Exposure Increases Depressive-Like Behavior and Central Estrogen Receptor α and Oxytocin Expressions in Adult Female Mandarin Voles. Zool. Stud. 2012, 51, 1–11. [Google Scholar]
- Spencer, J.L.; Waters, E.M.; Milner, T.A.; McEwen, B.S. Estrous cycle regulates activation of hippocampal Akt, LIM kinase, and neurotrophin receptors in C57BL/6 mice. Neuroscience 2008, 155, 1106–1119. [Google Scholar] [CrossRef]
- Ooishi, Y.; Kawato, S.; Hojo, Y.; Hatanaka, Y.; Higo, S.; Murakami, G.; Komatsuzaki, Y.; Ogiue-Ikeda, M.; Kimoto, T.; Mukai, H. Modulation of synaptic plasticity in the hippocampus by hippocampus-derived estrogen and androgen. J. Steroid Biochem. Mol. Biol. 2012, 131, 37–51. [Google Scholar] [CrossRef]
- Weniger, J.P.; Zeis, A.; Chouraqui, J. Estrogen production by fetal and infantile rat ovaries. Reprod. Nutr. Dev. 1993, 33, 129–136. [Google Scholar] [CrossRef]
- Vaillend, C.; Billard, J.M.; Laroche, S. Impaired long-term spatial and recognition memory and enhanced CA1 hippocampal LTP in the dystrophin-deficient Dmd(mdx) mouse. Neurobiol. Dis. 2004, 17, 10–20. [Google Scholar] [CrossRef]
- Jeffery, K.J. Paradoxical enhancement of long-term potentiation in poor-learning rats at low test stimulus intensities. Exp. Brain Res. 1995, 104, 55–69. [Google Scholar] [CrossRef]
- Weinberg, J.; Gallo, P.V. Prenatal ethanol exposure: Pituitary-adrenal activity in pregnant dams and offspring. Neurobehav. Toxicol. Teratol. 1982, 4, 515–520. [Google Scholar]
- Weinberg, J. Effects of ethanol and maternal nutritional status on fetal development. Alcohol. Clin. Exp. Res. 1985, 9, 49–55. [Google Scholar] [CrossRef]
- West, J.R.; Dewey, S.L.; Pierce, D.R.; Black, A.C., Jr. Prenatal and early postnatal exposure to ethanol permanently alters the rat hippocampus. Ciba Found. Symp. 1984, 105, 8–25. [Google Scholar]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates: Hard Cover Edition; Academic Press: Waltham, MA, USA, 2006. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Patten, A.R.; Gil-Mohapel, J.; Wortman, R.C.; Noonan, A.; Brocardo, P.S.; Christie, B.R. Effects of Ethanol Exposure during Distinct Periods of Brain Development on Hippocampal Synaptic Plasticity. Brain Sci. 2013, 3, 1076-1094. https://doi.org/10.3390/brainsci3031076
Patten AR, Gil-Mohapel J, Wortman RC, Noonan A, Brocardo PS, Christie BR. Effects of Ethanol Exposure during Distinct Periods of Brain Development on Hippocampal Synaptic Plasticity. Brain Sciences. 2013; 3(3):1076-1094. https://doi.org/10.3390/brainsci3031076
Chicago/Turabian StylePatten, Anna R., Joana Gil-Mohapel, Ryan C. Wortman, Athena Noonan, Patricia S. Brocardo, and Brian R. Christie. 2013. "Effects of Ethanol Exposure during Distinct Periods of Brain Development on Hippocampal Synaptic Plasticity" Brain Sciences 3, no. 3: 1076-1094. https://doi.org/10.3390/brainsci3031076