The Hepatocyte Growth Factor/c-Met Antagonist, Divalinal-Angiotensin IV, Blocks the Acquisition of Methamphetamine Dependent Conditioned Place Preference in Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Objectives and Hypotheses
3. Material and Methods
3.1. Animals
3.2. Surgery
3.2.1. Chronic Divalinal Infusion
3.2.2. Acute Divalinal Infusion
3.2.3. MA Reinstatement
3.3. Apparatus
3.4. Behavioral Training
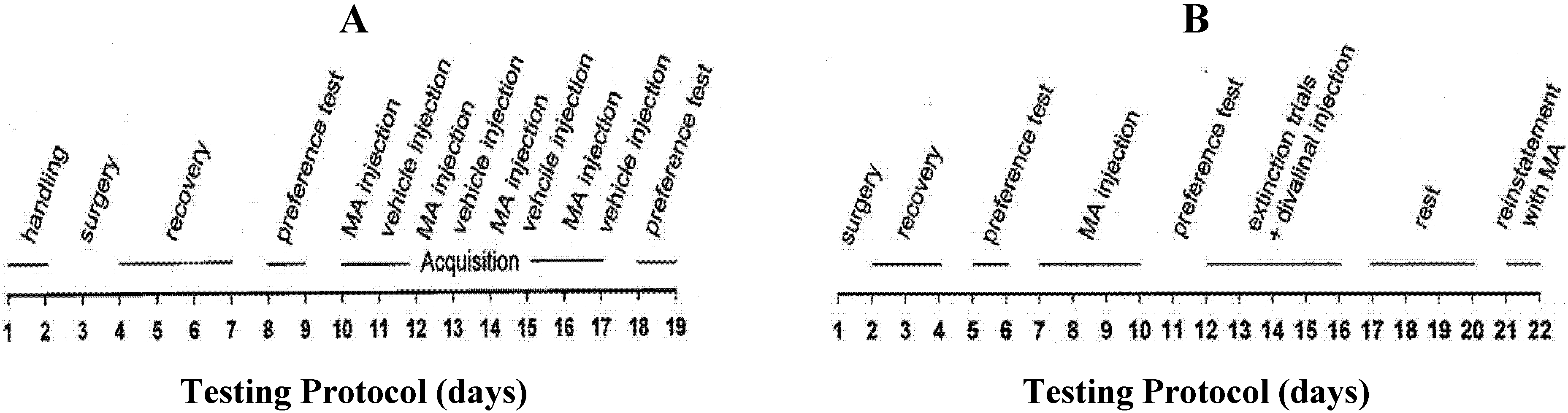
3.4.1. Experiment 1: Chronic Divalinal Infusion
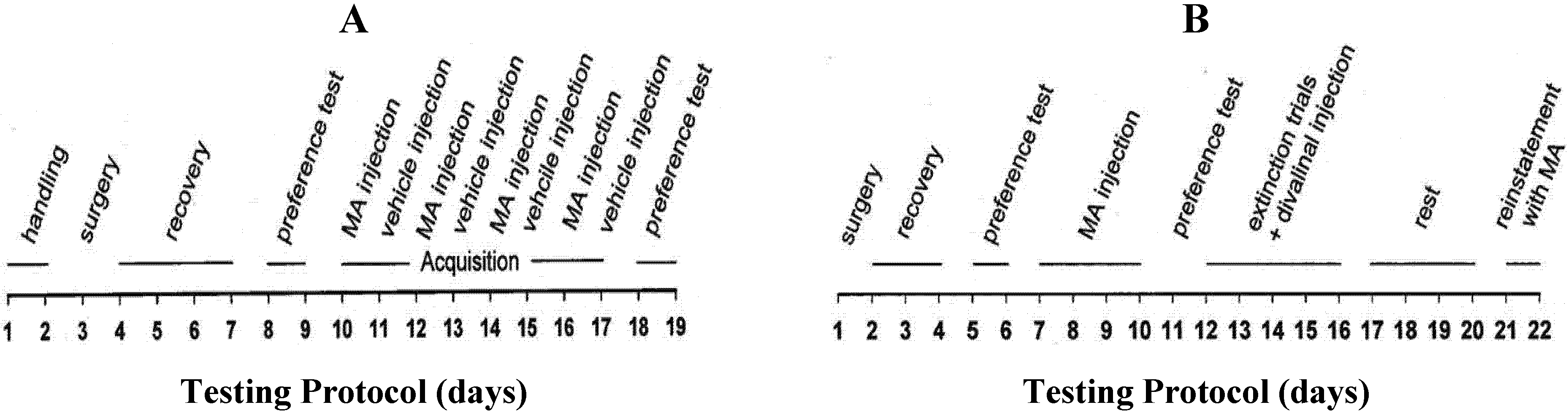
3.4.2. Experiment 2: Acute Divalinal Infusion
3.4.3. Experiment 3: MA Reinstatement
3.5. Phospho-Met Western Blots
3.6. Scattering Assay
3.7. Compounds
3.8. Statistical Analysis

4. Results
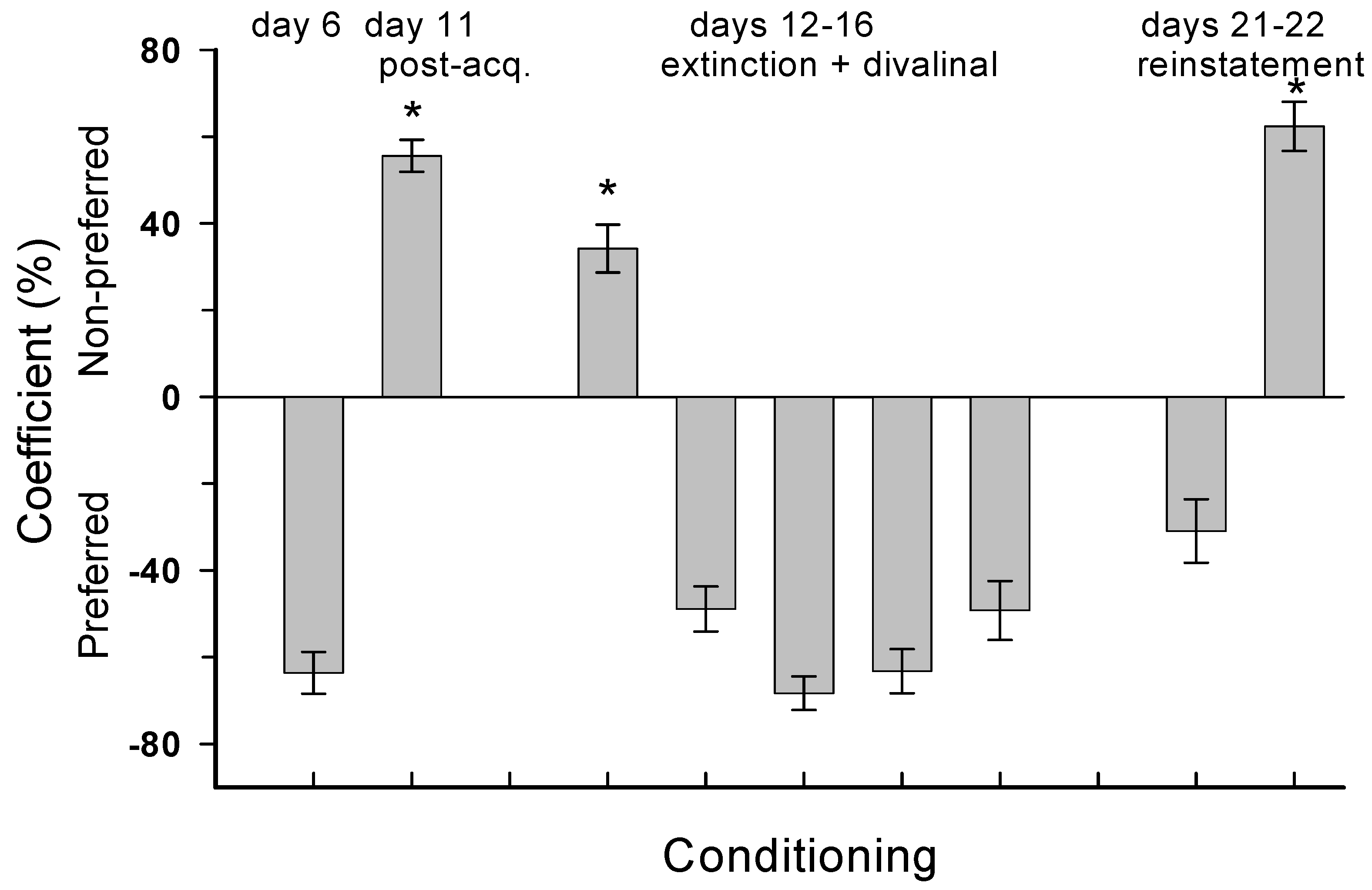
4.1. Experiment 1: Chronic Divalinal Infusion

4.2. Experiment 2: Acute Divalinal Infusion

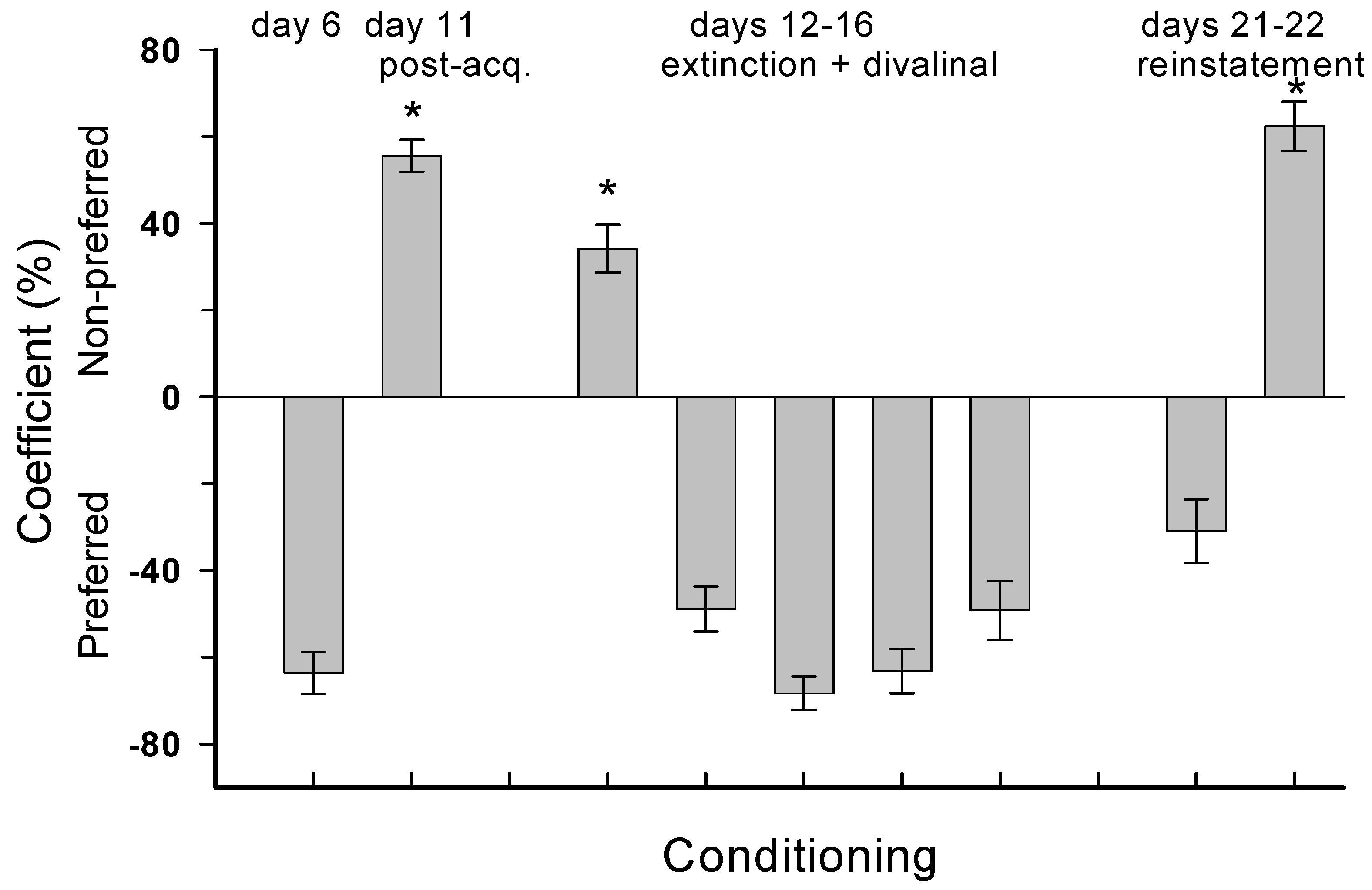
4.3. Experiment 3: MA Reinstatement
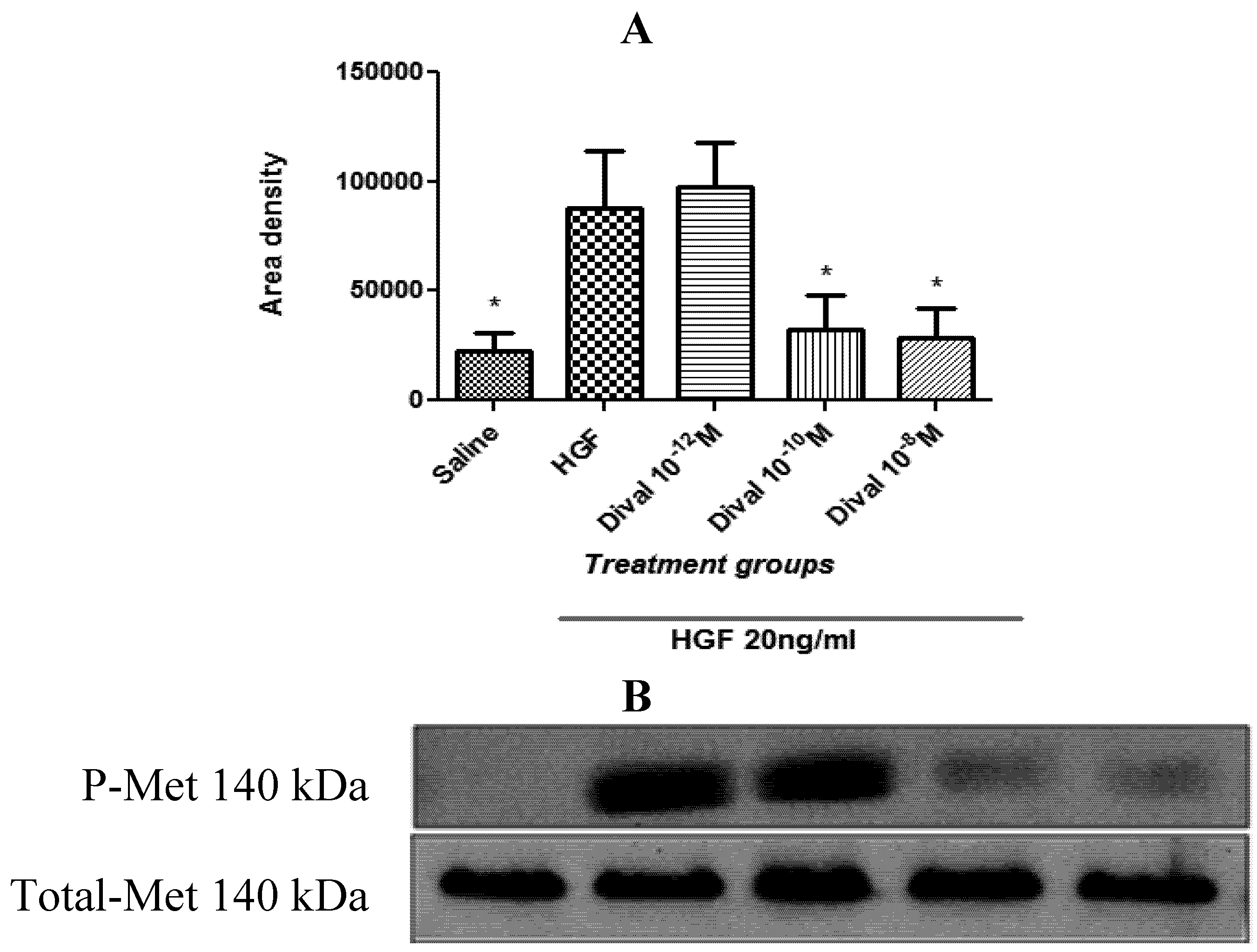
4.4. Divalinal Augments HGF-Dependent Met Signaling and Cellular Activity

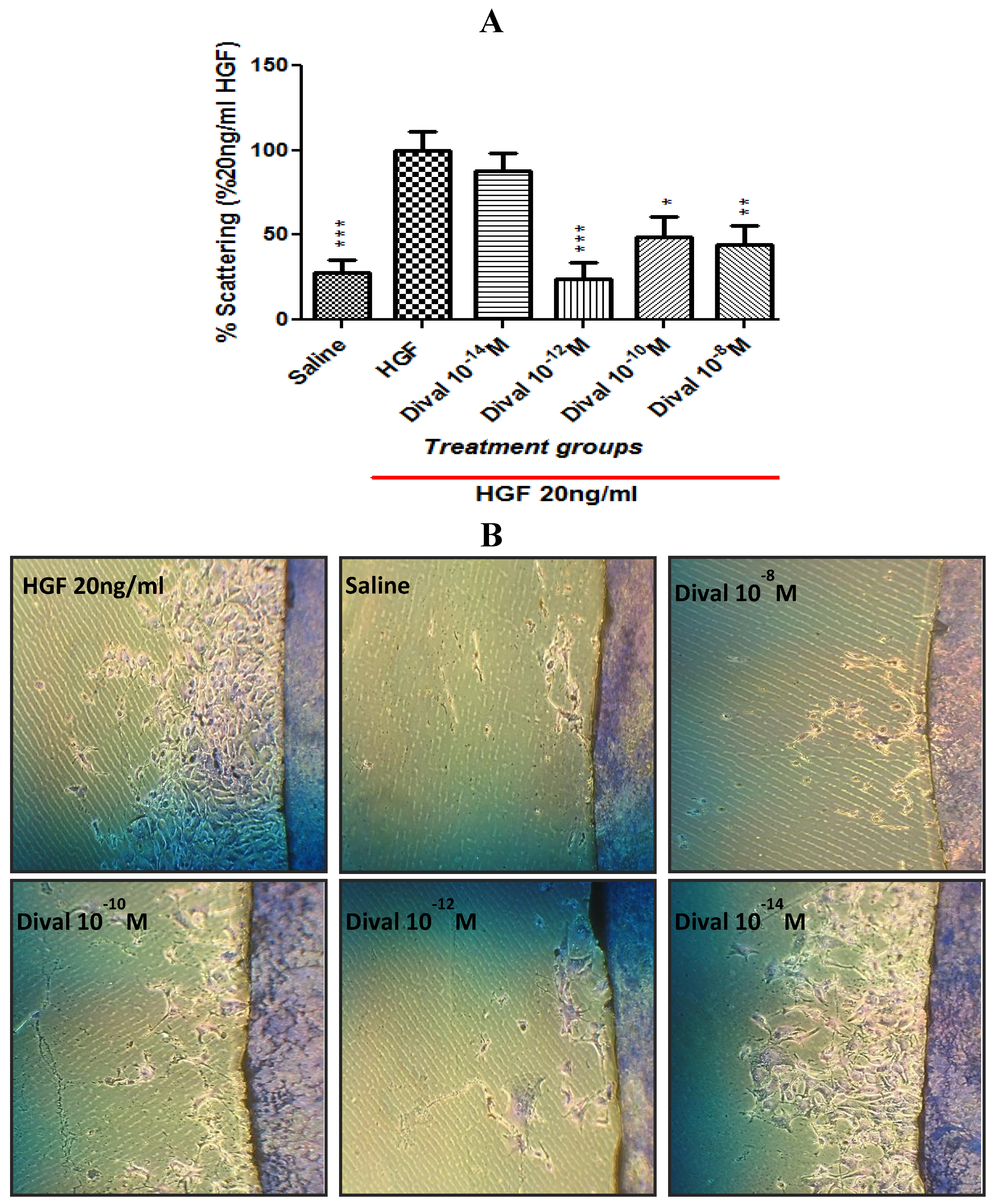
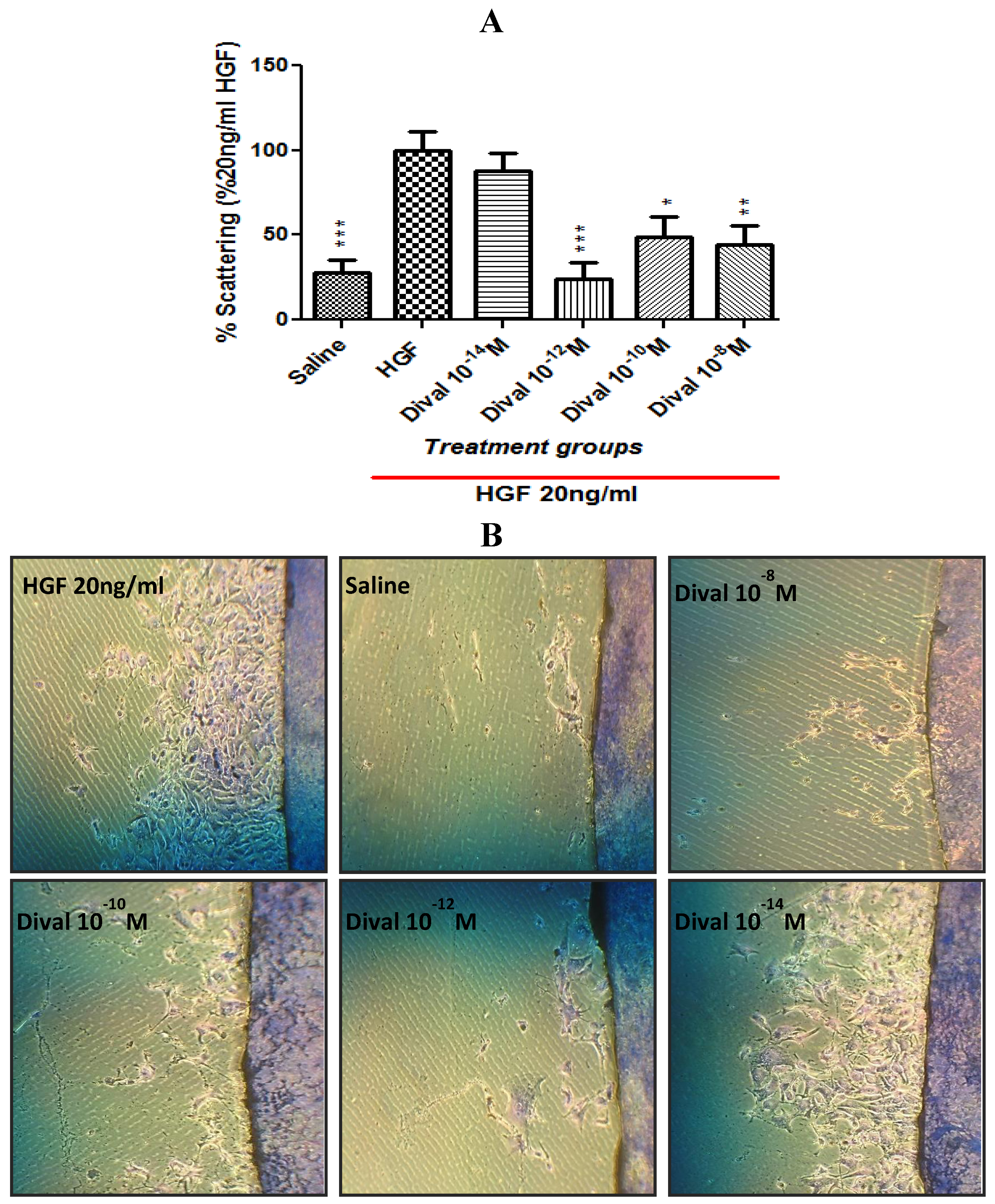
4.5. Scattering Assay
5. Discussion
6. Conclusion
Acknowledgements
References
- Cherng, C.G.; Tsai, C.W.; Tsai, Y.P.; Ho, M.C.; Kao, S.F.; Yu, L. Methamphetamine-disrupted processing mediates conditioned place preference performance. Behav. Brain Res. 2007, 182, 103–108. [Google Scholar] [CrossRef]
- Jefferson, D.J.; Shenfeld, H.; Murr, A.; Campo-Flores, A.; Childress, S.; Skipp, C.; Meadows, S.; Johnson, D.; Silver-Greenberg, J. America’s most dangerous drug. Newsweek 2005, 40–48. [Google Scholar]
- Howell, L.L.; Kimmel, H.L. Monoamine transporters and psychostimulant addiction. Biochem. Pharmacol. 2008, 75, 196–217. [Google Scholar] [CrossRef]
- Nordahl, T.E.; Salo, R.; Leamon, M. Neuropsychological effects of chronic methamphetamine use on neurotransmitters and cognition: A review. J. Neuropsychiatry Clin. Neurosci. 2003, 15, 317–325. [Google Scholar] [CrossRef]
- Schmitz, Y.; Lee, C.J.; Schmauss, D.; Gonon, F.; Sulzer, D. Amphetamine distorts stimulation-dependent dopamine overflow: Effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J. Neurosci. 2001, 21, 5916–5924. [Google Scholar]
- Sekine, Y.; Ivo, M.; Ouchi, Y.; Matsunaga, T.; Tsukada, H.; Okada, H.; Yoshikawa, E.; Futatsubashi, M.; Takei, N.; Mori, N. Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am. J. Psychiatry 2001, 158, 1206–1214. [Google Scholar] [CrossRef]
- Mantle, T.J.; Tipton, K.F.; Garrett, N.J. Inhibition of monoamine oxidase by amphetamine and related compounds. Biochem. Pharmacol. 1976, 25, 2073–2077. [Google Scholar] [CrossRef]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Yamada, K.; Mouri, A.; Niwa, M.; Mizuno, T.; Noda, Y.; Nitta, A.; Itohara, S.; Banno, Y.; Tabeshima, T. Role of matrix metalloproteinase and tissue inhibitor of MMP in methamphetamine-induced behavioral sensitization and reward: Implications for dopamine receptor down-regulation and dopamine release. J. Neurochem. 2007, 102, 1548–1560. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Yamada, K.; Nabeshima, T. Neuropsychotoxicity of abused drugs: Involvement of matrix metalloproteinase-2 and -9 and tissue inhibitor of matrix metalloproteinase-2 in methamphetamine-induced behavioral sensitization and reward in rodents. J. Pharmacol. Sci. 2008, 106, 9–14. [Google Scholar] [CrossRef]
- Rogers, J.L.; Ghee, S.; See, R.E. The neural circuitry underlying reinstatement of heroin-seeking behavior in an animal model of relapse. Neuroscience 2008, 151, 579–588. [Google Scholar] [CrossRef]
- Voigt, R.M.; Herrold, A.A.; Napler, T.C. Baclofen facilitates the extinction of methamphetamine-induced conditioned place preference in rats. Behav. Neurosci. 2011, 125, 261–267. [Google Scholar] [CrossRef]
- Voigt, R.M.; Herrold, A.A.; Riddle, J.L.; Napler, T.C. Administration of GABA(B) receptor positive allosteric modulators inhibit the expression of previously established methamphetamine-induced conditioned place preference. Behav. Brain Res. 2011, 216, 419–423. [Google Scholar] [CrossRef]
- Friedman, S.D.; Castaneda, E.; Hodge, G.K. Long-term monamine depletion, differential recovery, and subtle behavioral impairment following methamphetamine-induced neurotoxicity. Pharmacol. Biochem. Behav. 1998, 61, 35–44. [Google Scholar] [CrossRef]
- Rogers, J.L.; de Santis, S.; See, R.E. Extended methamphetamine self-administration enhances reinstatement of drug seeking and impairs novel object recognition in rats. Psychopharmacology 2008, 199, 615–624. [Google Scholar] [CrossRef]
- Schroder, N.; O’Dell, S.J.; Marshall, J.F. Neurotoxic methamphetamine regimen severely impairs recognition memory in rats. Synapse 2003, 49, 89–96. [Google Scholar] [CrossRef]
- Williams, M.T.; Morford, L.L.; Wood, S.L.; Wallace, T.L.; Fukumura, M.; Broening, H.W.; Vorhees, C.V. Developmental D-methamphetamine treatment selectively induces spatial navigation impairments in reference memory in the Morris water maze while sparing working memory. Synapse 2003, 48, 138–148. [Google Scholar] [CrossRef]
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858. [Google Scholar] [CrossRef]
- Sorg, B.A. Reconsolidation of drug memories. Neurosci. Biobehav. Rev. 2012, 36, 1400–1417. [Google Scholar] [CrossRef]
- Dacher, M.; Nugent, F.S. Opiates and plasticity. Neuropharmacology 2011, 61, 1088–1096. [Google Scholar] [CrossRef]
- Gerdeman, G.L.; Partridge, J.G.; Lupica, C.R.; Lovinger, D.M. It could be habit forming: Drugs of abuse and striatal synaptic plasticity. Trends Neurosci. 2003, 26, 184–192. [Google Scholar] [CrossRef]
- Hyman, S.E.; Malenka, R.C.; Nestler, E.J. Neural mechanisms of addiction: The role of reward-related learning and memory. Annu. Rev. Neurosci. 2006, 29, 565–598. [Google Scholar] [CrossRef]
- Kelley, A.E. Memory and addiction: Shared neural circuitry and molecular mechanisms. Neuron 2004, 44, 161–179. [Google Scholar] [CrossRef]
- Nestler, E.J. Common molecular and cellular substrates of addiction and memory. Neurobiol. Learn. Mem. 2002, 78, 637–647. [Google Scholar]
- Niehaus, J.L.; Cruz-Bermudez, N.D.; Kauer, J.A. Plasticity of addiction: A mesolimbic dopamine short-circuit? Am. J. Addict. 2009, 18, 259–271. [Google Scholar] [CrossRef]
- Robinson, T.E.; Kolb, B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 2004, 47, 33–46. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. Contributions of matrix metalloproteinases to neural plasticity, habituation, associative learning and drug addiction. Neural Plast. 2009. [Google Scholar] [CrossRef]
- Harding, J.W.; Cook, V.I.; Miller-Wing, A.V.; Hanesworth, J.M.; Sardinia, M.F.; Hall, K.L.; Stobb, J.W.; Swanson, G.N.; Coleman, J.K.; Wright, .JW.; Harding, E.C. Identification of an AII (3-8) [AIV] binding site in guinea pig hippocampus. Brain Res. 1992, 583, 340–343. [Google Scholar] [CrossRef]
- Gard, P.R. Cognitive—Enhancing effects of angiotensin IV. BMC Neurosci. 2008, 9, S15. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. The brain angiotensin system and extracellular matrix molecules in neural plasticity, learning, and memor. Prog. Neurobiol. 2004, 72, 263–293. [Google Scholar] [CrossRef]
- Recinto, P.; Samant, A.R.; Chavez, G.; Kim, A.; Yuan, C.J.; Soleiman, M.; Grant, Y.; Edwards, S.; Wee, S.; Koob, G.F.; et al. Levels of neural progenitors in the hippocampus predicts memory impairment and relapse to drug seeking as a function of excessive methamphetamine self-administration. Neuropsychopharmacology 2011, 37, 1275–1287. [Google Scholar]
- Ricoy, U.M.; Martinez, J.L. Local hippocampal methamphetamine-induced reinforcement. Behav. Neurosci. 2009, 3. [Google Scholar] [CrossRef]
- Kawas, L.H.; McCoy, A.T.; Yamamoto, B.J.; Wright, J.W.; Harding, J.W. Development of angiotensin IV analogs as hepatocyte growth factor/Met modifiers. J. Pharmacol. Exp. Ther. 2012, 340, 539–548. [Google Scholar] [CrossRef]
- Kawas, L.H.; Yamamoto, B.J.; Wright, J.W.; Harding, J.W. Mimics of the dimerization domain of hepatocyte growth factor exhibit anti-met and anticancer activity. J. Pharmacol. Exp. Ther. 2011, 339, 509–518. [Google Scholar] [CrossRef]
- Yamamoto, B.J.; Elias, P.D.; Masino, J.A.; Hudson, B.D.; McCoy, A.T.; Anderson, Z.J.; Varnum, M.D.; Sardinia, M.F.; Wright, J.W.; Harding, J.W. The angiotensin IV analog Nle-Tyr-Leu-ψ-(CH2-NH2)3-4-His-Pro-Phe (Norleual) can act as a hepatocyte growth factor/c-Met inhibitor. J. Pharmacol. Exp. Ther. 2010, 333, 161–173. [Google Scholar] [CrossRef]
- Akimoto, M.; Baba, A.; Ikeda-Matsuo, Y.; Yamada, M.K.; Itamura, R.; Nishiyama, N.; Ikegaya, Y.; Matsuki, N. Hepatocyte growth factor as an enhancer of NMDA currents and synaptic plasticity in the hippocampus. Neuroscience 2004, 128, 155–162. [Google Scholar] [CrossRef]
- Date, I.; Takagi, N.; Takagi, K.; Kago, T.; Matsumoto, K.; Nakamura, T.; Takeo, S. Hepatocyte growth factor attenuates cerebral ischemia-induced learning dysfunction. Biochem. Biophys. Res. Commun. 2004, 319, 1152–1158. [Google Scholar] [CrossRef]
- Date, I.; Takagi, N.; Takagi, K.; Kago, T.; Matsumoto, K.; Nakamura, T.; Takeo, S. Hepatocyte growth factor improved learning and memory dysfunction of microsphere-embolized rats. J. Neurosci. Res. 2004, 78, 442–453. [Google Scholar] [CrossRef]
- Takeo, S.; Takagi, N.; Takagi, K. Ischemic brain injury and hepatocyte growth factor. Yakugaku Zasshi 2007, 127, 1813–1823. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 2nd ed; Academic Press: New York, NY, USA, 1986. [Google Scholar]
- Roberts, K.A.; Krebs, L.T.; Kramár, E.A.; Shaffer, M.J.; Harding, J.W.; Wright, J.W. Autoradiographic identification of brain angiotensin IV binding sites and differential c-Fos expression following intracerebroventricular injection of angiotensin II and IV in rats. Brain Res. 1995, 682, 13–21. [Google Scholar] [CrossRef]
- Wright, J.W.; Stubley, L.A.; Pederson, E.S.; Kramar, E.A.; Hanesworth, J.M.; Harding, J.W. Contributions of the brain angiotensin IV-AT4 receptor subtype system to spatial learning. J. Neurosci. 1999, 19, 3952–3961. [Google Scholar]
- Vastola, B.J.; Douglas, L.A.; Varlinskaya, E.I.; Spear, L.P. Nicotine-induced conditioned place preference in adolescent and adult rats. Physiol. Behav. 2002, 77, 107–114. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Vande Woude, G.F. HGF/SF-Met signaling in the control of branching morphogenesis and invasion. J. Cell. Biochem. 2003, 88, 408–417. [Google Scholar] [CrossRef]
- Stella, M.C.; Comoglio, P.M. HGF: A multifunctional growth factor controlling cell scattering. Int. J. Biochem. Cell Biol. 1999, 31, 1357–1362. [Google Scholar] [CrossRef]
- Comoglio, P.; Boccaccio, C. Scatter factors and invasive growth. Semin. Cancer Biol. 2001, 11, 153–165. [Google Scholar] [CrossRef]
- Berke, J.D.; Hyman, S.E. Addiction, dopamine, and the molecular mechanisms of memory. Neuron 2000, 25, 515–532. [Google Scholar] [CrossRef]
- Luscher, C.; Malenka, R.C. Drug-evoked synaptic plasticity in addiction: From molecular changes to circuit remodeling. Neuron 2011, 69, 650–663. [Google Scholar] [CrossRef]
- Mameli, M.; Lüscher, C. Synaptic plasticity and addiction: Learning mechanisms gone awry. Neuropharmacology 2011, 61, 1052–1059. [Google Scholar] [CrossRef]
- Yamada, K. Endogenous modulators for drug dependence. Biol. Pharm. Bull. 2008, 31, 1635–1638. [Google Scholar] [CrossRef]
- Meighan, S.E.; Meighan, P.C.; Choudhury, P.; Davis, C.J.; Olson, M.L.; Zornes, P.A.; Wright, J.W.; Harding, J.W. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 2006, 96, 1227–1241. [Google Scholar] [CrossRef]
- Brown, T.E.; Forquer, M.R.; Cocking, D.L.; Jansen, H.T.; Harding, J.W.; Sorg, B.A. Role of matrix metalloproteinases in the acquisition and reconsolidation of cocaine-induced conditioned place preference. Learn. Mem. 2007, 14, 214–223. [Google Scholar] [CrossRef]
- Smith, A.W.; Nealey, K.A.; Wright, J.W.; Walker, B.M. Plasticity associated with escalated operant ethanol self-administration during acute withdrawal in ethanol-dependent rats requires intact matrix metalloproteinase systems. Neurobiol. Learn. Mem. 2011, 96, 199–206. [Google Scholar] [CrossRef]
- Samuels, I.S.; Saitta, S.C.; Landreth, G.E. MAP’ing CNS development and cognition: An ERKsome process. Neuron 2009, 61, 160–167. [Google Scholar] [CrossRef]
- Brown, T.E.; Lee, B.R.; Sorg, B.A. The NMDA antagonist MK-801 disrupts reconsolidation of a cocaine-associated memory for conditioned place preference but not for self-administration in rats. Learn. Mem. 2008, 15, 857–865. [Google Scholar] [CrossRef]
- Benoist, C.; Wright, J.W.; Wayman, G.A.; Harding, J.W. Facilitation of hippocampal synaptogenesis and spatial memory by C-terminal truncated Nle1-angiotensin IV analogs. J. Pharmacol. Exp. Ther. 2011, 339, 35–44. [Google Scholar] [CrossRef]
- Braszko, J.J.; Kupryszewski, G.; Witczuk, B.; Wisniewski, K. Angiotensin II (3-8)-hexapeptide affects motor activity, performance of passive avoidance, and a conditioned avoidance response in rats. Neuroscience 1988, 27, 777–783. [Google Scholar] [CrossRef]
- Braszko, J.J.; Wlasienko, J.; Koziolkiewicz, W.; Janecka, A.; Wisniewski, K. The 3-7 fragment of angiotensin II is probably responsible for its psychoactive properties. Brain Res. 1991, 542, 49–54. [Google Scholar] [CrossRef]
- Wright, J.W.; Miller-Wing, A.V.; Shaffer, M.J.; Higginson, C.; Wright, D.E.; Hanesworth, J.M.; Harding, J.W. Angiotensin II(3-8) [ANG IV] hippocampal binding: Potential role in the facilitation of memory. Brain Res. Bull. 1993, 32, 497–502. [Google Scholar] [CrossRef]
- Pederson, E.S.; Krishnan, R.; Harding, J.W.; Wright, J.W. A role for the angiotensin AT4 receptor subtype in overcoming scopolamine-induced spatial memory deficits. Regul. Pept. 2001, 102, 147–156. [Google Scholar] [CrossRef]
- Barak, S.; Hamida, S.B. Memory erasure, enhanced extinction and disrupted reconsolidation. J. Neurosci. 2012, 32, 2250–2251. [Google Scholar] [CrossRef]
- Krebs, L.T.; Kramár, E.A.; Hanesworth, J.M.; Sardinia, M.F.; Ball, A.E.; Wright, J.W.; Harding, J.W. Characterization of the binding properties and physiological action of divalinal-angiotensin IV, a putative AT4 receptor antagonist. Regul. Pept. 1996, 67, 123–130. [Google Scholar] [CrossRef]
- Tyndall, S.J.; Walikonis, R.S. The receptor tyrosine kinase Met and its ligand hepatocyte growth factor are clustered at excitatory synapses and can enhance clustering of synaptic proteins. Cell Cycle 2006, 5, 1560–1568. [Google Scholar] [CrossRef]
- Davis, C.J.; Kramár, E.A.; De, A.; Meighan, P.C.; Simasko, S.M.; Wright, J.W.; Harding, J.W. AT4 receptor activation increases intracellular calcium influx and induces a non-N-methyl-D-aspartate dependent form of long-term potentiation. Neuroscience 2006, 137, 1369–1379. [Google Scholar] [CrossRef]
- Kramár, E.A.; Armstrong, D.L.; Ikeda, S.; Wayner, M.J.; Harding, J.W.; Wright, J.W. The effects of angiotensin IV analogs on long-term potentiation within the CA1 region of the hippocampus in vitro. Brain Res. 2001, 897, 114–121. [Google Scholar] [CrossRef]
- Gutierrez, H.; Dolcet, X.; Tolcos, M.; Davies, A. HGF regulates the development of cortical pyramidal dendrites. Development 2004, 131, 3717–26. [Google Scholar] [CrossRef]
- Tyndall, S.J.; Patel, S.J.; Walikonis, R.S. Hepatocyte growth factor-induced enhancement of dendritic branching is blocked by inhibitors of N-methyl-D-aspartate receptors and calcium/calmodulin-dependent kinases. J. Neurosci. Res. 2007, 85, 2343–2351. [Google Scholar] [CrossRef]
- Ebens, A.; Brose, K.; Leonardo, E.D.; Hanson, M.G., Jr.; Bladt, F.; Birchmeier, C.; Barres, B.A.; Tessier-Lavigne, M. Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 1996, 17, 1157–1172. [Google Scholar] [CrossRef]
- Kato, N.; Nemoto, K.; Nakanishi, K. Efficacy of HGF gene transfer for various nervous injuries and disorders. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 300–306. [Google Scholar]
- Shang, J.; Deguchi, K.; Ohta, Y.; Liu, N.; Zhang, X.; Tian, F.; Yamashita, T.; Ikeda, Y.; Matsuura, T.; Abe, K.; et al. Strong neurogenesis, angiogenesis, synaptogenesis, and antifibrosis of hepatocyte growth factor in rats brain after transient middle cerebral artery occlusion. J. Neurosci. Res. 2011, 89, 86–95. [Google Scholar] [CrossRef]
- Wang, T.W.; Zhang, H.; Gyetko, M.R.; Parent, J.M. Hepatocyte growth factor acts as a mitogen and chemoattractant for postnatal subventricular zone-olfactory bulb neurogenesis. Mol. Cell. Neurosc. 2011, 48, 38–50. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. Brain renin-angiotensin—A new look at an old system. Prog. Neurobiol. 2011, 95, 49–67. [Google Scholar] [CrossRef]
- Lee, J.; Chai, S.Y.; Mendelsohn, F.A.; Morris, M.J.; Allen, A.M. Potentiation of cholinergic transmission in the rat hippocampus by angiotensin IV and LVV-hemorphin-7. Neuropharmacology 2001, 40, 618–623. [Google Scholar] [CrossRef]
- Deiana, S.; Platt, B.; Riedel, G. The cholinergic system and spatial learning. Behav. Brain Res. 2011, 221, 389–411. [Google Scholar] [CrossRef]
- Pepeu, G.; Giovannini, M.G. Cholinesterase inhibitors and memory. Chem. Biol. Interact. 2010, 187, 403–408. [Google Scholar] [CrossRef]
- Sofuoglu, M.; Mooney, M. Cholinergic functioning in stimulant addiction: Implications for medications development. CNS Drugs 2009, 23, 939–952. [Google Scholar] [CrossRef]
- Hiranita, T.; Anggadiredja, K.; Fujisaki, C.; Watanabe, S.; Yamamoto, T. Nicotine attenuates relapse to methamphetamine-seeking behavior (craving) in rats. Ann. N. Y. Acad. Sci. 1025, 504–507. [Google Scholar]
- Zanetti, L.; Picciotto, M.R.; Zoli, M. Differential effects of nicotinic antagonists perfused into the nucleus accumbens or the ventral tegmental area on cocaine-induced dopamine release in the nucleus accumbens of mice. Psychopharmacology 2007, 190, 189–199. [Google Scholar]
- DiChiara, G.; Bassareo, V.; Fenu, S.; DeLuca, M.A.; Spina, L.; Cadoni, C.; Acquas, E.; Carboni, E.; Valentini, V.; Lecca, D. Dopamine and drug addiction: The nucleus accumbens shell connection. Neuropharmacology 2004, 47, 227–241. [Google Scholar] [CrossRef]
- Gerasimov, M.R.; Franceschi, M.; Volkow, N.D.; Rice, O.; Schiffer, W.K.; Dewey, S.L. Synergistic interactions between nicotine and cocaine or methylphenidate depend on the dose of dopamine transporter inhibitor. Synapse 2000, 38, 432–437. [Google Scholar] [CrossRef]
- Braszko, J.J. Participation of D1-4 dopamine receptors in the pro-cognitive effects of angiotensin IV and des-Phe6 angiotensin IV. Neurosci. Biobehav. Rev. 2010, 34, 343–350. [Google Scholar] [CrossRef]
- Stragier, B.; Demaegdt, H.; de Bundel, D.; Smolders, I.; Sarre, S.; Vauquelin, G.; Ebinger, G.; Michotte, Y.; Vanderheyden, P. Involvement of insulin-regulated aminopeptidase and/or aminopeptidase N in the angiotensin IV-induced effect on dopamine release in the striatum of the rat. Brain Res. 1131, 97–105. [Google Scholar]
- Stragier, B.; Sarre, S.; Vanderheyden, P.; Vauquelin, G.; Fournie-Zaluski, M.C.; Ebinger, G.; Michotte, Y. Metabolism of angiotensin II is required for its in vivo effect on dopamine release in the striatum of the rat. J. Neurochem. 2004, 90, 1251–1257. [Google Scholar] [CrossRef]
- Hamanoue, M.; Takemoto, N.; Matsumoto, K.; Nakamura, T.; Nakajima, K.; Kohsaka, S. Neurotrophic effect of hepatocyte growth factor on central nervous system neurons in vitro. J. Neurosci. Res. 1996, 43, 554–564. [Google Scholar] [CrossRef]
- Guessous, F.; Zhang, F.; Marcinkiewicz, L.; Schiff, D.; Abounader, R.; DiPierro, C.; Sarkaria, J.; Buchanan, S. An orally bioavailable c-Met kinase inhibitor potently inhibits brain tumor malignancy and growth. Anticancer Agents Med. Chem. 2010, 10, 28–35. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wright, J.W.; Wilson, W.L.; Wakeling, V.; Boydstun, A.S.; Jensen, A.; Kawas, L.; Harding, J.W. The Hepatocyte Growth Factor/c-Met Antagonist, Divalinal-Angiotensin IV, Blocks the Acquisition of Methamphetamine Dependent Conditioned Place Preference in Rats. Brain Sci. 2012, 2, 298-318. https://doi.org/10.3390/brainsci2030298
Wright JW, Wilson WL, Wakeling V, Boydstun AS, Jensen A, Kawas L, Harding JW. The Hepatocyte Growth Factor/c-Met Antagonist, Divalinal-Angiotensin IV, Blocks the Acquisition of Methamphetamine Dependent Conditioned Place Preference in Rats. Brain Sciences. 2012; 2(3):298-318. https://doi.org/10.3390/brainsci2030298
Chicago/Turabian StyleWright, John W., Wendy L. Wilson, Vanessa Wakeling, Alan S. Boydstun, Audrey Jensen, Leen Kawas, and Joseph W. Harding. 2012. "The Hepatocyte Growth Factor/c-Met Antagonist, Divalinal-Angiotensin IV, Blocks the Acquisition of Methamphetamine Dependent Conditioned Place Preference in Rats" Brain Sciences 2, no. 3: 298-318. https://doi.org/10.3390/brainsci2030298