1. Introduction
The isotope
89Zr is one of several positron-emitting radiometals that are increasing in popularity as radiolabels for positron emission tomography (PET) [
1,
2,
3,
4,
5]. A search of the PubMed database [
6] found over 55 publications with
89Zr mentioned in the title, 75% of which had been published since 2009. In particular,
89Zr holds significant potential for immunoPET [
4,
7,
8,
9,
10], a growing technique that uses radiolabelled antibodies, antibody fragments, and peptides for the
in vivo molecular imaging of antigens using PET [
7,
11,
12,
13,
14].
89Zr is well-suited for this technique because it is a positron-emitter with a half-life (
t1/2 = 3.27 days) that is long enough to accommodate the targeting time for these relatively large imaging agents, which is on the order of days. So far, the most prevalent application for
89Zr has been cancer imaging, which is the focus of the majority of publications that used
89Zr for
in vivo PET imaging. The information that is obtained using
89Zr-immunoPET can be useful for cancer staging, therapy planning, and treatment monitoring, making
89Zr a potentially valuable tool for personalized medicine.
There are several properties of
89Zr that make it an attractive radiolabel for immunoPET. Most importantly, the half-life of
89Zr matches the biological half-life of antibodies. Additionally,
89Zr emits positrons with a sufficient branching ratio (
y = 23%) and provides good PET spatial resolution because it emits positrons at a low average energy (
Eβ+,avg = 396 keV), and therefore a lower positron range (1.2 mm [
15]) compared to several other PET isotopes. Unfortunately,
89Zr also emits a high energy (909 keV) gamma ray with a high branching ratio (99.0%). Thus,
89Zr has a high gamma factor,
Γ15 keV = 6.6 R∙cm
2∙mCi
−1∙h
−1 (weighted average of all gamma rays emitted) [
16], meaning that the dose rate for this isotope is significant. The Zr
4+ ion prefers a coordination state of 6 to form a stable complex, and, so far, the most prevalent chelator of
89Zr is desferrioxamine (DFO) [
8,
17].
Table 1 summarizes chemical and nuclear decay properties of
89Zr, including a simplified decay scheme.
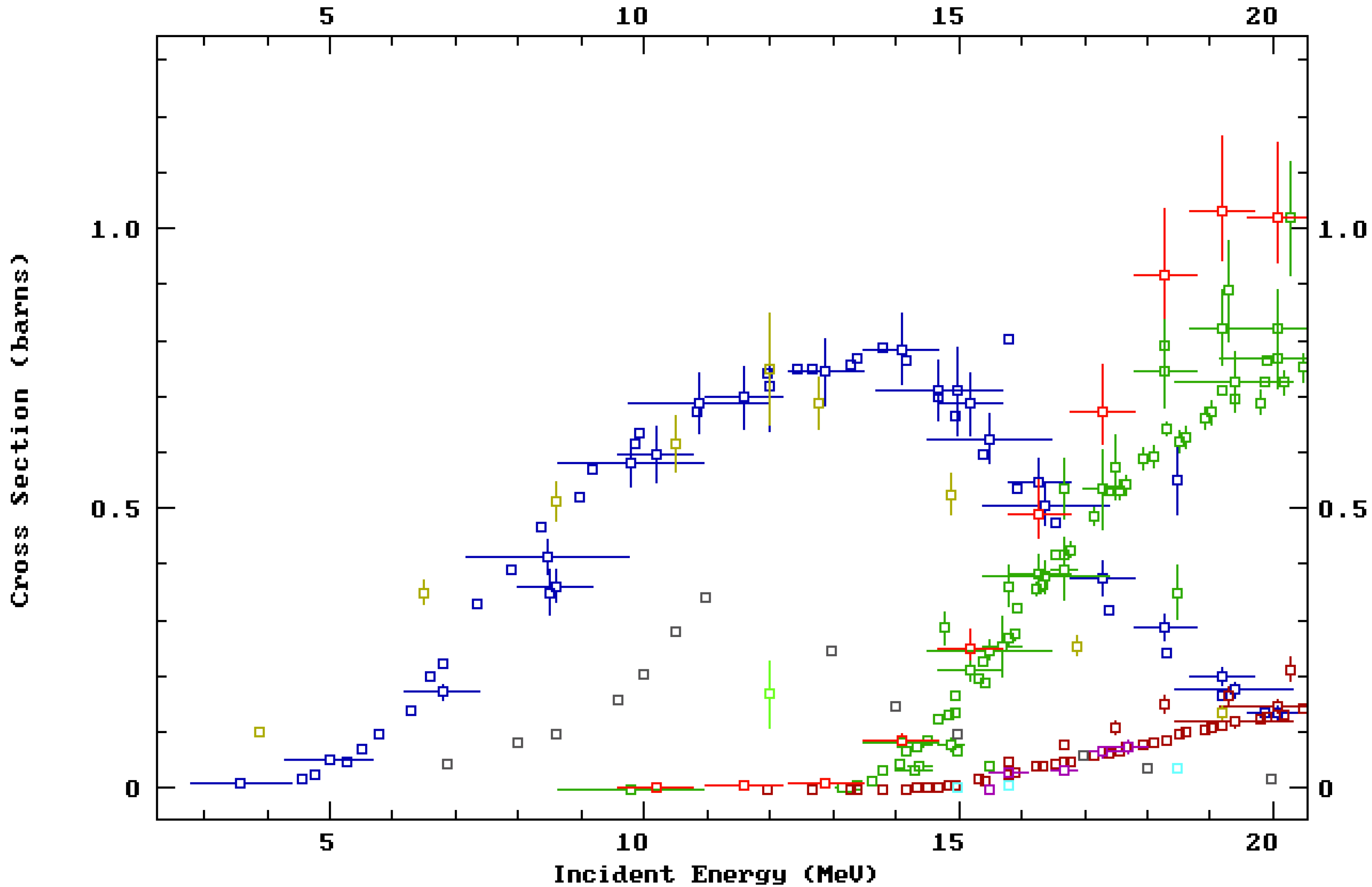
The vast majority of medical centers capable of producing medical isotopes do so using a low-energy “biomedical” cyclotron capable of bombarding a target with protons with energy (
Ep) < 20 MeV (and alternatively deuterons with energy (
Ed) <10 MeV). For these particles and energies, the highest cross-section is the (
p,
n) reaction with a peak cross-section of ~0.8 b at
Ep~14 MeV. The (
p,
n) reaction uses
89Y target material, which is 100% naturally abundant and thus relatively inexpensive. For the above reasons, the
natY(
p,
n)
89Zr reaction is the most common reaction route for
89Zr production [
18]. However, contaminants can also be produced in a
natY foil via the following common low-energy positron reactions: (
p,
n)
89mZr, (
p,2
n)
88Zr, (
p,
pn)
88Y, as shown in the Electronic Supplementary Information (ESI). Fortunately, high radionuclidic purity can still be achieved using ~15 MeV protons because
89mZr has a short half-life (
t1/2 = 4.2 m) and a high degree of isomeric transition (IT = 93.8%) and because the (
p,2
n) and (
p,
pn) reactions require greater proton beam energies, so they have low cross-sections at
Ep~15 MeV—less than 0.2 and 0.02 b, respectively [
19]. The important characteristics of
89Zr production in general are summarized in
Table 2, and the parameters and results from eight publications [
20,
21,
22,
23,
24,
25,
26,
27] about
89Zr production are shown in the ESI.
Table 1.
Summary of chemical and nuclear decay properties of 89Zr with a simplified decay scheme.
Table 1.
Summary of chemical and nuclear decay properties of 89Zr with a simplified decay scheme.
| Nuclear Decay: | | Simplified decay scheme: |
|---|
| Half-life, t1/2 | 3.27 days | ![Applsci 03 00593 i001]() |
|---|
| Daughter isotope | 100% 89Y (stable) |
| Decay modes | 23% β+ |
| | 77% EC |
| β+ energy, Eβ+, avg (range in water) | 396 keV (1.2 mm) |
| γ-ray energy, Eγ (intensity) | 909 keV (99.0%) |
| Gamma factor, Γ15 keV | 6.6 R∙cm2∙mCi−1∙h−1 |
| Theoretical specific activity | 40 Ci∙μmol−1 |
| Metastable isomer(s) | 89mZr |
Table 2.
Summary of characteristics for production of 89Zr using a low-energy, biomedical cyclotron.
Table 2.
Summary of characteristics for production of 89Zr using a low-energy, biomedical cyclotron.
| Target (natural abundance) | natY (100% 89Y) |
| Production reaction | (p,n)89Zr (t1/2 = 3.27 days) |
| Peak cross-section and energy, σ (Eβ+) | ~0.8 b (~15 MeV) |
| Reaction threshold, Q | 3.7 MeV |
| Stopping range of 14.7 MeV protons | 1.02 mm |
| Other possible reactions | (p,n)89mZr (t1/2 = 4.2 m) |
| | (p,2n)88Zr (t1/2 = 83.4 days) |
| | (p,pn)88Y (t1/2 = 107 days) |
| Optimum beam energy, Ep | 13 MeV |
| Target preparation | hot-rolled natY metal foil |
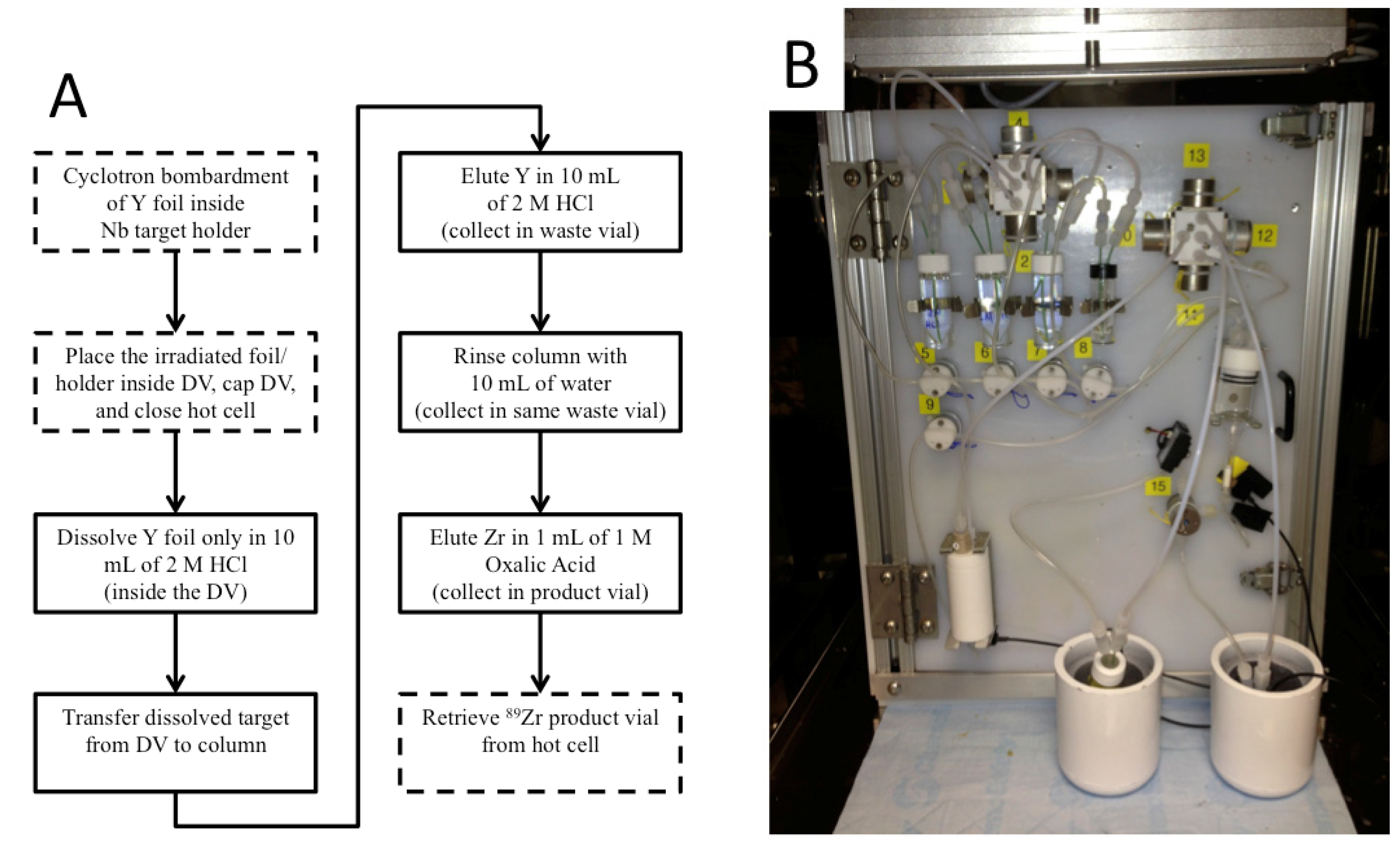
Out of eight papers that used the (
p,
n) reaction for
89Zr production, five used Y foil targets [
20,
21,
23,
26,
27], two used sputtered Y onto Cu [
24,
25], and one used Y
2O
3 pellets [
22]. Although the sputtered targets provided superior heat transfer and therefore allowed for higher beam currents, we chose to use Y foil for ease of use. For a hypothetical irradiated Y foil that contains 100 mCi of
89Zr, <0.3 μg of
89Zr atoms are present in the foil, and these product atoms must be extracted chemically to give the desired product—a solution containing
89Zr
4+ ions. This separation has been performed with mixed results using several different techniques [
23], including solvent extraction [
21,
22,
26,
27,
37], solid cation exchange [
21,
22], solid anion exchange [
22,
26,
27,
38], and solid hydroxamate resin [
23,
24,
25,
39,
40]. Hydroxamate resin separation has emerged as the preferred method following the publication of a standardized method for producing and separating
89Zr by Holland,
et al. [
23]. This separation can produce high recovery, radionuclidic purity, and effective specific activity (ESA). Our work does not contribute new separation chemistry; rather, the objective for this work was to automate the process that was reported by Holland,
et al. [
23] based on chemistry that was developed by Verel,
et al. [
24], Meijs,
et al. [
25], and Herscheid,
et al. [
39].
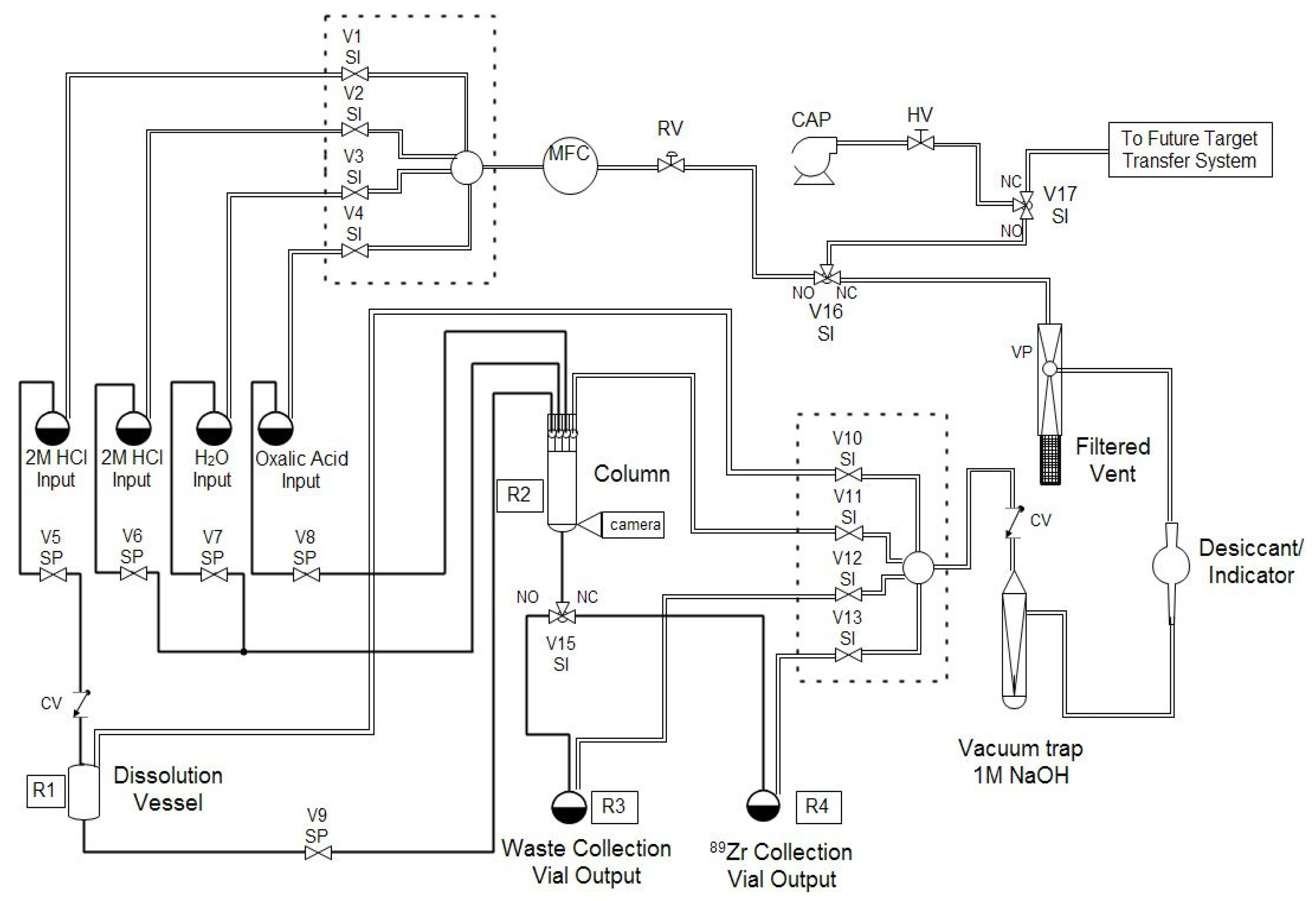
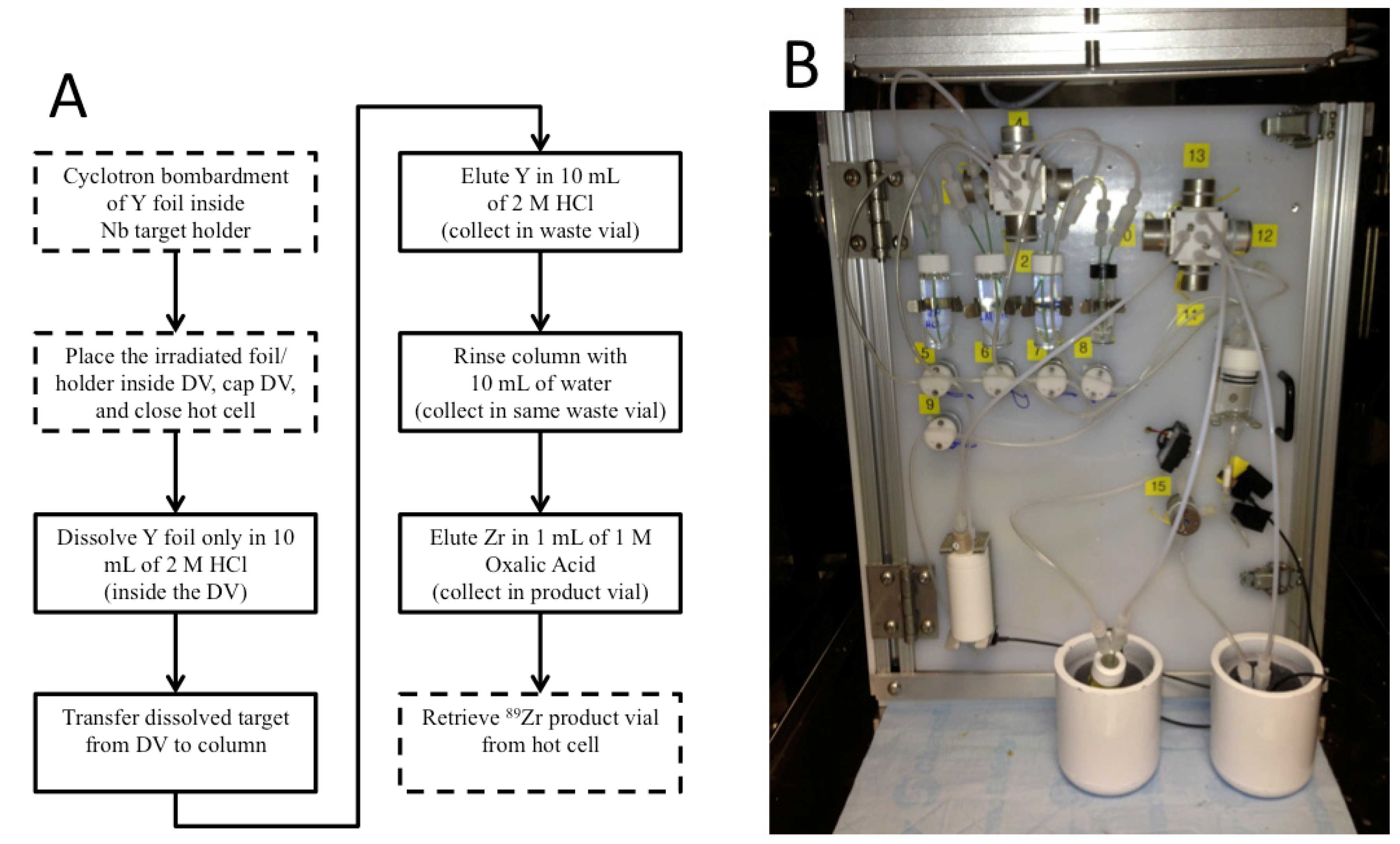
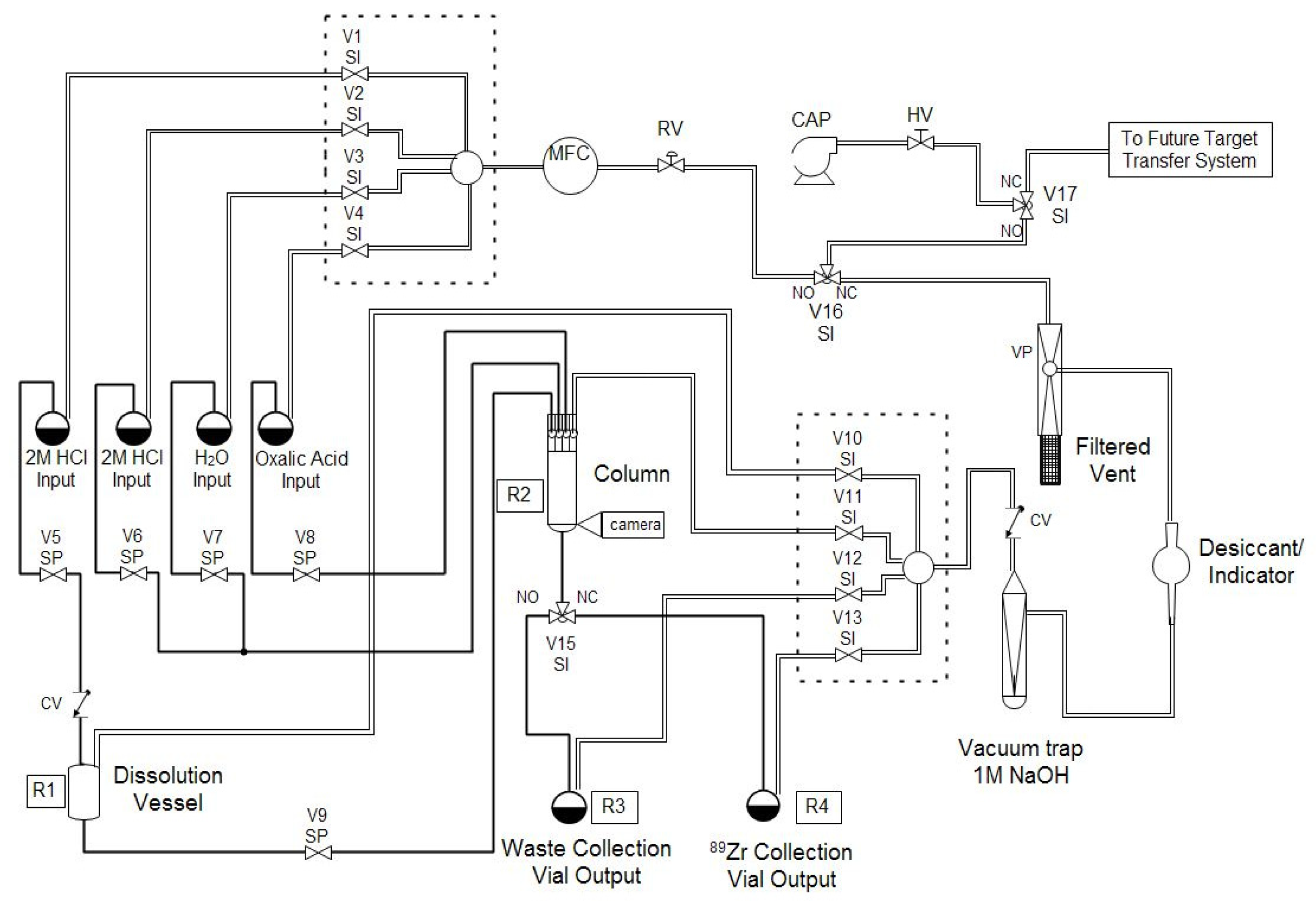
Automation is an important part of isotope production in particular and nuclear medicine in general because automated modules enable production centers to separate large activities of isotopes while still meeting institutional and federal regulations for radiation dose. Automated modules are ubiquitous at many PET centers, where they are typically used in good manufacturing practice (GMP) facilities for synthesizing patient doses of radiopharmaceuticals, such as [
18F]fluoro-deoxy-glucose ([
18F]FDG). Automated modules are essential to such processes because these syntheses are for human injection and because the radiolabels for these compounds have short half-lives, high activities, and high gamma factors. Therefore, automated modules are used to make the synthesis clean, reproducible, fast, and safe to production personnel. Although most PET radiometals are still at the research stage, automated separation modules are still useful for many of the same reasons, but principally to minimize dose to personnel. Several groups have published on custom-made automated modules for various radiometals, including
64Cu [
41,
42,
43,
44],
124I [
45],
99mTc [
46],
86Y [
47], and
89Zr [
48,
49]. In this work, we have designed, built and commissioned a module that has performed the chemical separation of
89Zr safely and routinely, at activities in excess of 50 mCi, with high radionuclidic purity and satisfactory ESA.
3. Results and Discussion
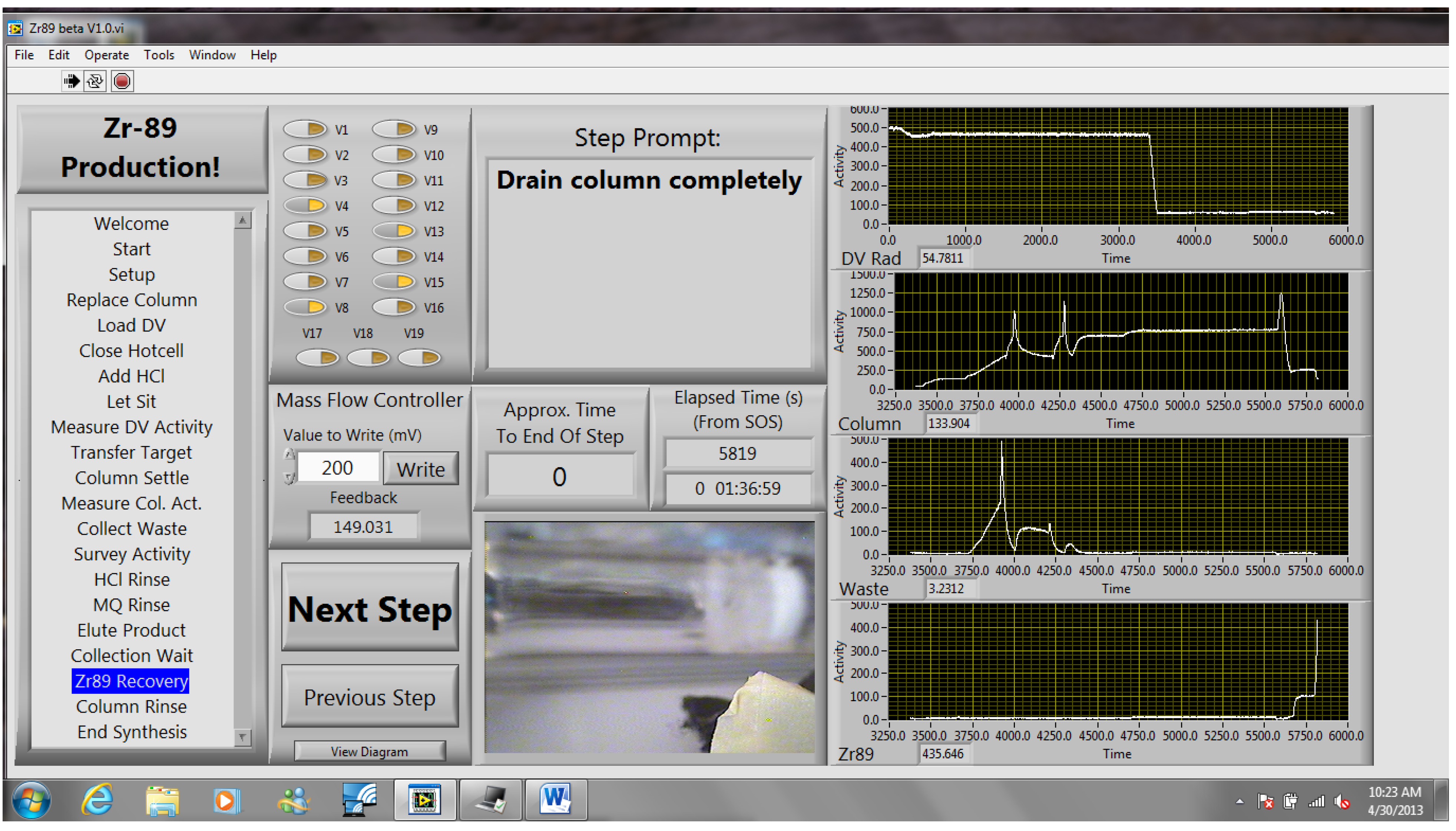
The methods described in this work have been used successfully for several months for the cyclotron production and chemical separation of
89Zr. Typically, after the chemical separation was completed using the automated module, a dose calibrator was used to measure the activity of
89Zr collected in the product and waste vials. To reduce radiation dose, the system was allowed to decay for one week before measuring the dissolution vessel, which still contained the irradiated Nb target holder, and the intact column assembly, which still included the resin, frits, and glass wool. The radioactivity measured for all four major module components was decay-corrected to the end-of-bombardment (EOB) time for each production. The decay-corrected activities were summed to give an estimated total radioactivity of
89Zr produced in each cyclotron bombardment. This estimate neglects any activity that was lost in tubing, valves,
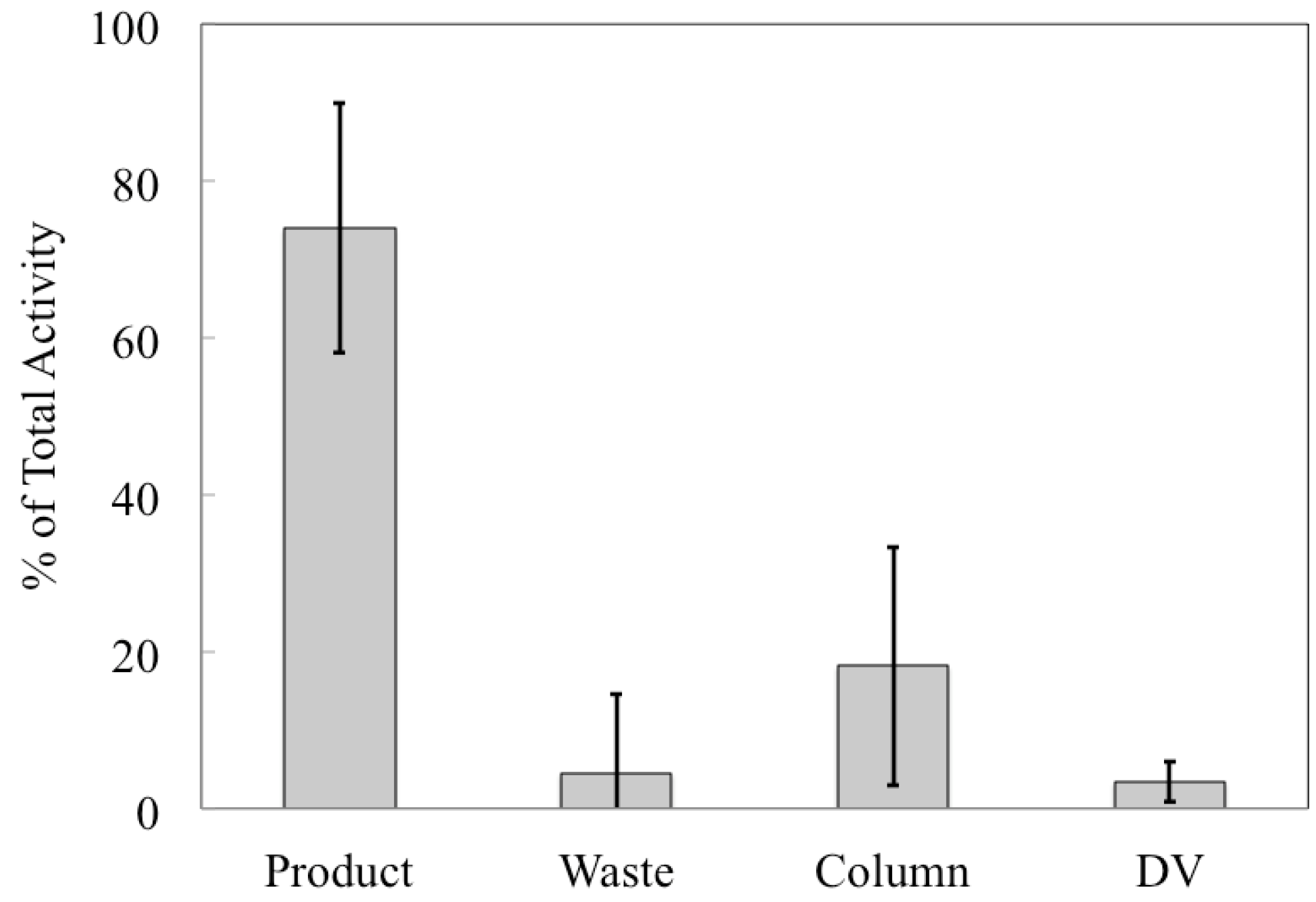
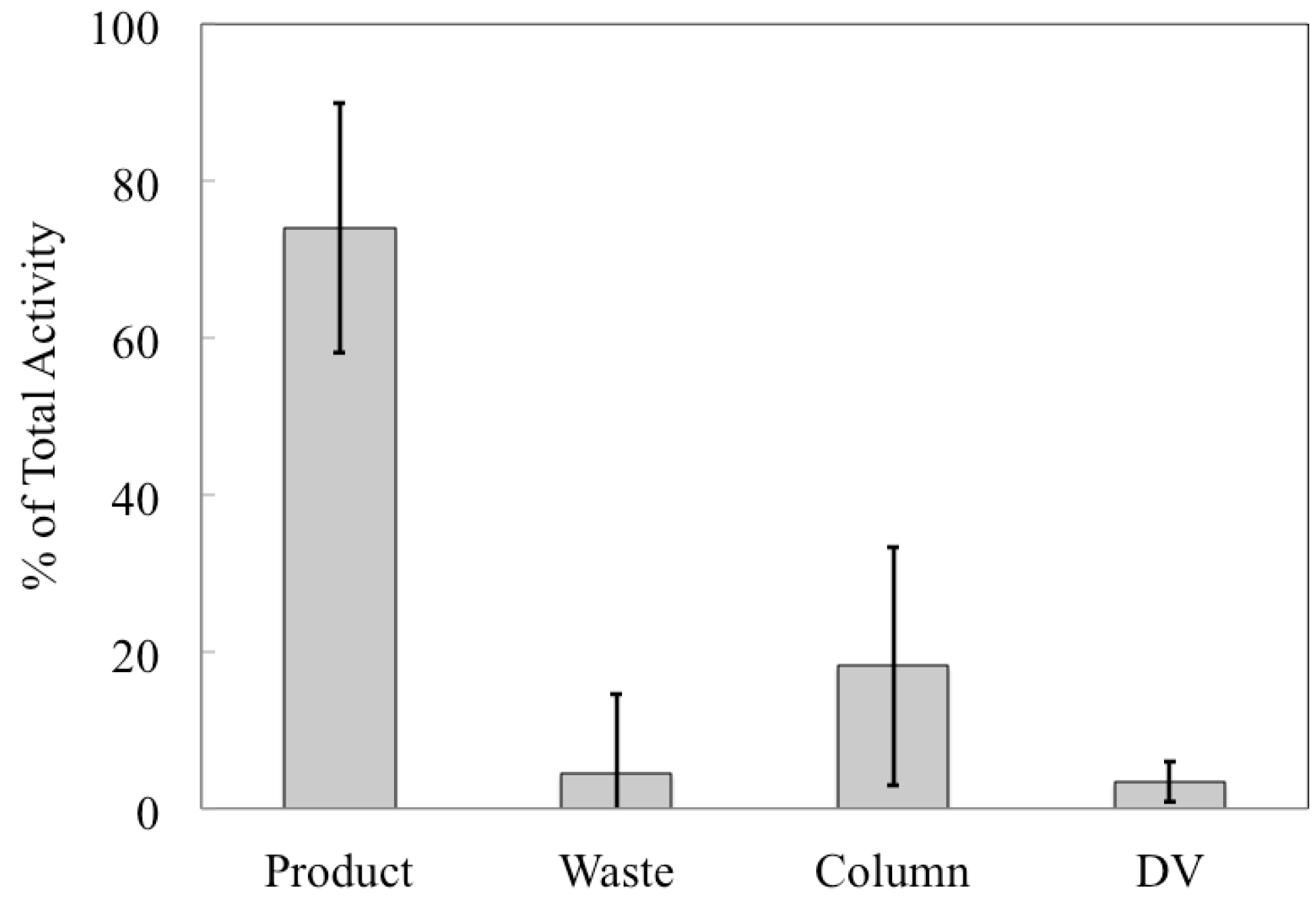
etc., but it allows us to compare the performance of the automated chemical separation for productions with different starting activities. The majority (74% ± 16%) of
89Zr was collected in the product vial and the largest loss of activity was to the column (18% ± 15%), which was greater than either the waste vial (4% ± 10%) or the dissolution vessel (3% ± 3%), as shown in
Figure 5. The percentage of
89Zr activity collected in the product vial is the recovery rate of the chemical separation that was carried out using the automated module and is an important performance indicator for the separation process. Our average recovery was significantly lower than other publications about
89Zr production (see ESI). In some cases this may be a result of us only collecting one elution instead of multiple fractions; otherwise, it may be because the stock of hydroxamate resin became less effective at binding Zr if it was stored for too long. We chose to make a stock of the resin for simplicity to avoid functionalizing a new batch of resin for each production.
Characteristics for ten selected productions are shown in
Table 3, including cyclotron parameters, predicted and estimated yields from the cyclotron, and ESA. All targets were bombarded with ~14.7 MeV protons. In this current work, Nb was selected as the material for the target holder primarily because of its chemical inertness toward HCl and its high melting temperature. The Nb target holders have been irradiated up to 15 μA without significant alterations of target or target holder. Since the same beam current, beam energy, and foil thickness were used for all productions in this work, the bombardment time (
t) was the only bombardment parameter that was adjusted to control the amount of activity produced in the cyclotron. The maximum bombardment time in this work was 4 h, which was equivalent to 60 μA∙h. The parameters for comparable publications varied widely. Excluding the Y
2O
3 pellet targets, only one publication [
26] used a foil that was close to ours in thickness. Also, the beam current, bombardment time, and μA∙h all varied widely across publications. Not surprisingly, the Y
2O
3 targets used lower currents, presumably because of reduced heat transfer, while the sputtered targets received high beam current for long bombardment times.
Figure 5.
Distribution of 89Zr in various components after each production. For each production, the activity in each of the four primary system components was decay-corrected to a common time point (end-of-bombardment in the cyclotron) and calculated as a percentage of the total activity. Displayed here are the average percentages for each component ± 1σ. N = 14.
Figure 5.
Distribution of 89Zr in various components after each production. For each production, the activity in each of the four primary system components was decay-corrected to a common time point (end-of-bombardment in the cyclotron) and calculated as a percentage of the total activity. Displayed here are the average percentages for each component ± 1σ. N = 14.
Table 3.
Results from ten productions of 89Zr selected for having the highest effective specific activities (sorted from highest to lowest).
Table 3.
Results from ten productions of 89Zr selected for having the highest effective specific activities (sorted from highest to lowest).
| Bombardment Time | Predicted Activityfrom Cyclotron | Estimated Total Activity | Effective Specific Activity |
|---|
| h (μA∙h) | mCi | mCi (% of Predicted) | mCi∙μmol−1 (% of
TSA) |
|---|
| 4 (60) | 79 | 62 (78%) | 353 (0.88%) |
| 2 (30) | 40 | 30 (74%) | 250 (0.63%) |
| 2 (30) | 40 | 30 (76%) | 134 (0.33%) |
| 2 (30) | 40 | 29 (74%) | 87 (0.22%) |
| 2 (30) | 40 | 27 (68%) | 73 (0.18%) |
| 2 (30) | 40 | 30 (75%) | 63 (0.16%) |
| 4 (60) | 79 | 37 (47%) | 59 (0.15%) |
| 2 (30) | 40 | 20 (50%) | 25 (0.06%) |
| 2 (30) | 40 | 29 (72%) | 21 (0.05%) |
| 1 (15) | 20 | 15 (75%) | 15 (0.04%) |
The predicted activity produced from each cyclotron bombardment is given in
Table 1 and was calculated using published empirical cross-section data for the
natY(
p,
n)
89Zr reaction [
29,
30,
31,
32,
33,
34,
35,
36] (see ESI) and the theoretical stopping ranges of protons in Y metal. The former data were accessed through the NNDC database [
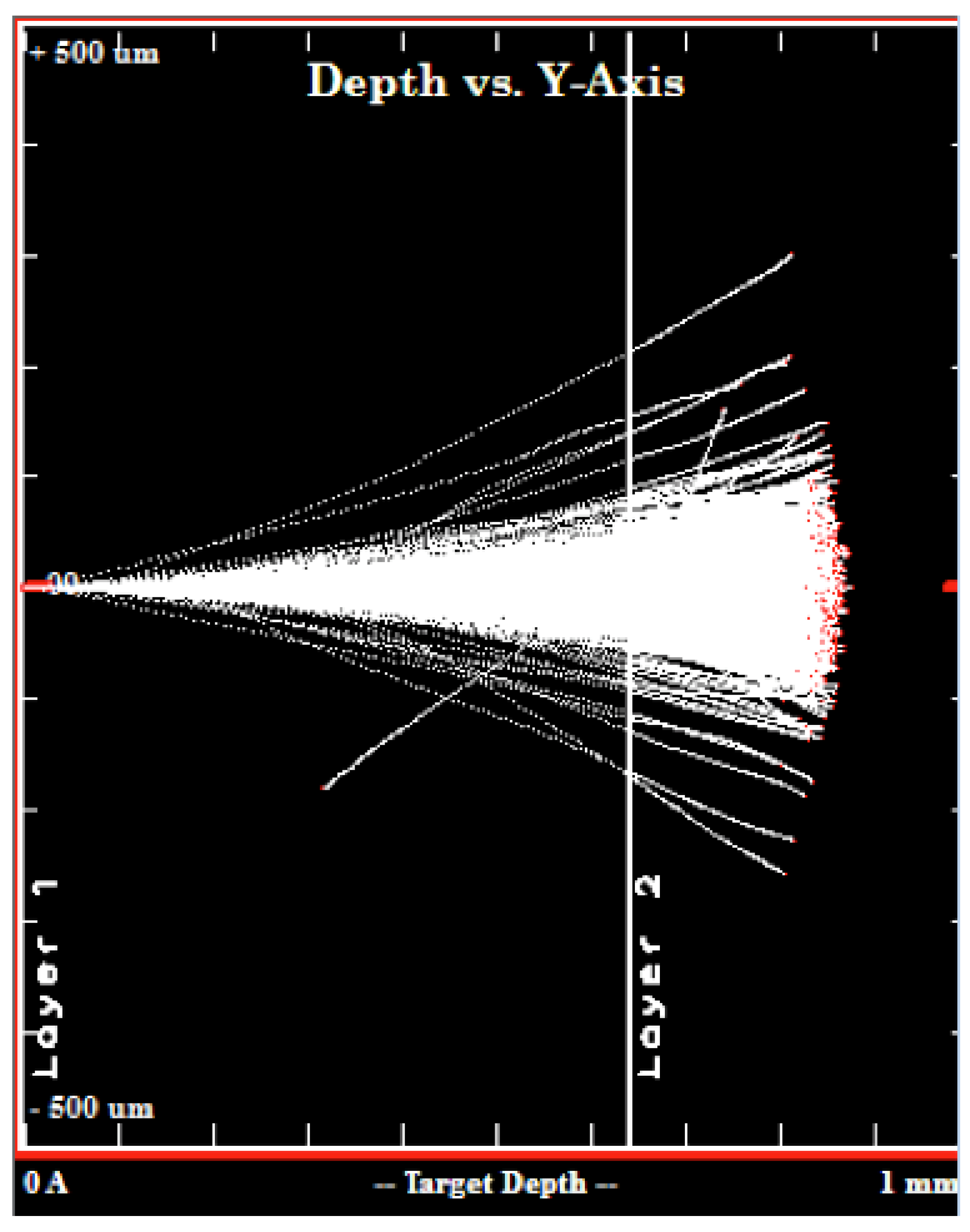
19] and the latter from calculations performed using the
SRIM software [
28]. By geometry, we estimated that 37% of the beam was lost on the lid of the target holder, instead of irradiating the exposed Y foil. The reaction rate (
R) was calculated, followed by the activity (
A), using the equation
A =
R(1−
e−λt) [
52], where
λ is the decay constant for
89Zr. The predicted values were also used to evaluate the total activity in the automated module as a percentage, also shown in
Table 1.
To confirm the radionuclidic purity of the solution in the product vial, gamma spectroscopy was performed. Typically, a short (10 m) scan of a diluted aliquot from the product vial was performed on the same day as each chemical separation, (see spectra in the ESI). For one production, a long (12 h) scan was performed after waiting for several half-lives (~22 days) of
89Zr to allow it to decay relative to longer-lived potential radiocontaminants, making them easier to detect by gamma spectroscopy. This spectrum indicated a radionuclidic purity of
89Zr equal to 99.998% of total activity (99.95% of total atoms) and identified
88Zr as the only radiocontaminant. Even though our recovery percentage was less than most other studies, our radionuclidic purity was on par with other publications, only one of which had a radionuclidic purity <99%. This is likely because of the minimal overlap of the excitation function for the
natY(
p,
n)
89Zr reaction with other
natY(
p,
x) reactions, especially the
natY(
p,2
n)
88Zr reaction at these irradiation energies. For most productions of
89Zr in this work, ESA was determined by DFO titration [
23]. ESA accounts for the presence of any metal isotope (stable or radioactive) that can be chelated by the selected chelator, in this case, DFO. For each production, the results of the DFO titration were plotted and fit with a sigmoidal dose-response curve to produce an EC50 value (see ESI), which was used to calculate ESA in mCi∙μmol
−1. ESA was also expressed as a percentage of TSA, with absolute and relative ESA values shown in
Table 1. Our results for ESA as a percentage of TSA were very low, although we expect when we produce and recover larger activities of
89Zr this will improve. Only one other publication [
23] reported ESA values, and our values were significantly less than their results, although typically our production activities were less as well. We did observe an improvement in ESA with increasing size of production batches, and we are saving samples to analyze for metal contaminants that would diminish the ESA. Elemental analysis will be performed on these samples by ion-coupled plasma mass spectrometry (ICP-MS).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}