1. Introduction
In earlier work we introduced a simple calculational model for predicting the primary substitution site for nucleophilic substitution involving the replacement of fluorine, initially in aromatic perfluorocarbons [
1] and subsequently in fluorinated aromatic systems containing both ring heteroatoms (principally nitrogen, but also oxygen, phosphorus and sulphur) and non-fluorine substituents [
2]. The model assumes the standard two-stage description of nucleophilic aromatic substitution involving the formation of a negatively charged intermediate, known as a Meisenheimer complex [
3], which is the anionic equivalent of the Wheland intermediate in electrophilic substitution [
4].
The negative charge in the Meisenheimer complex is delocalized into the aromatic π-system, which like the Wheland intermediate, can be considered as a resonance hybrid of multiple canonical forms.
Specifically, the model involves calculating the energy of all possible Meisenheimer complexes at a standard level of
ab initio theory (basic Hartree-Fock theory with the 6-31G* basis set [
5]: HF/6-31G*), using the fluoride ion as a “typical” nucleophile, with the preferred site for nucleophilic substitution corresponding to the most stable (lowest energy) complex.
This simple model has been remarkably successful in predicting not only the primary site in aromatic nucleophilic substitution reactions, but also secondary sites. In our two previous papers on this topic [
1,
2] we have presented correct predictions for primary and secondary substitution sites for over 70 different nucleophilic substitution reactions. Currently we are aware of only one system (perfluorophthalazine) where our model fails to predict the correct primary substitution site (although there are certainly likely to be more). Although the model is only meant to be qualitative we note a recent attempt to use a very similar model quantitatively [
6].
Our Meisenheimer model nominally involves a purely thermodynamic rationale, i.e., the thermodynamically most stable Meisenheimer complex is the one most likely to form, and the substitution pattern follows directly from that. The underlying assumptions are, first, that the substitution reaction is indeed two-step, involving an at least meta-stable Meisenheimer complex as intermediate; second, that barrier heights—both from reactant to intermediate and intermediate to product—are either more or less identical in all cases, and can therefore be ignored, or reflect the thermodynamic stability in the Meisenheimer complex so that considering only the latter gives the correct substitution pattern; and third, that varying the reaction conditions, principally the solvent and the actual nucleophile itself, makes no difference to the preferred substitution site, i.e., the mechanism is essentially the same for all nucleophiles in all solvents. Even for nucleophilic substitution reactions that are one-step, i.e., do not involve a Meisenheimer intermediate, the model can still be successfully applied provided the Meisenheimer complex can reliably predict the relative energies in the respective transition states.
In the last few years liquid ammonia has become a very useful reagent for aminating aromatic fluorocarbons. It has proven to be a useful synthetic method as it is capable of substituting the amino group (-NH
2) for fluorine site-specifically in high yield. This use of liquid ammonia has been pioneered primarily by the Malykhin group at Novosibirsk State University in Russia. They have published a number of papers from 2007 onwards involving amination of polyfluorinated benzenes and pyridines [
7,
8], perfluorobiphenyl [
7], perfluoronaphthalene [
9] and tetrafluorophthalic acid [
10]. Additionally, Page and coworkers have found that liquid ammonia aminates 1-nitro-2,4-difluorobenzene dominantly in the 2-position [
11]. Collectively these papers present around 20 different nucleophilic substitution reactions (all aminations), the vast majority of which we have not looked at in our previous work. They represent a further series of reactions to test the Meisenheimer model.
2. Results and Discussion
For all of the calculations presented in this work we used the PQS program package from Parallel Quantum Solutions together with the associated graphical user interface, PQSMol [
12,
13].
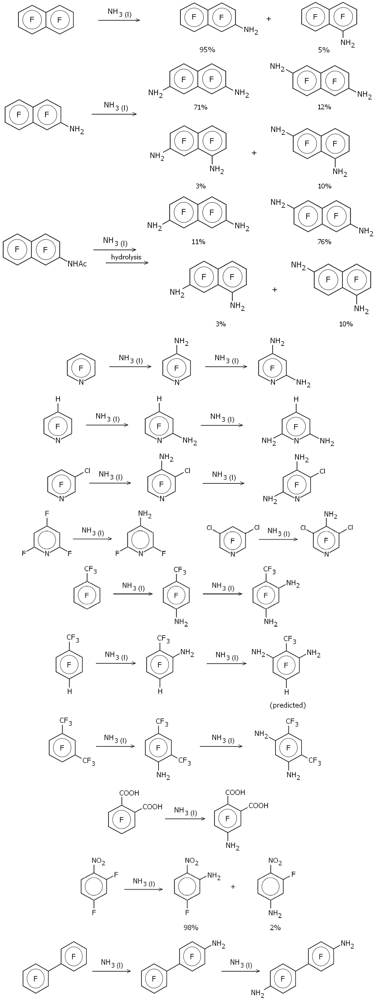
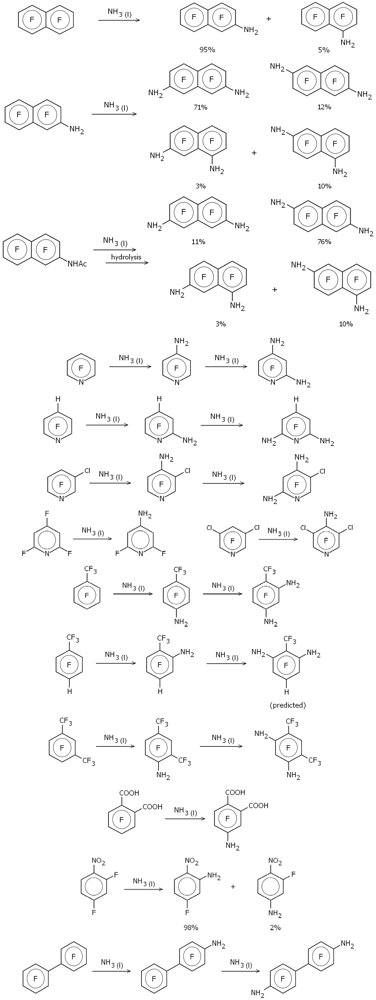
Figure 1 depicts schematically the various amination reactions examined (as previously noted, the majority from the Malykhin group [
7,
8,
9,
10]). Since all of the reactions
are aminations, we have made an important modification to our standard calculational approach: instead of using the fluoride ion as a generic attacking nucleophile we specifically use NH
2−. The use of the actual nucleophile instead of the fluoride ion as a possible extension to the basic model was mentioned in our last paper [
2], and it just seemed dogmatic to us to use fluorine when the substituting group was known to be NH
2 in every case.
Figure 1.
Amination reactions in liquid ammonia.
Figure 1.
Amination reactions in liquid ammonia.
The use of NH2− as the attacking nucleophile brings both advantages and disadvantages compared to F−. One advantage of using the single-atom fluoride ion as a generic attacking nucleophile is that there are no additional conformational issues in the resulting Meisenheimer complex, greatly simplifying the calculations. On the other hand it is known that most substituents to an aromatic ring direct para as far as replacement of fluorine is concerned, with any preferable ortho substitution nearly always resulting from direct interaction between the attacking nucleophile and the substituent, an interaction that cannot be modeled using fluorine. If we want to properly take into account interactions between an existing (already substituted) amino group and an attacking nucleophile, especially ortho to the existing amino substituent, then it seems pretty clear that the attacking nucleophile has to be represented more realistically, and so for these amination reactions we used NH2−.
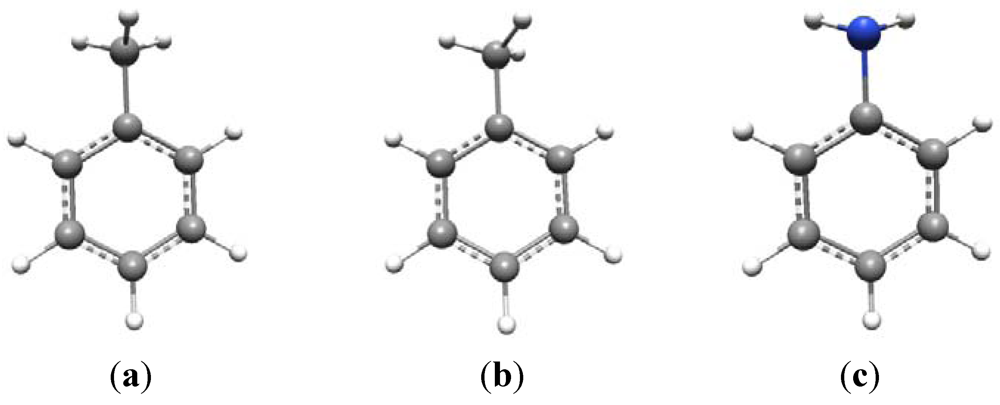
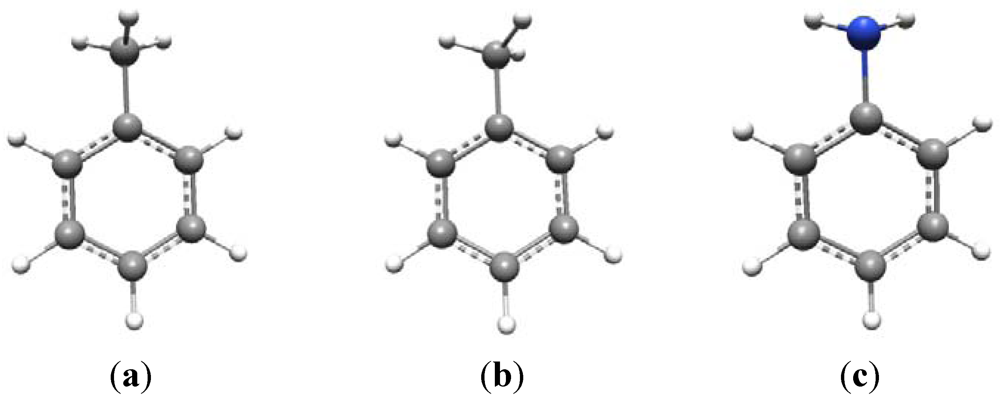
The conformational issue is more problematic than might at first be thought. For example, consider toluene (
Scheme 1a and
b). This is known to have C
s symmetry and its vibrational spectrum can be fully analyzed under this point group [
14]. Given C
s symmetry, there are two possible configurations of the methyl group—as shown in (a) and (b) —in which the symmetry plane is either perpendicular to, or in the plane of, the aromatic ring, respectively, bisecting a methyl HCH angle. The ground state geometry actually conforms to (a) with (b) being the transition state for methyl rotation. At room temperature the methyl group is essentially a free rotor with a 3-fold torsional potential.
The amino group is conformationally similar to the methyl group with one of the C-H bonds replaced by a lone pair, although the two N-H bonds in aniline are nowhere near as “bent” out of the plane of the aromatic ring as are the methyl C-H bonds in toluene. Aniline also has C
s symmetry in its ground state, with the lone pair occupying the position of the symmetry-unique C-H bond of the methyl group in toluene,
i.e., perpendicular to the benzene ring (
Scheme 1c).
Scheme 1.
Conformational preferences in toluene and aniline.
Scheme 1.
Conformational preferences in toluene and aniline.
Now as soon as you add, e.g., a second methyl group to toluene, the conformational issues increase significantly. Not only do you have the conformation of the new methyl group to consider, but the local three-fold symmetry of the original methyl group no longer holds globally and the three methyl hydrogen atoms are no longer equivalent. More to the point for this work, the same thing applies of course to the amino group.
We have no desire to carry out an exhaustive conformational analysis each time we calculate the energy of a given Meisenheimer complex. We have found that the following tactics when constructing the initial starting geometry of a Meisenheimer complex increases the likelihood of optimizing to the global or a near-global minimum:
One of the reactions studied by Malykhin and coworkers
was included in our previous work [
2], namely the amination of perfluoronaphthalene in liquid ammonia [
9]. This was done with the standard Meisenheimer model in which the attacking nucleophile was the fluoride ion. Malykhin and coworkers found that single amination of perfluoronapthalene gave dominantly (~95%) the 2-substituted isomer; further amination of this (or direct diamination of perfluoronaphthalene) gave a mixture of the 2,7-, 2,6-, 1,7- (2,8-) and 1,6- (2,5-) isomers with the former dominating (~70%). Virtually all known nucleophilic di-substitution reactions involving perfluoronaphthalene result primarily (>75%) in the 2,6-isomer and the amination in liquid ammonia was claimed to be “the first example of the predominant substitution at position 7 in 2-substituted polyfluoronaphthalenes” [
9].
These results were nicely reproduced with our standard Meisenheimer model and the first thing we are going to do here is to check that when the fluoride ion is replaced in our calculations by NH
2− we still reproduce the principal experimental results.
Table 1 gives the relative energies (in kcal/mol) of each possible Meisenheimer complex, with the lowest energy complex–and hence the predicted most favorable site for nucleophilic substitution–taken as the energy zero. Results are shown both for NH
2− (this work) and F- (taken from reference [
2]) as nucleophile.
Table 1.
Relative Energies (kcal/mol) of the various Meisenheimer complexes for 2-amino-heptafluoronaphthalene.
Table 1.
Relative Energies (kcal/mol) of the various Meisenheimer complexes for 2-amino-heptafluoronaphthalene.
| Site | Fluoride 1 | Amino 2 |
|---|
| 2,1- | 4.0 | 4.8 |
| 2,3- | 2.2 | 2.9 |
| 2,4- | 1.5 | 3.6 |
| 2,5- | 0.6 | 2.4 |
| 2,6- | 1.3 | 0.9 |
| 2,7- | 0.0 | 0.0 |
| 2,8- | 1.9 | 3.7 |
As can be seen, the results are similar, but not identical. Both sets of calculations correctly predict that 7-substitution is the most favorable with 5- and 6-substitution as the most likely secondary sites; however the energy ordering of the secondary sites is reversed. Neither set suggests that formation of the 1,7-isomer (the 2,8-) is particularly likely; however we have found that the most favorable substitution sites in 1-amino-heptafluoronapthalene (a by-product in the single amination reaction; see
Figure 1) are positions 6 and 7, so this may well be a major source of the 1,6- and 1,7-isomers, especially for the direct di-amination reaction. Either way, the results shown in
Table 1 certainly give us confidence in further predictions using NH2− as nucleophile.
Relative energies (in kcal/mol) of the various Meisenheimer complexes for the amination reactions shown in
Figure 1 are given in
Table 2. Comparison of the experimental results with the relative energies from
Table 2 show that for every reaction depicted in
Figure 1 the Meisenheimer model accurately predicts the primary substitution site. Furthermore, the large difference in relative energies between the lowest energy complex and the next lowest suggests that there is little secondary substitution in the majority of cases; again this is in full agreement with the experimental observations. For example, Malykhin and coworkers state that aminodefluorination of tetrafluorophthalic acid in anhydrous ammonia gives the 4-amino substituted isomer selectively and in high yield [
10], while Page and coworkers found that amination of 1-nitro-2,4-difluorobenzene gave 98% of the 2-substituted isomer and 2% of the 4-substituted [
11]. The relative energy differences between the two different Meisenheimer complexes in these two examples are 12.3 and 2.9 kcal/mol, respectively (see
Table 2). Note that this is a clear case of ortho (not para) substitution to the nitro group which is correctly predicted by the Meisenheimer model.
Table 2.
Relative energies (kcal/mol) of the various Meisenheimer complexes for the amination reactions shown in
Figure 1.
Table 2.
Relative energies (kcal/mol) of the various Meisenheimer complexes for the amination reactions shown in Figure 1.
| System | E (relative) | Subst. site | System | E (relative) | Subst. site |
|---|
| 2-acetylamidoheptafluoronaphthalene | perfluorotoluene | |
| 1-NH2 | 5.7 | | 2-NH2 | 6.9 | |
| 3-NH2 | 5.2 | | 3-NH2 | 17.5 | |
| 4-NH2 | 9.7 | | 4-NH2 | 0.0 | p |
| 5-NH2 | 7.8 | | | | |
| 6-NH2 | 0.0 | p | 4-aminoperfluorotoluene |
| 7-NH2 | 1.5 | s | 2-NH2 | 0.0 | p |
| 8-NH2 | 3.4 | | 3-NH2 | 12.5 | |
| perfluoropyridine | 1-(CF3)-2,3,5,6-tetrafluorobenzene |
| 2-NH2 | 9.9 | | 2-NH2 | 0.0 | p |
| 3-NH2 | 23.6 | | 3-NH2 | 9.1 | |
| 4-NH2 | 0.0 | p | | | |
| | | | 1-(CF3)-2-amino-3,5,6-trifluorobenzene |
| 4-amino-2,3,5,6-tetrafluoropyridine | 3-NH2 | 10.5 | |
| 2-NH2 | 0.0 | p | 5-NH2 | 6.0 | |
| 3-NH2 | 14.5 | | 6-NH2 | 0.0 | p |
| 2,3,5,6-tetrafluoropyridine | 1,3-di(CF3)-2,4,5,6-tetrafluorobenzene |
| 2-NH2 | 0.0 | p | 2-NH2 | 9.1 | |
| 3-NH2 | 0.8 | | 4-NH2 | 0.0 | p |
| | | | 5-NH2 | 26.0 | |
| 2-amino-3,5,6-trifluoropyridine | | | |
| 3-NH2 | 12.2 | | 1,3-di(CF3)-4-amino-2,5,6-trifluorobenzene |
| 5-NH2 | 7.9 | | 2-NH2 | 8.9 | |
| 6-NH2 | 0.0 | p | 5-NH2 | 30.5 | |
| | | | 6-NH2 | 0.0 | p |
| 3-chloro-2,4,5,6-tetrafluoropyridine | | | |
| 2-NH2 | 10.3 | | tetrafluorophthalic acid |
| 4-NH2 | 0.0 | p | 3-NH2 | 12.3 | |
| 5-NH2 | 27.7 | | 4-NH2 | 0.0 | p |
| 6-NH2 | 6.3 | | | | |
| | | | 1-nitro-2,4-difluorobenzene |
| 3-chloro-4-amino-2,5,6-trifluoropyridine | 2-NH2 | 0.0 | p |
| 2-NH2 | 3.1 | | 4-NH2 | 2.9 | |
| 5-NH2 | 22.4 | | | | |
| 6-NH2 | 0.0 | p | perfluorobiphenyl |
| | | | 2-NH2 | 8.7 | |
| 2,4,6-trifluoropyridine | 3-NH2 | 16.5 | |
| 2-NH2 | 3.2 | | 4-NH2 | 0.0 | p |
| 4-NH2 | 0.0 | p | | | |
| | | | 4-aminoperfluorobiphenyl |
| 3,5-dichloro-2,4,6-trifluoropyridine | 2-NH2 | 11.4 | |
| 2-NH2 | 6.2 | | 3-NH2 | 20.8 | |
| 4-NH2 | 0.0 | p | 2’-NH2 | 8.8 | |
| | | | 3’-NH2 | 15.2 | |
| | 4’-NH2 | 0.0 | p |
One reaction for which we do predict significant secondary substitution (
i.e., more than a few percent) is for the amination of 2-acetylamidoheptafluoronaphthalene (
Table 2). This was one of the reactions studied in reference [
9] along with 2-aminoheptafluoronaphthalene (see
Table 1). Experimentally amination of 2-acetylamidoheptafluoronaphthalene gives mainly the 6- and 7-substituted isomers in a 5:1 ratio (similar to the amination of 2-aminoheptafluoronaphthalene except that the ratio is reversed). Our calculations, in which the 6- and 7-substituted Meisenheimer complexes have the lowest energy, with the 7-substituted complex 1.5 kcal/mol higher in energy than the 6-substituted, fully concur with the experimental observations.
Note that no experimental data was reported for the amination of 1-trifluoromethyl-2-amino-3,5,6-trifluorobenzene and the depiction in
Figure 1—substitution at position 6—is theoretical only, derived from the calculational results shown in
Table 2. This is thus a prediction based on our model which is open to experimental confirmation.




{kind=link}
{kind=link}