
Revised Taxonomy of Rhabdoviruses Infecting Fish and Marine Mammals


1
School of Chemistry and Molecular Biosciences, The University of Queensland, St. Lucia, QLD 4067, Australia
2
Laboratory of Ploufragan-Plouzané-Niort, Technopole Brest Iroise, ANSES, 29280 Plouzané, France
3
Western Fisheries Research Center, US Geological Survey, 6505 NE 65th Street, Seattle, WA 98115, USA
4
Unit Lyssavirus Epidemiology and Neuropathology, Université Paris Cité, Institut Pasteur, 28 Rue du Docteur Roux, CEDEX 15, 75724 Paris, France
*
Author to whom correspondence should be addressed.
Animals 2022, 12(11), 1363; https://doi.org/10.3390/ani12111363
Submission received: 24 April 2022
/
Revised: 15 May 2022
/
Accepted: 24 May 2022
/
Published: 26 May 2022
(This article belongs to the Special Issue Emerging and Re-emerging Fish and Shellfish Viruses - Special Issue Dedicated to Dr. James R. Winton)
Abstract
:Simple Summary
The Rhabdoviridae is a family of viruses that includes some important pathogens of fish and marine mammals. Aspects of the taxonomic classification of fish viruses assigned to this family have recently been reviewed by the International Committee on Taxonomy of Viruses (ICTV). This paper describes the newly approved taxonomy, including the assignment of new subfamilies and new virus species. The paper also considers a taxonomic conundrum presented by viruses assigned to one group of fish rhabdoviruses (genus Novirhabdovirus) for which assignment to the family Rhabdoviridae may not be appropriate.
Abstract
The Rhabdoviridae is a large family of negative-sense (-) RNA viruses that includes important pathogens of ray-finned fish and marine mammals. As for all viruses, the taxonomic assignment of rhabdoviruses occurs through a process implemented by the International Committee on Taxonomy of Viruses (ICTV). A recent revision of taxonomy conducted in conjunction with the ICTV Rhabdoviridae Study Group has resulted in the establishment of three new subfamilies (Alpharhabdovirinae, Betarhabdovirinae, and Gammarhabdovirinae) within the Rhabdoviridae, as well as three new genera (Cetarhavirus, Siniperhavirus, and Scophrhavirus) and seven new species for viruses infecting fish or marine mammals. All rhabdovirus species have also now been named or renamed to comply with the binomial format adopted by the ICTV in 2021, comprising the genus name followed by a species epithet. Phylogenetic analyses of L protein (RNA-dependent RNA polymerase) sequences of (-) RNA viruses indicate that members of the genus Novirhabdovirus (subfamily Gammarhabdovirinae) do not cluster within the Rhabdoviridae, suggesting the need for a review of their current classification.
1. Introduction
The Rhabdoviridae is a large and ecologically diverse family of viruses, members of which infect plants, vertebrates, and/or invertebrates; many rhabdoviruses are transmitted by arthropod vectors in which they replicate [1,2,3]. The negative-sense, single-stranded RNA ([-] ssRNA) genome of rhabdoviruses typically features five structural protein genes (N, P, M, G, and L) but commonly contains additional genes encoding nonstructural accessory proteins. For animal rhabdoviruses, the enveloped virions are typically bullet-shaped with prominent surface projections and contain a rigidly assembled tubular nucleocapsid with helical symmetry. Rhabdoviruses infecting fish and marine mammals include important pathogens that can have significant economic impacts on fish farming and environmental impacts on wild fish populations [4,5].
2. Virus Classification and the Virus Species Concept
The taxonomic classification of viruses is the responsibility of the International Committee on Taxonomy of Viruses (ICTV) through the authority conferred by the International Union of Microbiological Societies (IUMS) [6]. Viruses are classified into hierarchical taxonomic ranks based primarily on their evolutionary relationships with a demarcation of ranks reflecting distinguishable genetic and phenotypic characteristics. To accommodate the rapidly growing and highly diverse nature of the known virosphere, the ICTV has recently expanded the scope of virus classification to include 15 taxonomic ranks extending from realms at the highest level to the lowest level of virus species [7]. There are minimum requirements that a virus species must be assigned to a genus and that all RNA viruses employing an RNA-directed RNA polymerase (RdRP) must be assigned to the realm Riboviria; the assignment of viruses to intermediate taxonomic levels varies according to the availability of data on the relevant evolutionary relationships [8].
Central (but often poorly understood) aspects of virus classification are the concept of virus species and the difference between a virus and a virus species [9]. Viruses are the concrete entities that virologists study and work with daily. They are named according to common practice in the scientific literature, usually following the lead of the first report of the virus. Viruses can be isolated and purified, and their genomes can be sequenced and can infect a host in which they may cause disease. A virus species, on the other hand, is an abstract taxonomic category. One cannot isolate, purify, or sequence a virus species as they do not physically exist. Therefore, one can have an isolate or strain of a virus but not of a virus species. Virus species are named according to the rules set recently by the ICTV; the species name now must be binomial, the first word being the genus name and the second word (the species epithet) being a unique freeform identifier [10]. Unlike virus names that should never include italics (even when adopting the host species name) and are commonly abbreviated, virus species names are always italicized and should not be abbreviated. A common misconception is that the virus species name is the formal or scientific name of a virus. Unlike the practice used in other branches of biology, a virus species name is not a formal substitute for a virus name. A virus is not assigned as a species; a virus is assigned taxonomically to the rank of species.
The ICTV has also now abandoned the concept of the “type species” that was used previously to identify a virus representing a typical member of a genus [10]. The requirement for a type species was largely historical; however, it is now evident that, although viruses selected to represent the type species may have been either the first discovered or most studied member of a genus, they were quite often not necessarily typical of all members of the genus. With the increasing importance of phylogenetic relationships in defining genera, it is the relationship between the sequences of the member viruses, and not the sequence of any particular member, that defines a genus. Consequently, rather than a “type species”, the ICTV now requires the identification of at least one “exemplar” virus sample for each virus species and a GenBank nucleotide sequence deposition for each exemplar virus. Typically, a complete or near-complete coding sequence is required for classification of a virus.
3. The Taxonomic Structure of the Family Rhabdoviridae
Rhabdoviruses were named originally for the apparently unique and characteristic morphology of the virions (rhabdos [Greek] = rod). Animal rhabdoviruses were described as bullet- or cone-shaped, and plant rhabdoviruses as rod-shaped with two rounded ends. The family Rhabdoviridae was established by the ICTV in 1976 comprising only two formally approved genera (Vesiculovirus and Lyssavirus) and 15 viruses assigned to species [11]. Following ratification by the ICTV membership in February 2022, the family Rhabdoviridae currently comprises some 45 genera and 265 species approved for viruses infecting or detected in plants, invertebrates (arthropods, nematodes), or vertebrates (mammals, amphibians, reptiles, birds, fish) [12]. Within the Rhabdoviridae, there are now three subfamilies (Alpharhabdovirinae, Betarhabdovirinae, Gammarhabdovirinae), two of which contain viruses of fish and marine mammals as detailed below. The family is also now assigned to higher taxonomic ranks: realm Riboviria, kingdom Orthornavirae, phylum Negarnaviricota, subphylum Haploviricotina, class Monojiviricetes, order Mononegavirales [13].
The taxonomic structure within the Rhabdoviridae is based primarily on evolutionary relationships determined by phylogenetic analysis of rhabdovirus L protein (RdRP) amino acid sequences [12]. The L protein is the most highly conserved of the rhabdovirus proteins, allowing relationships to be determined across the extent of this diverse family, and indeed beyond to include other [-] ssRNA viruses. The use of a single genetic marker for the identification of evolutionary relationships is justified by the absence or extremely rare occurrence of genetic recombination in rhabdoviruses [1].
The demarcation of taxa within the family generally reflects the ecological context of rhabdovirus evolution through which clusters of closely related viruses usually share similar categories of host and/or arthropod vector. The inherently dynamic nature of rhabdovirus genome evolution is also a consideration in the demarcation of genera [1]. Although core structural protein genes (particularly N, P, M, and L) are invariably retained, accessory genes may be gained de novo and subsequently lost during rhabdovirus genome evolution and, as a consequence, viruses clustering at the genus level usually share similar genome architectures [1,14]. Ultimately, viruses assigned to each subfamily or genus must be monophyletic based on L protein sequences but the points of demarcation can be somewhat arbitrary, depending on whether the chosen approach is based on “lumping” or ”splitting”.
The demarcation of rhabdovirus species requires consideration of several criteria typically based on amino acid sequence identities of structural proteins, natural host/vector associations, and, when available, the virus neutralization phenotype. Pathogenicity is considered to be too much dependent on environmental and host factors, and too easily modified by mutation, to be a useful consideration in species demarcation. Nevertheless, despite the variable nature of this approach as applied to viruses assigned to different genera, it can be argued that clusters of viruses representing the species taxon do occur naturally and can be identified by clear discontinuities in the amino acid or nucleotide sequence identity profiles of virus isolates. Although potentially influenced by the variation in the rates of evolution across the family, isolates of viruses assigned to individual rhabdovirus species typically display the protein amino acid sequence divergence of <10% in the L and N proteins, and <15% in the G protein.
4. The Subfamily Alpharhabdovirinae
The Alpharhabdoviridae is the largest subfamily currently assigned within the Rhabdoviridae, comprising 31 genera and 189 approved species for viruses infecting mammals, amphibians, reptiles, birds, fish, insects, ticks, or nematodes [12]. Viruses assigned to the Alpharhabdovirinae have sometimes been referred to informally as dimarhabdoviruses (dipteran-mammalian rhabdoviruses), but this term does not correctly capture the extent of ecological diversity within the clade [15]. The subfamily now includes five genera for viruses infecting teleost fish and marine mammals (Table 1). Each genus forms a monophyletic clade of viruses based on the alignment of L protein sequences (Figure 1). Demarcation of the genera is based on considerations of the different ecological contexts in which the viruses have evolved and the genetic distances between the clades. The assignment of genera may potentially change in the future as more viruses are discovered and evolutionary relationships are more clearly delineated.
The genus Sprivivirus currently includes two species: Sprivivirus cyprinus and Sprivivirus esox. The species Sprivivirus cyprinus is assigned for spring viraemia of carp virus (SVCV). SVCV causes a lethal hemorrhagic disease in cyprinids, particularly common carp (Cyprinus carpio). The exemplar sample (VR-1390; GenBank U18101) was isolated from diseased common carp in former Yugoslavia more than 50 years ago [16]. Various genetic lineages of SVCV have since been reported from Europe, Asia, and the Americas [17]. The species Sprivivirus esox is assigned for pike fry rhabdovirus (PFRV). The exemplar sample (F4; Genbank FJ872827) was isolated in 1972 during an outbreak of hemorrhagic disease (red disease) in cultured northern pike (Esox lucius) in the Netherlands [18]. Two other viruses are classified as members of the species Sprivivirus esox. Grass carp rhabdovirus (GCRV V76; GenBank KC113518) and tench rhabdovirus (TRV S64; GenBank KC113517) were each isolated in Germany in 1982 from cyprinids of two species (Ctenopharyngodon idella and Tinca tinca, respectively).The amino acid sequence divergence (p-distance) amongst 48 SVCV isolates sampled from fish of different species in Europe, Asia, and North America over a period of more than 50 years is ≤2.7% in the L protein, ≤3.4% in the N protein, and ≤8.7% in the G protein (Table 1). In contrast, the amino acid sequence divergence between all SVCV isolates and the three isolates assigned to the species Sprivivirus esox (PFRV, GCRV, and TRV) is ≥11.8% in L, ≥8.2% in N, and ≥25.4% in G (Table 1). The amino acid sequence divergence amongst the three viruses assigned to the species Sprivivirus esox is ≤6.7% in L, ≤5.6% in N, and ≤16.7% in G (Table 1). Although this range is greater than observed for SVCV isolates, it falls below the sequence divergence threshold separating viruses representing the two different species, justifying the assignment of all three isolates to the same sprivivirus species. Although virus neutralization and host range/susceptibility data are not available, the limited sequence divergence suggests that PFRV, GCRV, and TRV may be considered to be isolates of the same virus. Divergence data for all available sprivivirus L, N, and G amino acid sequences are shown in Supplementary Data File S1.
The genus Perhabdovirus currently includes four species: Perhabdovirus perca, Perhabdovirus trutta, Perhabdovirus anguilla, and Perhabdovirus leman. The species Perhabdovirus perca is assigned perch rhabdovirus (PRV). The exemplar sample (Dorson; GenBank JX679246) was isolated in 1981 from European perch (Perca fluviatilis) in France [19]. PRV has also been isolated from pikeperch (Sander lucioperca) in Belgium [20]. The species Perhabdovirus trutta is assigned for lake trout rhabdovirus (LTRV). The exemplar sample (903/87; GenBank AF434991) was isolated in 1987 from moribund brown trout (Salmo trutta) fingerlings from Finland [21]. Several other isolates of this virus, but sometimes named sea trout rhabdovirus, have been reported from France and Switzerland. The species Perhabdovirus anguilla is assigned for eel virus European X (EVEX), which was first isolated in 1976 in Japan from a shipment of European eels (Anguilla anguilla) from France [22,23]. The exemplar sample of EVEX (CV1153311; GenBank FN557213) was isolated from farmed European eel in the Netherlands in 1992. In 1974, a rhabdovirus was isolated from young American eel (Anguilla rostrata) imported from Cuba to Japan and named eel virus American (EVA) [23,24]. EVA and EVEX are morphologically, serologically, and genetically highly similar and are considered to be strains of the same virus [23,25]. Multiple isolates of EVEX from European eel in Europe and Japan have been sequenced. The new species Perhabdovirus leman is assigned for Leman virus (LEMV). The exemplar sample (LEMV 18/193; GenBank MN963996) was isolated in 1999 from symptomatic wild young perch collected from Lake Leman in France [26]. The amino acid sequence divergence amongst viruses assigned to the same perhabdovirus species is ≤5.2% in L, ≤6.1% in N, and ≤11.6% in G. The amino acid sequence divergence between viruses assigned to different perhabdovirus species is ≥13.1% in L, ≥15.9% in N, and ≥16.7% in G (Table 1; Tables S1–S3).
The new genus Cetarhavirus includes two species for viruses infecting aquatic mammals (Cetacea): Cetarhavirus lagenorhynchus and Cetarhavirus phocoena. The new species Cetarhavirus lagenorhynchus is assigned for dolphin rhabdovirus (DRV). The exemplar sample (pxV1; GenBank KF958252) was isolated from a white-beaked dolphin (Lagenorhynchus albirostris) stranded on the Dutch island of Schiermonnikoog in 1992 [27]. A neutralizing antibody to DRV has been detected in various cetaceans (dolphins, porpoises, whales) and pinnipeds (seals) sampled from the coast of northwest Europe or the Mediterranean Sea [27,28]. The new species Cetarhavirus phocoena is assigned for harbor porpoise rhabdovirus (HPRV). The exemplar sample (WVL17017A; GenBank MN103537) was isolated from a harbor porpoise (Phocoena phocoena) stranded off the coast of Alaska in 2013 [29]. These are the only reported samples of the viruses at this time. The amino acid sequence divergence between the exemplar samples of DRV and HPRV is 31.3% in L, 31.8% in N, and 56.1% in G (Table 1; Tables S1–S3).
The new genus Siniperhavirus currently includes two species: Siniperhavirus zoarces and Siniperhavirus chuatsi. The new species Siniperhavirus zoarces is assigned for eelpout rhabdovirus (EPRV). The exemplar sample (FSK0523; GenBank KR612230) was detected by high-throughput sequencing in samples of eelpout (Zoarces viviparous) collected during mass fish mortalities near Stockholm, Sweden, in 2014 [30]. This is the only virus currently assigned to this species. The new species Siniperhavirus chuatsi is assigned for Siniperca chuatsi rhabdovirus (SCRV). The exemplar sample (GenBank DQ399789) was isolated in 1997 from mandarin fish (Siniperca chuatsi) collected in Guangdong Province, China [31,32]. Several other viruses have been assigned as members of the species Siniperhavirus chuatsi. Hybrid snakehead rhabdovirus (C1207; GenBank KC519324) was isolated in 2012 from a moribund hybrid snakehead fish (Channa maculata × Channa argus cross) collected in Guangdong Province, China [33]. HSRV (Xingtan; GenBank KP876483) was subsequently isolated from hybrid snakehead fish from the same province of China in 2014 and shown to infect mandarin fish [34]. A third HSRV isolate (SHVV-02019; GenBank MW291462) was obtained from snakehead fish (Channa argus) in China in 2019. Chinese rice-field eel rhabdovirus (CrERV) (GenBank MH319839) was isolated from diseased Asian swamp eels (Monopterus albus) collected from Hubei Province, China, in 2017 [35]. Micropterus salmoides rhabdovirus (MSRV) was first discovered in juvenile mandarin fish collected in Guangdong Province, China, in 2011 [36]. A second sample (YH01; GenBank MK397811) was isolated from moribund largemouth bass (Micropterus salmoides) collected in Zhejiang Province, China, in 2017 [37]. A third isolate of the virus (FJ985; GenBank MT818233) was obtained from China in 2019. The amino acid sequence divergence amongst all of these isolates (SCRV, HSHV, CrERV, MSRV) is low (≤3.6% in L, ≤6.8% in N, and ≤10.6% in G) and they should all be considered to be variants of the same virus. The amino acid sequence divergence between viruses assigned to the two different siniperhavirus species is ≥28.8% in L, ≥33.7% in N, and ≥50.0% in G (Table 1; Tables S1–S3).
The new genus Scophrhavirus currently includes two species: Scophrhavirus maximus and Scophrhavirus chanodichthys. The new species Scophrhavirus maximus is assigned for Scophthalmus maximus rhabdovirus (SMRV). The exemplar sample (GenBank HQ003891) was isolated from turbot fish (Scophthalmus maximus) with signs of hemorrhagic disease collected from Shandong Province, China [38]. The new species Scophrhavirus chanodichthys is assigned for Wuhan redfin culter dimarhabdovirus (WhRCDRV). The exemplar sample (DSYS6218; GenBank MG600013) was detected by high-throughput sequencing in redfin culter (Chanodichthys erythropterus) collected in Hubei Province, China [39]. These are the only reported samples of the viruses at this time. The amino acid sequence divergence between the exemplar samples of SMRV and WhRCDRV is 46.5% in L, 62.6% in N, and 71.0% in G (Table 1; Tables S1–S3).
5. Subfamily Gammarhabdovirinae
The Gammarhabdovirinae is the smallest subfamily in the Rhabdoviridae, currently comprising only a single genus for viruses infecting teleost fish. The genus Novirhabdovirus currently includes four species: Novirhabdovirus salmonid, Novirhabdovirus piscine, Novirhabdovirus hirame, and Novirhabdovirus snakehead.
The species Novirhabdovirus salmonid is assigned for infectious hematopoietic necrosis virus (IHNV). The exemplar sample (WRAC; GenBank L40883) was isolated in 1982 from diseased rainbow trout (Oncorhynchus mykiss) in Idaho, USA [40]. IHNV causes economically important disease in a wide variety of salmonid fish species. The virus is enzootic in coastal areas and river systems throughout western North America but has spread to Asia and Europe through translocation of infected stock [41]. IHNV is resolved globally into five major genogroups (U, M, L, J, and E) and several subgroups that vary in geography and host specificity [42]. The amino acid sequence divergence across all available IHNV isolates of all genotypes is ≤2.4% in L, ≤12.6% in N, and ≤10.2% in G (Table 1).
The species Novirhabdovirus piscine is assigned for viral hemorrhagic septicaemia virus (VHSV). The exemplar sample (Fil3; GenBank Y18263) was isolated in 1962 from diseased rainbow trout (Oncorhynchus mykiss) in Denmark [43]. VHSV is considered to be a pathogen of major economic and environmental importance. It has been isolated from or detected in a wide range of teleost fish in Europe, Asia, and North America [44]. VHSV falls into four major genotypes (I–IV) and various sub-types with naturally confined geographic distributions [45,46,47]. The amino acid sequence divergence across all available VHSV isolates of all genotypes is ≤7.1% in L, ≤11.1% in N, and ≤11.5% in G (Table 1).
The species Novirhabdovirus hirame is assigned for hirame rhabdovirus (HIRRV). First detected in Japan in 1984, HIRRV causes a hemorrhagic disease characterized by congestion of the gonads and the accumulation of ascitic fluid [48]. The exemplar sample (CA9703; GenBank AF104985) was isolated in 1997 from Japanese flounder (Paralichthys olivaceus) cultured in the Republic of Korea [49,50]. HIRRV occurs in a wide range of marine fish in several countries in East Asia [49,51,52]. HIRRV has also caused mortalities in freshwater fish in Europe, possibly as a result of translocation from East Asia [53]. The amino acid sequence divergence across the small number of available HIRRV isolates is 0.4% in L, 0.8% in N, and ≤1.2% in G (Table 1).
The species Novirhabdovirus snakehead is assigned for snakehead rhabdovirus (SHRV). The exemplar sample (GenBank AF147498) was isolated in 1986 from snakehead fish (Ophicephalus struatus) in Thailand [54,55] with a disease characterized by necrotic ulcerations. The disease was reported in wild and cultured snakehead fish in several countries of Southeast Asia and various other organisms (viruses, bacteria, fungi, and parasites) have been found in association with diseased fish [55,56]. Sequences are currently available only for the exemplar sample of SHRV.
The amino acid sequence divergence between viruses assigned to different novirhabdovirus species is ≥15.1% in L, ≥35.8% in N, and ≥22.9% in G (Table 1). Divergence data for all available novirhabdovirus L, N, and G amino acid sequences are shown in Supplementary Data File S2 and S3.
Classification of the genus Novirhabdovirus presents somewhat of a taxonomic conundrum. Historically, the novirhabdoviruses have been placed taxonomically within the Rhabdoviridae as they share many of the characteristics of classical rhabdoviruses. Virions are enveloped, bullet-shaped particles with clear surface projections and a helical nucleocapsid [57,58,59]. The novirhabdovirus genome comprises homologs of the five rhabdovirus structural protein genes (N, P, M, G, and L) as well as an additional gene (NV) encoding a unique nonstructural protein that has been shown to be involved in pathogenesis and evasion of host immune responses [60,61,62]. Conserved transcription initiation and transcription termination sequences flank each gene and, as occurs typically in all alpharhabdoviruses, the transcription termination sequences feature a conserved run of seven uridine residues [40,63]. Importantly, novirhabdoviruses encode a single type I transmembrane glycoprotein (G) that is structurally homologous with the G proteins of other animal rhabdoviruses, featuring a unique set of cysteine residues that stabilize the pre- and post-fusion-folded structures of the protein [64,65,66,67]. Nevertheless, phylogenetically, the novirhabdoviruses sit separately from all other members of the Rhabdoviridae.
Evolutionary analysis using the complete L protein (RdRP) sequences of viruses representing families in the order Mononegavirales indicates that novirhabdoviruses do not cluster monophyletically with other rhabdoviruses but tend to cluster with members of the families Paramyxoviridae, Pneumoviridae, and Filoviridae (Figure 2; Figure S1) [68]. The RdRP is considered to be the most useful marker for mapping viral evolutionary history as it the most highly conserved sequence element and is indicative of the core replicating lineage. Indeed, there is evidence that all viral RdRPs and reverse transcriptases are monophyletic [69,70], leading to the creation by the ICTV of the realm Riboviria to accommodate all viruses with RNA genomes, the phylum Negarnaviricota for all [-] ssRNA viruses, and various other intermediate and subsidiary taxonomic ranks [7]. Evolutionary analyses based on the phylogeny of mononegavirus L proteins cannot, therefore, be disregarded.
A possible explanation for the apparent dichotomy between the novirhabdovirus virion structure and RdRP-based phylogeny is that paramyxovirus, pneumovirus, and filovirus glycoprotein genes were introduced into an ancestral [-] ssRNA virus by recombination or lateral gene transfer after the novirhabdovirus lineage had diverged from that of other rhabdoviruses. Indeed, this appears to be supported by alignments of the type I transmembrane glycoprotein sequences of members of the family Chuviridae with the vesicular stomatitis Indiana virus (VSIV) and novirhabdovirus G proteins (Figure 3), which all share subsets of the same conserved set of cysteine residues. Although genetic recombination occurs very rarely in mononegaviruses, several other possible recombination events have been reported previously [72,73,74,75,76]. Indeed, the structural homology between the VSIV G protein and herpes simplex virus 1 glycoprotein gB indicates that they also have a common evolutionary origin involving an ancient recombination event [77]. Whatever the exact mechanism, it appears that some form of either recombination or lateral gene transfer would be required to arrive at the phylogenetic relationships determined using RdRP-based sequence alignments.
Nevertheless, we suggest that removal of the novirhabdoviruses from the Rhabdoviridae requires careful consideration. For example, do we know that alignments used to generate phylogenetic trees from such distantly related RdRP sequences are sufficiently reliable to be confident of the deep nodes? Certainly, alignments do vary according to the algorithms employed and parameters selected, and the reliability of sequence alignments used to infer deeply rooted phylogenies linking all RNA viruses has been questioned [69,78]. Moreover, it has been recognized that “even a correct and informative alignment does not guarantee correct phylogenetic reconstruction due to the technical limitations of the software, systematic biases of the available evolutionary models, and the fundamentally random nature of sequence divergence” [79] and that evolutionary relationships inferred from phylogenetic analysis need to take account of associated biological data [79]. The structural homology of hallmark genes such as capsid proteins has been used to define taxonomic relationships for higher taxonomic ranks of some DNA viruses for which there are no common genes displaying evident sequence homology [80,81]. In the absence of confidently reliable sequence alignments, should virion structural homology also be a consideration in the demarcation of some lower taxa? A global view of the molecular and structural properties of novirhabdoviruses may provide a more informative and useful guide to their taxonomic classification than an analysis based only on RdRP sequence alignments.
6. Conclusions
As the result of proposals approved by the ICTV in February 2022, all currently known rhabdoviruses infecting fish or marine mammals for which complete or near-complete genome sequences are available have now been assigned taxonomically to the ranks of genus and species. This is a significant advance as many of these viruses and their complete coding sequences have been known for years but they were not accommodated in the previous taxonomic structures. All rhabdovirus species, including those infecting fish or marine mammals, have also been named or renamed to comply with the binomial format approved by the ICTV in 2021. Importantly, these taxonomic assignments and the renaming of species have no bearing on the names of the viruses themselves, which will continue to follow the long-standing practice of reflecting the common usage adopted in the scientific literature.
In a practical sense, changes to virus taxonomy can be very disruptive to governments and industry as the taxonomic classification is often embedded in regulations and legislation that are intended to ensure timely reporting of detected pathogens and limit the spread of viral diseases. Nevertheless, virus taxonomy is by nature a complex tapestry that develops and evolves as knowledge of the virosphere expands. To remain useful and relevant, taxonomic classifications must adapt as new viruses are discovered and the evolutionary and ecological relationships between viruses are better understood. This is particularly so in the age of metagenomic sampling and high-throughput sequencing that has seen very rapid growth in the number of viral genome sequences and concomitant expansion in the number of newly assigned taxa. It is hoped that this review will assist by promulgating the current taxonomic assignments and by explaining some underlying principles of virus taxonomy that are often misunderstood by members of the scientific community.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ani12111363/s1, Figure S1: Maximum-likelihood (ML) trees inferred from the same trimmed L protein sequence alignment (916 amino acids) as that used in Figure 2 but using different methods to evaluate branch support; Table S1: Percentage amino acid sequence divergence (p-distances) estimated from a CLUSTAL W alignment of perhabdovirus, cetarhavirus, siniperhavirus, and scophrhavirus L proteins; Table S2: Percentage amino acid sequence divergence (p-distances) estimated from a CLUSTAL W alignment of perhabdovirus, cetarhavirus, siniperhavirus, and scophrhavirus N proteins; Table S3: Percentage amino acid sequence divergence (p-distances) estimated from a CLUSTAL W alignment of perhabdovirus, cetarhavirus, siniperhavirus, and scophrhavirus G proteins; Supplementary data file S1: Percentage amino acid sequence divergence (p-distances) estimated from CLUSTAL W alignments of sprivivirus L, N, and G proteins; Supplementary data file S2: Percentage amino acid sequence divergence (p-distances) estimated from CLUSTAL W alignments of novirhabdovirus L and N proteins; Supplementary data file S3: Percentage amino acid sequence divergence (p-distances) estimated from CLUSTAL W alignments of novirhabdovirus G proteins.
Author Contributions
Conceptualization, P.J.W.; methodology, P.J.W., L.B., G.K., L.D. and L.P.; formal analysis, P.J.W. and L.B.; investigation; writing—original draft preparation, P.J.W.; writing—review and editing, L.B., G.K. and L.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
References
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of genome size and complexity in the Rhabdoviridae. PLoS Path. 2015, 11, e1004664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, A.E.; Huot, O.B.; Martin, K.M.; Kondo, H.; Dietzgen, R.G. Plant rhabdoviruses-their origins and vector interactions. Curr. Opin. Virol. 2018, 33, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Winton, J.R. Emerging viral diseases of fish and shrimp. Vet. Res. 2010, 41, 51. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, B.; Beer, M.; Schutze, H.; Mettenleiter, T.C. Fish rhabdoviruses: Molecular epidemiology and evolution. Curr. Top. Microbiol. Immunol. 2005, 292, 81–117. [Google Scholar]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. 50 years of the International Committee on Taxonomy of Viruses: Progress and prospects. Arch. Virol. 2017, 162, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; Harrach, B.; et al. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; Junglen, S.; et al. Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019). Arch. Virol. 2019, 164, 2417–2429. [Google Scholar] [CrossRef] [Green Version]
- Zerbini, F.M.; Siddell, S.G.; Mushegian, A.; Walker, P.J.; Lefkowitz, E.J.; Adriaenssens, E.; Alfenas-Zerbini, P.; Duthil, B.; García, M.L.; Junglen, S.; et al. Differentiating between viruses and virus species by writing their names correctly. Arch. Virol. 2022, 167, 1231–1234. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef]
- Fenner, F. Classification and nomenclature of viruses. Second report of the International Committee on Taxonomy of Viruses. Intervirology 1976, 7, 1–115. [Google Scholar] [CrossRef]
- Walker, P.J.; Blasdell, K.R.; Calisher, C.H.; Dietzgen, R.G.; Kondo, H.; Kurath, G.; Longdon, B.; Stone, D.M.; Tesh, R.B.; Tordo, N.; et al. ICTV taxonomy profile: Rhabdoviridae. J. Gen. Virol. 2018, 99, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Adkins, S.; Agwanda, B.R.; Al Kubrusli, R.; Alkhovsky, S.V.; Amarasinghe, G.K.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2021 Taxonomic update of phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch. Virol. 2021, 166, 3513–3566. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K.R. Rhabdovirus accessory genes. Virus Res. 2011, 162, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Bourhy, H.; Cowley, J.A.; Larrous, F.; Holmes, E.C.; Walker, P.J. Phylogenetic relationships among rhabdoviruses inferred using the L polymerase gene. J. Gen. Virol. 2005, 86, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Fijan, N.; Petrinec, Z.; Sulimanovic, D.; Zwillenberg, L.O. Isolation of the viral causative agent from the acute form of infectious dropsy of carp. Veterinarsky Arhiv 1971, 41, 125–138. [Google Scholar]
- Ashraf, U.; Lu, Y.; Lin, L.; Yuan, J.; Wang, M.; Liu, X. Spring viraemia of carp virus: Recent advances. J. Gen. Virol. 2016, 97, 1037–1051. [Google Scholar] [CrossRef]
- De Kinkelin, P.; Galinard, B. Isolation and identification of the causative agent of “red disease” on pike (Esox licius L. 1766). Nature 1972, 241, 465–467. [Google Scholar] [CrossRef]
- Dorson, M.; Torchy, C.; Chilmonczyk, S.; de Kinkelin, P.; Michel, C. A rhabdovirus pathogenic for perch, Perca fluviatilis L.:Isolation and preliminary study. J. Fish. Dis. 1984, 7, 241–245. [Google Scholar] [CrossRef]
- Bigarré, L.; Plassiart, G.; de Boisséson, C.; Pallandre, L.; Pozet, F.; Ledoré, Y.; Fontaine, P.; Lieffrig, F. Molecular investigations of outbreaks of Perch perhabdovirus infections in pike-perch. Dis. Aquat. Org. 2017, 127, 19–27. [Google Scholar] [CrossRef]
- Koski, P.; Hill, B.J.; Way, K.; Neuvonen, E.; Rintamaki, P. A rhabdovirus isolated from brown trout [Salmo trutta m. lacustris (L.)] with lesions in parenchymatous organs. Bull. Eur. Assn Fish. Pathol. 1992, 12, 177–180. [Google Scholar]
- Sano, T.; Nishimura, T.; Okamoto, N.; Fukuda, H. Studies on viral diseases of Japanese fishes. VII A rhabdovirus isolated from European eel, Anguilla anguilla. Bull. Jap. Soc. Sci. Fish. 1977, 43, 491–495. [Google Scholar] [CrossRef]
- Galinier, R.; van Beurden, S.; Amilhat, E.; Castric, J.; Schoehn, G.; Verneau, O.; Fazio, G.; Allienne, J.F.; Engelsma, M.; Sasal, P.; et al. Complete genomic sequence and taxonomic position of eel virus European X (EVEX), a rhabdovirus of European eel. Virus Res. 2012, 166, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sano, T. Viral diseases of cultured fishes in Japan. Fish. Pathol. 1976, 19, 221–226. [Google Scholar] [CrossRef]
- Nishimura, T.; Toba, M.; Ban, F.; Okamoto, N.; Sano, T. Eel rhabdovirus, EVA, EXEX and their infectivity to fishes. Fish. Path. 1981, 15, 173–184. [Google Scholar] [CrossRef]
- Pallandre, L.; Luo, D.; Feuvrier, C.; Lieffrig, F.; Pozet, F.; Dacheux, L.; Bigarré, L. Revisiting the classification of percid perhabdoviruses using new full-length genomes. Viruses 2020, 12, 649. [Google Scholar] [CrossRef] [PubMed]
- Osterhaus, A.D.; Broeders, H.W.; Teppema, J.S.; Kuiken, T.; House, J.A.; Vos, H.W.; Visser, I.K. Isolation of a virus with rhabdovirus morphology from a white-beaked dolphin (Lagenorhynchus albirostris). Arch. Virol. 1993, 133, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegers, J.Y.; van de Bildt, M.W.; van Elk, C.E.; Schurch, A.C.; Tordo, N.; Kuiken, T.; Bodewes, R.; Osterhaus, A.D. Genetic relatedness of dolphin rhabdovirus with fish rhabdoviruses. Emerg. Inf. Dis. 2014, 20, 1081–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emelianchik, A.; Rodrigues, T.C.S.; Subramaniam, K.; Nielsen, O.; Burek-Huntington, K.A.; Rotstein, D.; Popov, V.L.; Stone, D.; Waltzek, T.B. Characterization of a novel rhabdovirus isolated from a stranded harbour porpoise (Phocoena phocoena). Virus Res. 2019, 273, e197742. [Google Scholar] [CrossRef] [PubMed]
- Axen, C.; Hakhverdyan, M.; Boutrup, T.S.; Blomkvist, E.; Ljunghager, F.; Alfjorden, A.; Hagstrom, A.; Olesen, N.J.; Juremalm, M.; Leijon, M.; et al. Emergence of a new rhabdovirus associated with mass mortalities in eelpout (Zoarces viviparous) in the Baltic Sea. J. Fish. Dis. 2017, 40, 219–229. [Google Scholar] [CrossRef]
- Tao, J.J.; Gui, J.F.; Zhang, Q.Y. Isolation and characterization of a rhabdovirus from co-infection of two viruses in mandarin fish. Aquaculture 2007, 262, 1–9. [Google Scholar] [CrossRef]
- Tao, J.J.; Zhou, G.Z.; Gui, J.F.; Zhang, Q.Y. Genomic sequence of mandarin fish rhabdovirus with an unusual small non-transcriptional ORF. Virus Res. 2008, 132, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wang, Q.; Wang, Y.; Liu, C.; Liang, H.; Fang, X.; Wu, S. Genomic characterization and taxonomic position of a rhabdovirus from a hybrid snakehead. Arch. Virol. 2014, 159, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wen, Y.; Hu, X.; Wang, W.; Liang, X.; Li, J.; Vakharia, V.; Lin, L. Breaking the host range: Mandarin fish is susceptible to a vesiculovirus derived from snakehead fish. J. Gen. Virol. 2015, 96, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Fan, Y.; Li, Z.; Zhao, J.; Zhou, Y.; Jiang, N.; Zeng, J.; Cain, K.; Zeng, L. Isolation, identification, and classification of a novel rhabdovirus from diseased Chinese rice-field eels (Monopterus albus). Arch. Virol. 2019, 164, 105–116. [Google Scholar] [CrossRef]
- Ma, D.; Deng, G.; Bai, J.; Li, S.; Yu, L.; Quan, Y.; Yang, X.; Jiang, X.; Zhu, Z.; Ye, X. A strain of Siniperca chuatsi rhabdovirus causes high mortality among cultured largemouth bass in South China. J. Aquat. Anim. Health 2013, 25, 197–204. [Google Scholar] [CrossRef]
- Lyu, S.J.; Yuan, X.M.; Zhang, H.Q.; Shi, W.D.; Hang, X.Y.; Liu, L.; Wu, Y.L. Isolation and characterization of a novel strain (YH01) of Micropterus salmoides rhabdovirus and expression of its glycoprotein by the baculovirus expression system. J. Zhejiang Uni. Sci. B 2019, 20, 728–739. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Tao, J.J.; Gui, L.; Zhou, G.Z.; Ruan, H.M.; Li, Z.Q.; Gui, J.F. Isolation and characterization of Scophthalmus maximus rhabdovirus. Dis. Aquat. Org. 2007, 74, 95–105. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Morzunov, S.P.; Winton, J.R.; Nichol, S.T. The complete genome structure and phylogenetic relationship of infectious hematopoietic necrosis virus. Virus Res. 1995, 38, 175–192. [Google Scholar] [CrossRef]
- Dixon, P.; Paley, R.; Alegria-Moran, R.; Oidtmann, B. Epidemiological characteristics of infectious hematopoietic necrosis virus (IHNV): A review. Vet. Res. 2016, 47, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurath, G. Fish novirhabdoviruses. In Rhabdoviruses: Molecular Taxonomy, Evolution, Genomics, Ecology, Cytopathology, and Control; Dietzgen, R.G., Kuzmin, I.V., Eds.; Caister Academic Press: Norfolk, UK, 2012; pp. 98–116. [Google Scholar]
- Jensen, M.H. Research on the virus of Egtved disease. Ann. N. Y. Acad. Sci. 1965, 126, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Skall, H.F.; Olesen, N.J.; Mellergaard, S. Viral haemorrhagic septicaemia virus in marine fish and its implications for fish farming—A review. J. Fish. Dis. 2005, 28, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Einer-Jensen, K.; Ahrens, P.; Forsberg, R.; Lorenzen, N. Evolution of the fish rhabdovirus viral haemorrhagic septicaemia virus. J. Gen. Virol. 2004, 85, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Snow, M.; Bain, N.; Black, J.; Taupin, V.; Cunningham, C.O.; King, J.A.; Skall, H.F.; Raynard, R.S. Genetic population structure of marine viral haemorrhagic septicaemia virus (VHSV). Dis. Aquat. Org. 2004, 61, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Emmenegger, E.J.; Moon, C.H.; Hershberger, P.K.; Kurath, G. Virulence of viral hemorrhagic septicemia virus (VHSV) genotypes Ia, IVa, IVb, and IVc in five fish species. Dis. Aquat. Org. 2013, 107, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.-J.; Choi, T.-J. A new rhabdovirus (HRV-like) isolated in Korea from cultured Japanese flounder (Paralichthys olivaceus). J. Fish. Pathol. 1998, 11, 129–136. [Google Scholar]
- Kim, W.S.; Oh, M.J. Hirame rhabdovirus (HIRRV) as the cause of a natural disease outbreak in cultured black seabream (Acanthopagrus schlegeli) in Korea. Arch. Virol. 2015, 160, 3063–3066. [Google Scholar] [CrossRef]
- Kim, D.H.; Oh, H.K.; Eou, J.I.; Seo, H.J.; Kim, S.K.; Oh, M.J.; Nam, S.W.; Choi, T.J. Complete nucleotide sequence of the hirame rhabdovirus, a pathogen of marine fish. Virus Res. 2005, 107, 1–9. [Google Scholar] [CrossRef]
- Seo, H.G.; Do, J.W.; Jung, S.H.; Han, H.J. Outbreak of hirame rhabdovirus infection in cultured spotted sea bass Lateolabrax maculatus on the western coast of Korea. J. Fish. Dis. 2016, 39, 1239–1246. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Yoshimizu, M.; Gorie, S. A new rhabdovirus isolated in Japan from cultured hirame (Japanese flounder) Paralichthys olivaceus and ayu Plecoglossus altivelis. Dis. Aquat. Org. 1986, 1, 209–217. [Google Scholar] [CrossRef]
- Borzym, E.; Matras, M.; Maj-Paluch, J.; Baud, M.; De Boisseson, C.; Talbi, C.; Olesen, N.J.; Bigarré, L. First isolation of hirame rhabdovirus from freshwater fish in Europe. J. Fish. Dis. 2014, 37, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Kasornchandra, J.; Engelking, H.M.; Lannan, C.N.; Rohovec, J.S.; Fryer, J.L. Characteristics of three rhabdoviruses from snakehead fish Ophicephalus striatus. Dis. Aquat. Org. 1992, 13, 89–94. [Google Scholar] [CrossRef]
- Johnson, M.C.; Maxwell, J.M.; Loh, P.C.; Leong, J.A. Molecular characterization of the glycoproteins from two warm water rhabdoviruses: Snakehead rhabdovirus (SHRV) and rhabdovirus of penaeid shrimp (RPS)/spring viremia of carp virus (SVCV). Virus Res. 1999, 64, 95–106. [Google Scholar] [CrossRef]
- Frerichs, G.N.; Millar, S.D.; Roberts, R.J. Ulcerative rhabdovirus in fish in South-East Asia. Nature 1986, 322, 216. [Google Scholar] [CrossRef] [PubMed]
- Olberding, K.P.; Frost, J.W. Electron microscopical observations of the structure of the virus of viral haemorrhagic septicaemia (VHS) of rainbow trout (Salmo gairdneri). J. Gen. Virol. 1975, 27, 305–312. [Google Scholar] [CrossRef]
- Zwillenberg, L.O.; Jensen, H.M.; Zwillenberg, H.H.L. Electron microscopy of the virus of viral haemorrhagie septicaemia of rainbow trout (Egtved virus). Arch. Gesamte Virusforsch. 1965, 17, 1–19. [Google Scholar] [CrossRef]
- Armend, D.F.; Chambers, V.C. Morphology of certain viruses of salmonid fishes. I. In vitro studies of sorne viruses causing hematopoietic necrosis. J. Fish. Res. Board Canada 1970, 27, 1285–1293. [Google Scholar] [CrossRef]
- Thoulouze, M.-I.; Bouguyon, E.; Carpentier, C.; Bremont, M. Essential role of the NV protein of novirhabdovirus for pathogenicity in rainbow trout. J. Virol. 2004, 78, 4098–4107. [Google Scholar] [CrossRef] [Green Version]
- Brémont, M. Reverse genetics on fish rhabdoviruses: Tools to study the pathogenesis of fish rhabdoviruses. Curr. Top. Microbiol, Immunol. 2005, 292, 119–141. [Google Scholar] [CrossRef]
- Kurath, G.; Leong, J.C. Characterization of infectious hematopoietic necrosis virus mRNA species reveals a nonvirion rhabdovirus protein. J. Virol. 1985, 53, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schütze, H.; Mundt, E.; Mettenleiter, T.C. Complete genomic sequence of viral hemorrhagic septicemia virus, a fish rhabdovirus. Virus Genes 1999, 19, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Kongsuwan, K. Deduced structural model for animal rhabdovirus glycoproteins. J. Gen. Virol. 1999, 80, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einer-Jensen, K.; Krogh, T.N.; Roepstorff, P.; Lorenzen, N. Characterization of intramolecular disulfide bonds and secondary modifications of the glycoprotein from viral hemorrhagic septicemia virus, a fish rhabdovirus. J. Virol. 1998, 72, 10189–10196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar] [CrossRef]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucia-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and evolution of the global RNA virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Eickbush, T.H. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9, 3353–3362. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Deviatkin, A.A.; Lukashev, A.N. Recombination in the rabies virus and other lyssaviruses. Inf. Genet. Evol. 2018, 60, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.J.; Biek, R.; Hassanin, A.; Rouquet, P.; Reed, P.; Yaba, P.; Pourrut, X.; Real, L.A.; Gonzalez, J.-P.; Leroy, E.M. Isolates of Zaire ebolavirus from wild apes reveal genetic lineage and recombinants. Proc. Natl. Acad. Sci. USA 2007, 104, 17123–17127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, P.J.; Kim, L.M.; Ip, H.S.; Afonso, C.L. Evolutionary dynamics of Newcastle disease virus. Virology 2009, 391, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.-Z.; Worobey, M. Homologous recombination in negative sense RNA viruses. Viruses 2011, 3, 1358–1373. [Google Scholar] [CrossRef]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar] [CrossRef]
- Roche, S.; Albertini, A.A.; Lepault, J.; Bressanelli, S.; Gaudin, Y. Structures of vesicular stomatitis virus glycoprotein: Membrane fusion revisited. Cell Mol. Life Sci. 2008, 65, 1716–1728. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C.; Duchene, S. Can sequence phylogenies safely infer the origin of the global virome? mBio 2019, 10, e00289-19. [Google Scholar] [CrossRef] [Green Version]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucia-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Reply to Holmes and Duchene, "Can sequence phylogenies safely infer the origin of the global virome?": Deep phylogenetic analysis of RNA viruses is highly challenging but not meaningless. mBio 2019, 10, e00542-19. [Google Scholar] [CrossRef] [Green Version]
- Abrescia, N.G.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure unifies the viral universe. Ann. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origin of viruses: Primordial replicators recruiting capsids from hosts. Nat. Rev. Microbiol. 2019, 17, 449–458. [Google Scholar] [CrossRef]
- Walker, P.J.; Blasdell, K.R.; Joubert, D.A. Ephemeroviruses: Arthropod-borne rhabdoviruses of ruminants, with large and complex genomes. In Rhabdoviruses: Molecular Taxonomy, Evolution, Genomics, Ecology, Cytopathology and Control; Dietzgen, R.G., Kuzman, I.V., Eds.; Horizon Scientific Press: Norwich, UK, 2012. [Google Scholar]
Figure 1.
The evolutionary history was inferred from a Clustal Omega alignment of 191 complete L protein sequences of animal rhabdoviruses currently assigned to species in the subfamily Alpharhabdovirinae. Phylogenetically informative sites were selected from the alignment using Gblocks resulting in 1029 positions in the final dataset. The tree was inferred in MEGA7 by using the maximum likelihood method based on the WAG + Γ amino acid substitution model. The tree with the highest log likelihood (−151,963.70) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Joining and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with the superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Bootstrap values (100 iterations) are shown for each node. Newly assigned genera and species are shown in blue font.
Figure 1.
The evolutionary history was inferred from a Clustal Omega alignment of 191 complete L protein sequences of animal rhabdoviruses currently assigned to species in the subfamily Alpharhabdovirinae. Phylogenetically informative sites were selected from the alignment using Gblocks resulting in 1029 positions in the final dataset. The tree was inferred in MEGA7 by using the maximum likelihood method based on the WAG + Γ amino acid substitution model. The tree with the highest log likelihood (−151,963.70) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Joining and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with the superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Bootstrap values (100 iterations) are shown for each node. Newly assigned genera and species are shown in blue font.

Figure 2.
Maximum clade credibility (MCC) tree inferred from MAFFT alignments of full-length rhabdovirus L sequences of viruses representing all four members of the genus Novirhabdovirus, as well as all other genera in the Rhabdoviridae (85 sequences), and members of the families Paramyxoviridae (4 sequences), Pneumoviridae (4 sequences), Filoviridae (4 sequences), Bornaviridae (4 sequences), Nyamiviridae (3 sequences), Atroviridae (2 sequences), and Chuviridae (6 sequences). Ambiguously aligned regions were removed from the alignment using TrimAl [71], resulting in a final alignment length of 916 amino acids. The MCC tree was inferred in BEAST.v1.10.4 by using the Whelan and Goldman (WAG) model of amino acid substitutions, the gamma + invariant sites model of site heterogeneity, and a strict molecular clock (coalescent: constant size) with a random starting tree to perform 10 million MCMC runs. The analysis was sampled at every 10000 states. Tree Annotator v1.10.4 was used to output the results of the MCC tree model and calculate posterior probabilities with a burn-in of 1 million states. FigTree was then used to plot the MCC phylogenetic tree. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site and rooted on the chuvirus clade. Posterior probability values are shown for each branch. Maximum-likelihood trees inferred from the same amino acid sequence alignment are shown in Figure S1. Family, subfamily and genus level taxonomic assignments are shown in bold.
Figure 2.
Maximum clade credibility (MCC) tree inferred from MAFFT alignments of full-length rhabdovirus L sequences of viruses representing all four members of the genus Novirhabdovirus, as well as all other genera in the Rhabdoviridae (85 sequences), and members of the families Paramyxoviridae (4 sequences), Pneumoviridae (4 sequences), Filoviridae (4 sequences), Bornaviridae (4 sequences), Nyamiviridae (3 sequences), Atroviridae (2 sequences), and Chuviridae (6 sequences). Ambiguously aligned regions were removed from the alignment using TrimAl [71], resulting in a final alignment length of 916 amino acids. The MCC tree was inferred in BEAST.v1.10.4 by using the Whelan and Goldman (WAG) model of amino acid substitutions, the gamma + invariant sites model of site heterogeneity, and a strict molecular clock (coalescent: constant size) with a random starting tree to perform 10 million MCMC runs. The analysis was sampled at every 10000 states. Tree Annotator v1.10.4 was used to output the results of the MCC tree model and calculate posterior probabilities with a burn-in of 1 million states. FigTree was then used to plot the MCC phylogenetic tree. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site and rooted on the chuvirus clade. Posterior probability values are shown for each branch. Maximum-likelihood trees inferred from the same amino acid sequence alignment are shown in Figure S1. Family, subfamily and genus level taxonomic assignments are shown in bold.

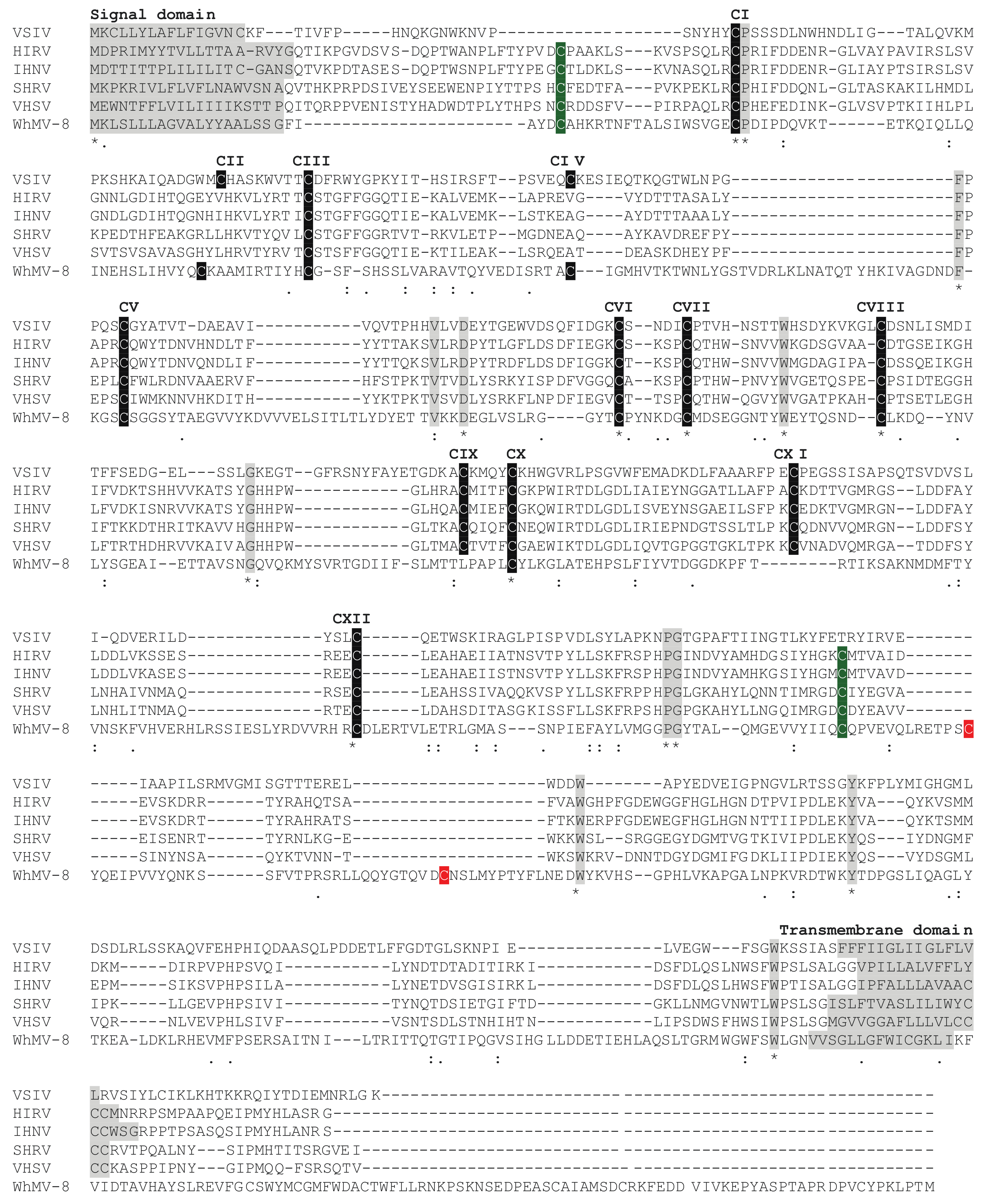
Figure 3.
Clustal Omega alignment of the G proteins of vesicular stomatitis Indiana virus (VSIV), hirame rhabdovirus (HIRV), infectious hematopoietic necrosis virus (IHNV), snakehead rhabdovirus (SHRV), viral hemorrhagic septicemia virus (VHSV), and the chuvirus Wuhan mosquito virus 8 (WhMV-8). The signal domain and transmembrane domains of these class I transmembrane glycoproteins are indicated. Twelve cysteine residues in VSIV (CI-CXII) form six disulfide bridges (CI-CXII, CII-CIV, CIII-CV, CVI-CVII, CVIII-CX, and CIX-CXI), one of which (CII-CIV) is absent in the novirhabdovirus G proteins [64,67,82]. The WhMV-8 G protein lacks the CIX-CXI disulfide bridge, shares two additional cysteine residues with the novirhabdoviruses (shaded green), and has two unique cysteine residues (shaded red) in the ectodomain that may also form a disulfide bridge.
Figure 3.
Clustal Omega alignment of the G proteins of vesicular stomatitis Indiana virus (VSIV), hirame rhabdovirus (HIRV), infectious hematopoietic necrosis virus (IHNV), snakehead rhabdovirus (SHRV), viral hemorrhagic septicemia virus (VHSV), and the chuvirus Wuhan mosquito virus 8 (WhMV-8). The signal domain and transmembrane domains of these class I transmembrane glycoproteins are indicated. Twelve cysteine residues in VSIV (CI-CXII) form six disulfide bridges (CI-CXII, CII-CIV, CIII-CV, CVI-CVII, CVIII-CX, and CIX-CXI), one of which (CII-CIV) is absent in the novirhabdovirus G proteins [64,67,82]. The WhMV-8 G protein lacks the CIX-CXI disulfide bridge, shares two additional cysteine residues with the novirhabdoviruses (shaded green), and has two unique cysteine residues (shaded red) in the ectodomain that may also form a disulfide bridge.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Taxonomic classification of rhabdoviruses infecting fish or marine mammals (2022).
| Subfamily | Genus | Species | Exemplar Virus | Abbrev. | Sample § | GenBank Accession | Intra-Species Divergence (%) # | Inter-Species Divergence (%) # | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L | N | G | L | N | G | |||||||
| Alpharhabdovirinae | Sprivivirus | Sprivivirus cyprinus | spring viremia of carp virus | SVCV | VR-1390 | U18101 | ≤2.7 | ≤3.4 | ≤8.7 | ≥11.8 | ≥8.2 | ≥25.4 |
| Sprivivirus esox | pike fry rhabdovirus | PFRV | F4 | FJ872827 | ≤6.7 | ≤5.6 | ≤16.7 | |||||
| Perhabdovirus | Perhabdovirus perca | perch rhabdovirus | PRV | Dorson | JX679246 | ≤4.5 | ≤2.7 | ≤3.3 | ≥13.1 | ≥15.9 | ≥16.7 | |
| Perhabdovirus trutta | lake trout rhabdovirus | LTRV | 903/87 | AF434991 | ≤5.2 | ≤6.1 | ≤11.6 | |||||
| Perhabdovirus anguilla | eel virus European X | EVEX | CV1153311 | FN557213 | ≤2.2 | ≤1.7 | ≤4.1 | |||||
| Perhabdovirus leman | Leman virus | LEMV | 18/193 | MN963996 | n.a. | n.a. | n.a. | |||||
| Cetarhavirus | Cetarhavirus lagenorhynchus | dolphin rhabdovirus | DRV | pxV1 | KF958252 | n.a. | n.a. | n.a. | 31.3 | 31.8 | 56.1 | |
| Cetarhavirus phocoena | harbour porpoise rhabdovirus | HPRV | WVL17017A | MN103537 | n.a. | n.a. | n.a. | |||||
| Siniperhavirus | Siniperhavirus zoarces | eelpout rhabdovirus | EPRV | FSK0523 | KR612230 | n.a. | n.a. | n.a. | ≥28.8 | ≥33.7 | ≥50.0 | |
| Siniperhavirus chuatsi | Siniperca chuatsi rhabdovirus | SCRV | n.a. | DQ399789 | ≤3.6 | ≤6.8 | ≤10.6 | |||||
| Scophrhavirus | Scophrhavirus maximus | Scophthalmus maximus rhabdovirus | SMRV | n.a. | HQ003891 | n.a. | n.a. | n.a. | 46.5 | 62.6 | 71.0 | |
| Scophrhavirus chanodichthys | Wuhan redfin culter dimarhabdovirus | WhRCDRV | DSYS6218 | MG600013 | n.a. | n.a. | n.a. | |||||
| Gammarhabdovirinae | Novirhabdovirus | Novirhabdovirus salmonid | infectious haematopoietic necrosis virus | IHNV | WRAC | L40883 | ≤2.4 | ≤12.6 | ≤10.2 | ≥15.1 | ≥35.8 | ≥22.9 |
| Novirhabdovirus piscine | viral haemorrhagic septicaemia virus | VHSV | Fil3 | Y18263 | ≤7.1 | ≤11.1 | ≤11.5 | |||||
| Novirhabdovirus hirame | hirame rhabdovirus | HIRRV | CA9703 | AF104985 | 0.4 | 0.8 | ≤1.2 | |||||
| Novirhabdovirus snakehead | snakehead rhabdovirus | SHRV | n.a. | AF147498 | n.a. | n.a. | n.a. | |||||
§ Samples of exemplar viruses listed by ICTV (https://talk.ictvonline.org/taxonomy/vmr/m/vmr-file-repository/13181 (accessed on 18 October 2021)). # p-distances estimated in MEGA 6.0 using all available full-length amino acid sequences. n.a. = not applicable (only one complete sequence currently available).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Walker, P.J.; Bigarré, L.; Kurath, G.; Dacheux, L.; Pallandre, L. Revised Taxonomy of Rhabdoviruses Infecting Fish and Marine Mammals. Animals 2022, 12, 1363. https://doi.org/10.3390/ani12111363
AMA Style
Walker PJ, Bigarré L, Kurath G, Dacheux L, Pallandre L. Revised Taxonomy of Rhabdoviruses Infecting Fish and Marine Mammals. Animals. 2022; 12(11):1363. https://doi.org/10.3390/ani12111363
Chicago/Turabian StyleWalker, Peter J., Laurent Bigarré, Gael Kurath, Laurent Dacheux, and Laurane Pallandre. 2022. "Revised Taxonomy of Rhabdoviruses Infecting Fish and Marine Mammals" Animals 12, no. 11: 1363. https://doi.org/10.3390/ani12111363
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.