Antimicrobial Human β-Defensins in the Colon and Their Role in Infectious and Non-Infectious Diseases


{kind=link}
Abstract
:1. Introduction
2. Characteristics of β-Defensin Peptides
3. Gene Expression of β-Defensins in the Intestinal Mucosa
4. Antimicrobial and Chemotactic Functions of β-Defensin
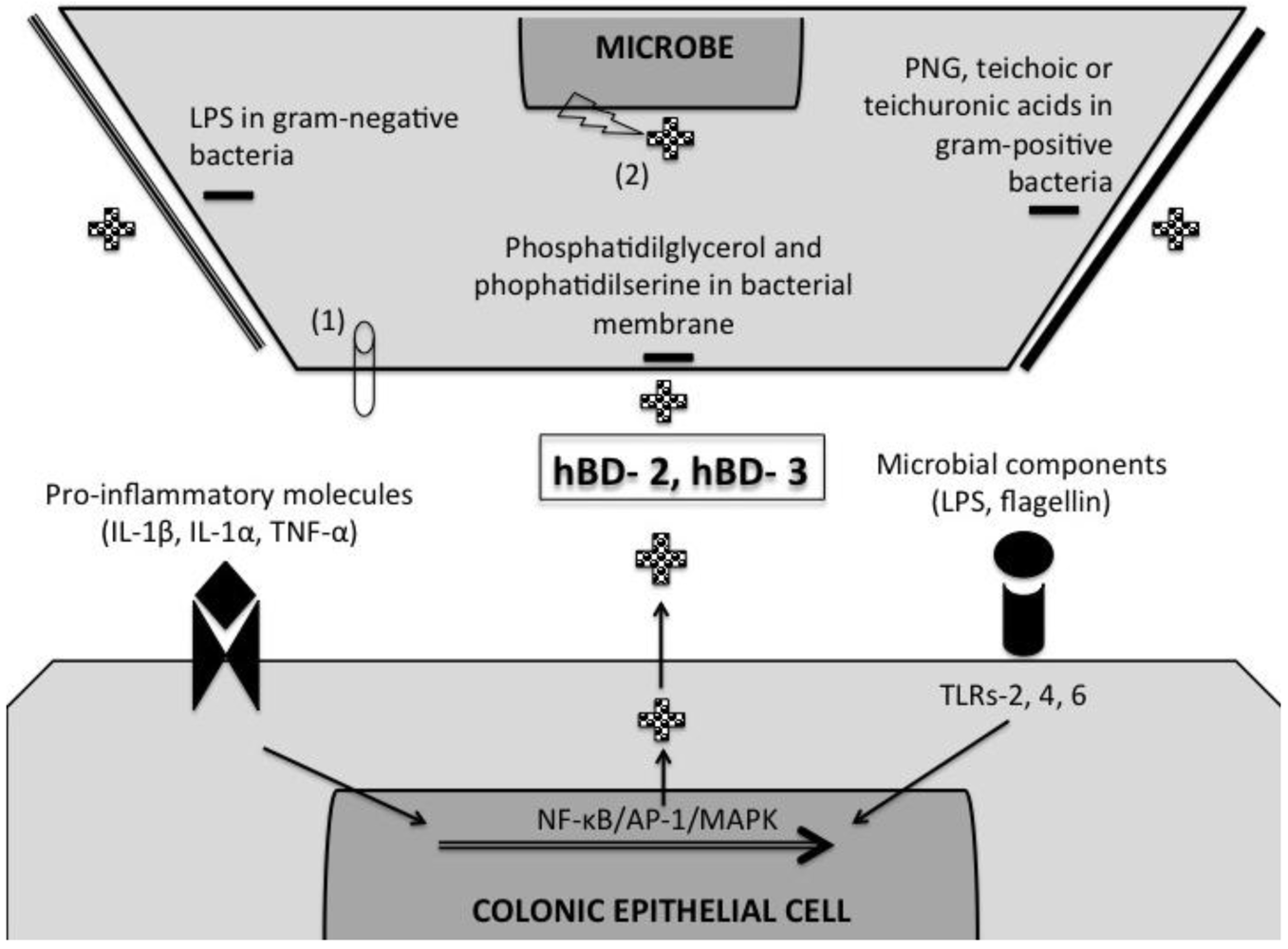
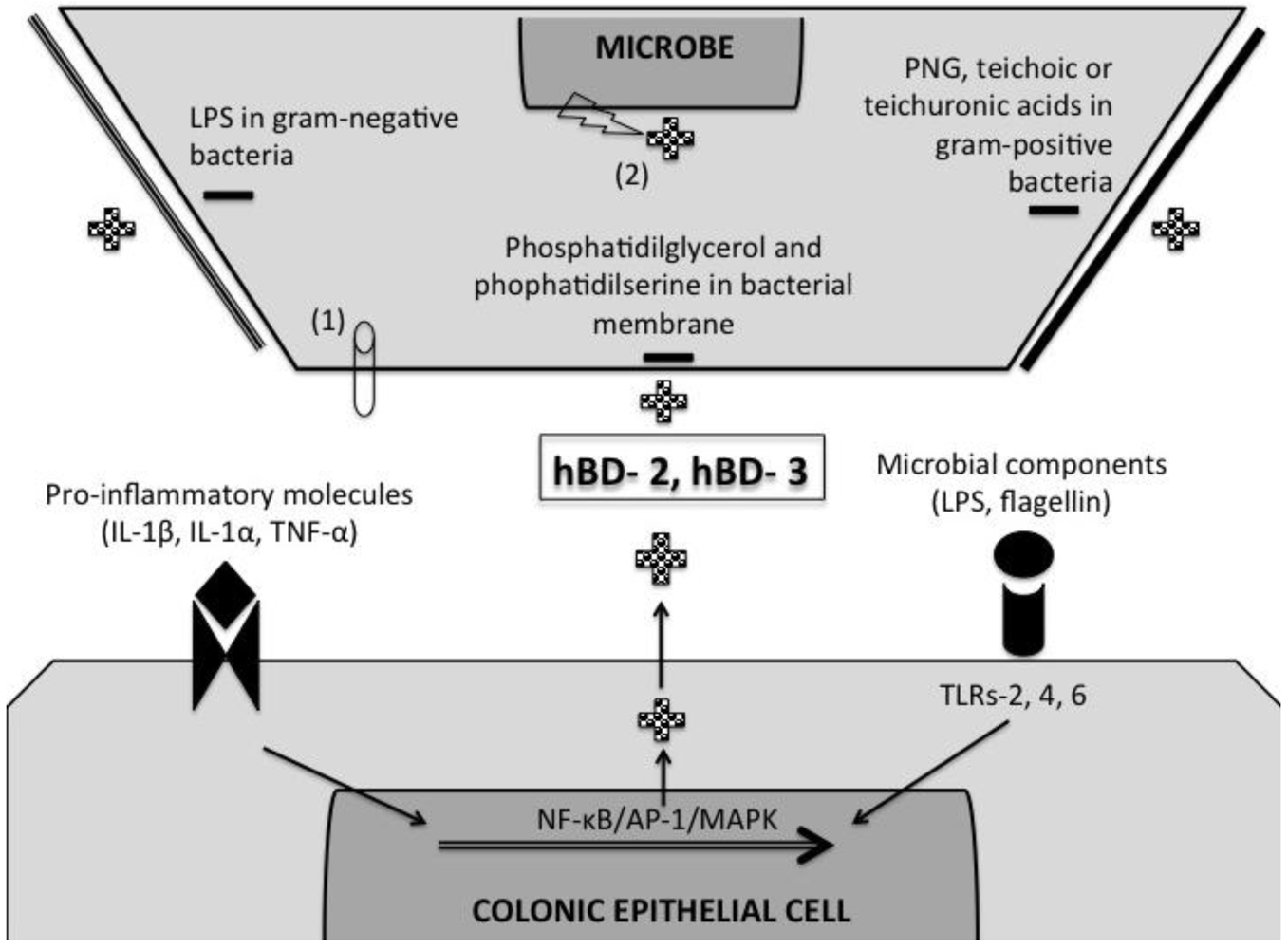
5. Induction of β-Defensins by Pro-Inflammatory and Microbial Stimuli
6. β-Defensin Response in Infectious and Non-Infectious Inflammation in the Colon
6.1. Cryptosporidium Parvum
6.2. Toxoplasma Gondii
6.3. Trypanosoma brucei
6.4. Giardia lamblia
6.5. Candida albicans and Other Fungal Infections
6.6. Clostridium difficile
6.7. Shigella spp.
6.8. Viral Infections
6.9. Inflammatory Bowel Diseases (IBD)
7. Concluding Remarks
References
- Bensch, K.W.; Raida, M.; Magert, H.J.; Schulz-Knappe, P.; Forssmann, W.G. Hbd-1: A novel beta-defensin from human plasma. FEBS Lett. 1995, 368, 331–335. [Google Scholar] [CrossRef]
- O’Neil, D.A.; Porter, E.M.; Elewaut, D.; Anderson, G.M.; Eckmann, L.; Ganz, T.; Kagnoff, M.F. Expression and regulation of the human beta-defensins hbd-1 and hbd-2 in intestinal epithelium. J. Immunol. 1999, 163, 6718–6724. [Google Scholar]
- Fahlgren, A.; Hammarstrom, S.; Danielsson, A.; Hammarstrom, M.L. Beta-defensin-3 and -4 in intestinal epithelial cells display increased mrna expression in ulcerative colitis. Clin. Exp. Immunol. 2004, 137, 379–385. [Google Scholar] [CrossRef]
- Jenke, A.C.; Zilbauer, M.; Postberg, J.; Wirth, S. Human beta-defensin 2 expression in elbw infants with severe necrotizing enterocolitis. Pediatr. Res. 2012, 72, 513–520. [Google Scholar] [CrossRef]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Mueller, O.; Herrlinger, K.R.; Fellermann, K.; Schroeder, J.M.; Stange, E.F. Inducible and constitutive beta-defensins are differentially expressed in crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2003, 9, 215–223. [Google Scholar] [CrossRef]
- Zilbauer, M.; Jenke, A.; Wenzel, G.; Postberg, J.; Heusch, A.; Phillips, A.D.; Noble-Jamieson, G.; Torrente, F.; Salvestrini, C.; Heuschkel, R.; et al. Expression of human beta-defensins in children with chronic inflammatory bowel disease. PLoS One 2010, 5, e15389. [Google Scholar] [CrossRef]
- Chen, X.M.; O’Hara, S.P.; Nelson, J.B.; Splinter, P.L.; Small, A.J.; Tietz, P.S.; Limper, A.H.; LaRusso, N.F. Multiple tlrs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappab. J. Immunol. 2005, 175, 7447–7456. [Google Scholar]
- McGwire, B.S.; Olson, C.L.; Tack, B.F.; Engman, D.M. Killing of african trypanosomes by antimicrobial peptides. J. Infect. Dis. 2003, 188, 146–152. [Google Scholar]
- Morampudi, V.; Braun, M.Y.; D’Souza, S. Modulation of early beta-defensin-2 production as a mechanism developed by type 1 Toxoplasma gondii to evade human intestinal immunity. Infect. Immun. 2011, 79, 2043–2050. [Google Scholar] [CrossRef]
- Zaalouk, T.K.; Bajaj-Elliott, M.; George, J.T.; McDonald, V. Differential regulation of beta-defensin gene expression during Cryptosporidium parvum infection. Infect. Immun. 2004, 72, 2772–2779. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Ganz, T. Defensins of vertebrate animals. Curr. Opin. Immunol. 2002, 14, 96–102. [Google Scholar] [CrossRef]
- Pazgier, M.; Hoover, D.M.; Yang, D.; Lu, W.; Lubkowski, J. Human beta-defensins. Cell. Mol. Life Sci. 2006, 63, 1294–1313. [Google Scholar] [CrossRef]
- Taylor, K.; Barran, P.E.; Dorin, J.R. Structure-activity relationships in beta-defensin peptides. Biopolymers 2008, 90, 1–7. [Google Scholar] [CrossRef]
- Ramasundara, M.; Leach, S.T.; Lemberg, D.A.; Day, A.S. Defensins and inflammation: The role of defensins in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2009, 24, 202–208. [Google Scholar] [CrossRef]
- Sallenave, J.M. Antimicrobial activity of antiproteinases. Biochem. Soc. Trans. 2002, 30, 111–115. [Google Scholar] [CrossRef]
- Yang, D.; Chen, Q.; Hoover, D.M.; Staley, P.; Tucker, K.D.; Lubkowski, J.; Oppenheim, J.J. Many chemokines including ccl20/mip-3alpha display antimicrobial activity. J. Leukoc. Biol. 2003, 74, 448–455. [Google Scholar] [CrossRef]
- Rodriguez-Jimenez, F.J.; Krause, A.; Schulz, S.; Forssmann, W.G.; Conejo-Garcia, J.R.; Schreeb, R.; Motzkus, D. Distribution of new human beta-defensin genes clustered on chromosome 20 in functionally different segments of epididymis. Genomics 2003, 81, 175–183. [Google Scholar]
- Schutte, B.C.; Mitros, J.P.; Bartlett, J.A.; Walters, J.D.; Jia, H.P.; Welsh, M.J.; Casavant, T.L.; McCray, P.B., Jr. Discovery of five conserved beta-defensin gene clusters using a computational search strategy. Proc. Natl. Acad. Sci. USA 2002, 99, 2129–2133. [Google Scholar] [CrossRef]
- Frye, M.; Bargon, J.; Lembcke, B.; Wagner, T.O.; Gropp, R. Differential expression of human alpha- and beta-defensins mrna in gastrointestinal epithelia. Eur. J. Clin. Investig. 2000, 30, 695–701. [Google Scholar] [CrossRef]
- Mathews, M.; Jia, H.P.; Guthmiller, J.M.; Losh, G.; Graham, S.; Johnson, G.K.; Tack, B.F.; McCray, P.B., Jr. Production of beta-defensin antimicrobial peptides by the oral mucosa and salivary glands. Infect. Immun. 1999, 67, 2740–2745. [Google Scholar]
- Zhao, C.; Wang, I.; Lehrer, R.I. Widespread expression of beta-defensin hbd-1 in human secretory glands and epithelial cells. FEBS Lett. 1996, 396, 319–322. [Google Scholar] [CrossRef]
- Wehkamp, J.; Harder, J.; Wehkamp, K.; Wehkamp-von Meissner, B.; Schlee, M.; Enders, C.; Sonnenborn, U.; Nuding, S.; Bengmark, S.; Fellermann, K.; et al. Nf-kappab- and ap-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli nissle 1917: A novel effect of a probiotic bacterium. Infect. Immun. 2004, 72, 5750–5758. [Google Scholar] [CrossRef]
- Bals, R.; Wang, X.; Wu, Z.; Freeman, T.; Bafna, V.; Zasloff, M.; Wilson, J.M. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J. Clin. Investig. 1998, 102, 874–880. [Google Scholar] [CrossRef]
- Harder, J.; Siebert, R.; Zhang, Y.; Matthiesen, P.; Christophers, E.; Schlegelberger, B.; Schroder, J.M. Mapping of the gene encoding human beta-defensin-2 (defb2) to chromosome region 8p22-p23.1. Genomics 1997, 46, 472–475. [Google Scholar]
- Otte, J.M.; Neumann, H.M.; Brand, S.; Schrader, H.; Schmidt, W.E.; Schmitz, F. Expression of beta-defensin 4 is increased in human gastritis. Eur. J. Clin. Investig. 2009, 39, 126–138. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef]
- Lasch, P.; Schultz, C.P.; Naumann, D. The influence of poly-(l-lysine) and porin on the domain structure of mixed vesicles composed of lipopolysaccharide and phospholipid: An infrared spectroscopic study. Biophys. J. 1998, 75, 840–852. [Google Scholar] [CrossRef]
- Yang, L.; Weiss, T.M.; Lehrer, R.I.; Huang, H.W. Crystallization of antimicrobial pores in membranes: Magainin and protegrin. Biophys. J. 2000, 79, 2002–2009. [Google Scholar] [CrossRef]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin ii: Buforin ii kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef]
- Yan, H.; Hancock, R.E. Synergistic interactions between mammalian antimicrobial defense peptides. Antimicrob. Agents Chemother. 2001, 45, 1558–1560. [Google Scholar] [CrossRef]
- Meyer-Hoffert, U.; Hornef, M.W.; Henriques-Normark, B.; Axelsson, L.G.; Midtvedt, T.; Putsep, K.; Andersson, M. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 2008, 57, 764–771. [Google Scholar] [CrossRef]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. A peptide antibiotic from human skin. Nature 1997, 387, 861. [Google Scholar]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. [Google Scholar]
- Schroeder, B.O.; Wu, Z.; Nuding, S.; Groscurth, S.; Marcinowski, M.; Beisner, J.; Buchner, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human beta-defensin 1. Nature 2011, 469, 419–423. [Google Scholar]
- Ganz, T. Fatal attraction evaded. How pathogenic bacteria resist cationic polypeptides. J. Exp. Med. 2001, 193, F31–F34. [Google Scholar]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schroder, J.M.; Wang, J.M.; Howard, O.M.; et al. Beta-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar] [CrossRef]
- Rohrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Specific binding and chemotactic activity of mbd4 and its functional orthologue HBD2 to CCR6-expressing cells. J. Biol. Chem. 2010, 285, 7028–7034. [Google Scholar]
- Rohrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J. Immunol. 2010, 184, 6688–6694. [Google Scholar] [CrossRef]
- Funderburg, N.; Lederman, M.M.; Feng, Z.; Drage, M.G.; Jadlowsky, J.; Harding, C.V.; Weinberg, A.; Sieg, S.F. Human -defensin-3 activates professional antigen-presenting cells via toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. USA 2007, 104, 18631–18635. [Google Scholar] [CrossRef]
- Otte, J.M.; Werner, I.; Brand, S.; Chromik, A.M.; Schmitz, F.; Kleine, M.; Schmidt, W.E. Human beta defensin 2 promotes intestinal wound healing in vitro. J. Cell. Biochem. 2008, 104, 2286–2297. [Google Scholar] [CrossRef]
- Witthoft, T.; Pilz, C.S.; Fellermann, K.; Nitschke, M.; Stange, E.F.; Ludwig, D. Enhanced human beta-defensin-2 (hbd-2) expression by corticosteroids is independent of nf-kappab in colonic epithelial cells (Caco2). Dig. Dis. Sci. 2005, 50, 1252–1259. [Google Scholar] [CrossRef]
- Vora, P.; Youdim, A.; Thomas, L.S.; Fukata, M.; Tesfay, S.Y.; Lukasek, K.; Michelsen, K.S.; Wada, A.; Hirayama, T.; Arditi, M.; et al. Beta-defensin-2 expression is regulated by tlr signaling in intestinal epithelial cells. J. Immunol. 2004, 173, 5398–5405. [Google Scholar]
- Schlee, M.; Harder, J.; Koten, B.; Stange, E.F.; Wehkamp, J.; Fellermann, K. Probiotic lactobacilli and vsl#3 induce enterocyte beta-defensin 2. Clin. Exp. Immunol. 2008, 151, 528–535. [Google Scholar] [CrossRef]
- Schlee, M.; Wehkamp, J.; Altenhoefer, A.; Oelschlaeger, T.A.; Stange, E.F.; Fellermann, K. Induction of human beta-defensin 2 by the probiotic Escherichia coli nissle 1917 is mediated through flagellin. Infect. Immun. 2007, 75, 2399–2407. [Google Scholar] [CrossRef]
- Schwab, M.; Reynders, V.; Loitsch, S.; Steinhilber, D.; Schroder, O.; Stein, J. The dietary histone deacetylase inhibitor sulforaphane induces human beta-defensin-2 in intestinal epithelial cells. Immunology 2008, 125, 241–251. [Google Scholar] [CrossRef]
- Carryn, S.; Schaefer, D.A.; Imboden, M.; Homan, E.J.; Bremel, R.D.; Riggs, M.W. Phospholipases and cationic peptides inhibit Cryptosporidium parvum sporozoite infectivity by parasiticidal and non-parasiticidal mechanisms. J. Parasitol. 2012, 98, 199–204. [Google Scholar] [CrossRef]
- Tarver, A.P.; Clark, D.P.; Diamond, G.; Russell, J.P.; Erdjument-Bromage, H.; Tempst, P.; Cohen, K.S.; Jones, D.E.; Sweeney, R.W.; Wines, M.; et al. Enteric beta-defensin: Molecular cloning and characterization of a gene with inducible intestinal epithelial cell expression associated with Cryptosporidium parvum infection. Infect. Immun. 1998, 66, 1045–1056. [Google Scholar]
- Nyakundi, J.N.; Pentreath, V.W. Preliminary observations on the intestinal pathology of mice infected with Trypanosoma brucei brucei. Trans. R. Soc. Trop. Med. Hyg. 1999, 93, 628–630. [Google Scholar] [CrossRef]
- Aley, S.B.; Zimmerman, M.; Hetsko, M.; Selsted, M.E.; Gillin, F.D. Killing of Giardia lamblia by cryptdins and cationic neutrophil peptides. Infect. Immun. 1994, 62, 5397–5403. [Google Scholar]
- Kiehne, K.; Brunke, G.; Meyer, D.; Harder, J.; Herzig, K.H. Oesophageal defensin expression during candida infection and reflux disease. Scand. J. Gastroenterol. 2005, 40, 501–507. [Google Scholar] [CrossRef]
- Pahl, R.; Brunke, G.; Steubesand, N.; Schubert, S.; Bottner, M.; Wedel, T.; Jurgensen, C.; Hampe, J.; Schafer, H.; Zeissig, S.; et al. Il-1beta and adam17 are central regulators of beta-defensin expression in candida esophagitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G547–G553. [Google Scholar] [CrossRef]
- Steubesand, N.; Kiehne, K.; Brunke, G.; Pahl, R.; Reiss, K.; Herzig, K.H.; Schubert, S.; Schreiber, S.; Folsch, U.R.; Rosenstiel, P.; et al. The expression of the beta-defensins hbd-2 and hbd-3 is differentially regulated by NF-kappab and MAPK/AP-1 pathways in an in vitro model of candida esophagitis. BMC Immunol. 2009, 10, 36. [Google Scholar] [CrossRef]
- Jiang, Y.; Yi, X.; Li, M.; Wang, T.; Qi, T.; She, X. Antimicrobial activities of recombinant mouse beta-defensin 3 and its synergy with antibiotics. J. Mater. Sci. 2012, 23, 1723–1728. [Google Scholar]
- Didier, E.S.; Weiss, L.M. Microsporidiosis: Current status. Curr. Opin. Infect. Dis. 2006, 19, 485–492. [Google Scholar] [CrossRef]
- Leitch, G.J.; Ceballos, C. A role for antimicrobial peptides in intestinal microsporidiosis. Parasitology 2009, 136, 175–181. [Google Scholar] [CrossRef]
- Giesemann, T.; Guttenberg, G.; Aktories, K. Human alpha-defensins inhibit clostridium difficile toxin b. Gastroenterology 2008, 134, 2049–2058. [Google Scholar] [CrossRef]
- Islam, M.S.; Hossain, M.A.; Khan, S.I.; Khan, M.N.; Sack, R.B.; Albert, M.J.; Huq, A.; Colwell, R.R. Survival of shigella dysenteriae type 1 on fomites. J. Health Popul. Nutr. 2001, 19, 177–182. [Google Scholar]
- Sperandio, B.; Regnault, B.; Guo, J.; Zhang, Z.; Stanley, S.L., Jr.; Sansonetti, P.J.; Pedron, T. Virulent shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. J. Exp. Med. 2008, 205, 1121–1132. [Google Scholar] [CrossRef]
- Crough, T.; Khanna, R. Immunobiology of human cytomegalovirus: From bench to bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef]
- Paparone, P.P.; Paparone, P.A. Cytomegalovirus colitis in a human immunodeficiency virus-positive patient with a normal cd4 count. Am. J. Med. Sci. 2012, 344, 508–510. [Google Scholar] [CrossRef]
- Dieterich, D.T.; Rahmin, M. Cytomegalovirus colitis in aids: Presentation in 44 patients and a review of the literature. J. Acquir. Immune Defic. Syndr. 1991, 4 Suppl 1, S29–S35. [Google Scholar]
- Lawlor, G.; Moss, A.C. Cytomegalovirus in inflammatory bowel disease: Pathogen or innocent bystander? Inflamm. Bowel Dis. 2010, 16, 1620–1627. [Google Scholar] [CrossRef]
- Freguja, R.; Gianesin, K.; Zanchetta, M.; de Rossi, A. Cross-talk between virus and host innate immunity in pediatric HIV-1 infection and disease progression. New Microbiol. 2012, 35, 249–257. [Google Scholar]
- Seidel, A.; Ye, Y.; de Armas, L.R.; Soto, M.; Yarosh, W.; Marcsisin, R.A.; Tran, D.; Selsted, M.E.; Camerini, D. Cyclic and acyclic defensins inhibit human immunodeficiency virus type-1 replication by different mechanisms. PLoS One 2010, 5, e9737. [Google Scholar] [CrossRef]
- Gaudreault, E.; Gosselin, J. Leukotriene b4-mediated release of antimicrobial peptides against cytomegalovirus is blt1 dependent. Viral Immunol. 2007, 20, 407–420. [Google Scholar] [CrossRef]
- Zapata, W.; Rodriguez, B.; Weber, J.; Estrada, H.; Quinones-Mateu, M.E.; Zimermman, P.A.; Lederman, M.M.; Rugeles, M.T. Increased levels of human beta-defensins mrna in sexually hiv-1 exposed but uninfected individuals. Curr. HIV Res. 2008, 6, 531–538. [Google Scholar] [CrossRef]
- Weinberg, A.; Quinones-Mateu, M.E.; Lederman, M.M. Role of human beta-defensins in hiv infection. Adv. Dent. Res. 2006, 19, 42–48. [Google Scholar] [CrossRef]
- Mohan, T.; Sharma, C.; Bhat, A.A.; Rao, D.N. Modulation of HIV peptide antigen specific cellular immune response by synthetic alpha- and beta-defensin peptides. Vaccine 2013, Corrected proof in press. [Google Scholar]
- Voss, E.; Wehkamp, J.; Wehkamp, K.; Stange, E.F.; Schroder, J.M.; Harder, J. Nod2/card15 mediates induction of the antimicrobial peptide human beta-defensin-2. J. Biol. Chem. 2006, 281, 2005–2011. [Google Scholar]
- Fellermann, K.; Stange, D.E.; Schaeffeler, E.; Schmalzl, H.; Wehkamp, J.; Bevins, C.L.; Reinisch, W.; Teml, A.; Schwab, M.; Lichter, P.; et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to crohn disease of the colon. Am. J. Hum. Genet. 2006, 79, 439–448. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cobo, E.R.; Chadee, K. Antimicrobial Human β-Defensins in the Colon and Their Role in Infectious and Non-Infectious Diseases. Pathogens 2013, 2, 177-192. https://doi.org/10.3390/pathogens2010177
Cobo ER, Chadee K. Antimicrobial Human β-Defensins in the Colon and Their Role in Infectious and Non-Infectious Diseases. Pathogens. 2013; 2(1):177-192. https://doi.org/10.3390/pathogens2010177
Chicago/Turabian StyleCobo, Eduardo R., and Kris Chadee. 2013. "Antimicrobial Human β-Defensins in the Colon and Their Role in Infectious and Non-Infectious Diseases" Pathogens 2, no. 1: 177-192. https://doi.org/10.3390/pathogens2010177