The Significance of Matrix Metalloproteinases in Parasitic Infections Involving the Central Nervous System



Abstract
:1. Introduction
2. The matrix
3. The Role of MMPs in Normal and Pathological Conditions
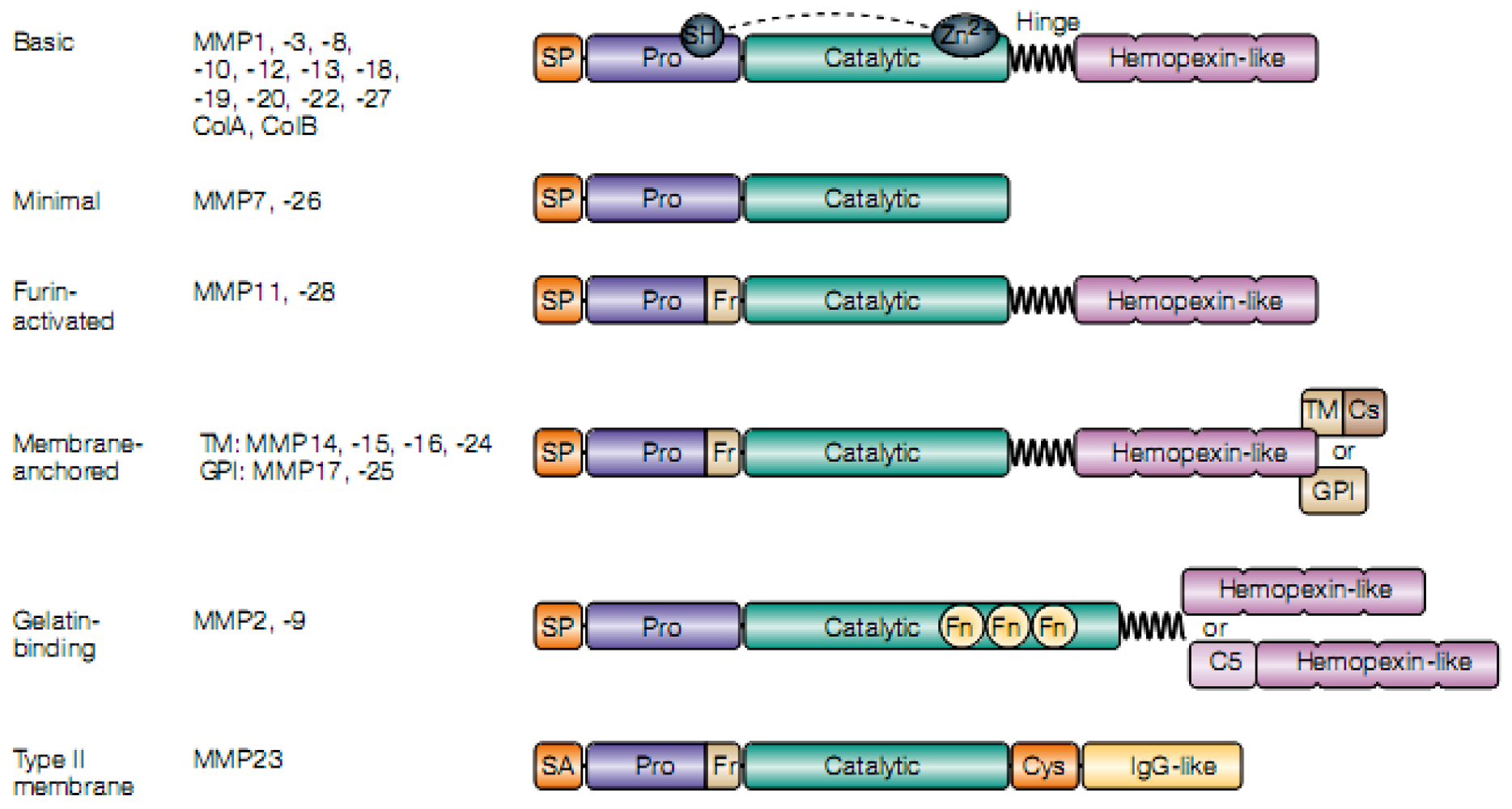
3.1. The MMP Family

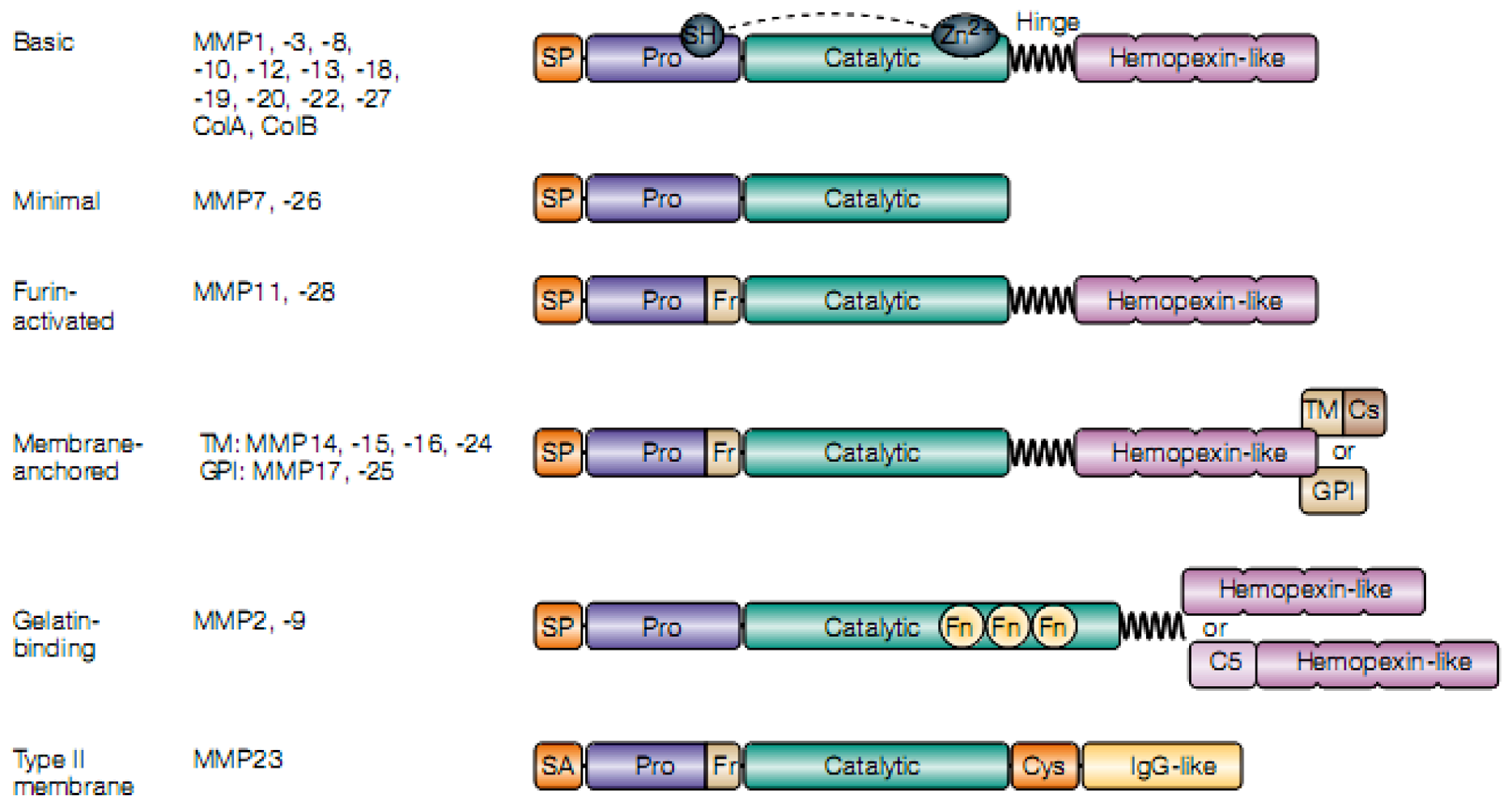{kind=link}
{kind=link}
{kind=link}
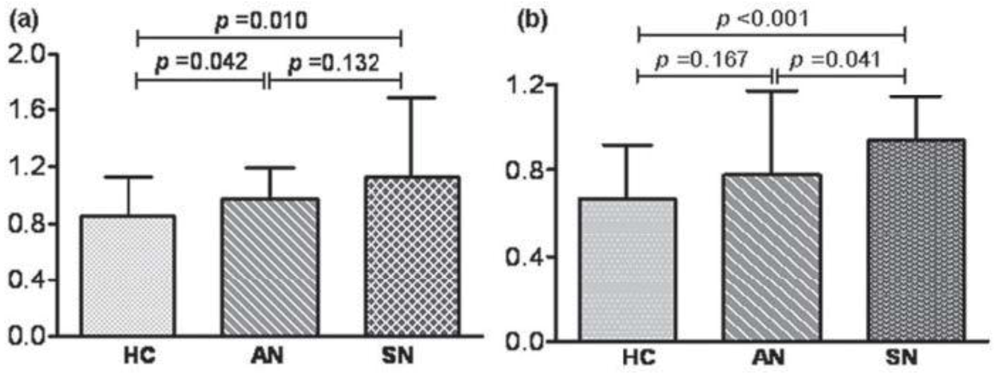{kind=link}
| Common name | MMP | Chromosomal location (human) | M.W. (kDa) | Collagen substrates | Some additional substrates* |
|---|---|---|---|---|---|
| Collagenases | |||||
| Collagenase-1 | MMP-1 | 11q22-q23 | 55/45 | I, II,III, VII, VIII, X, | Aggrecan, gelatin |
| Collagenase-2 | MMP-8 | 11q21-q22 | 75/58 | I, II, III, VII, VIII X | Aggrecan, gelatin, fibronectin |
| Collagenase-3 | MP-13 | 11q22.3 | 60/48 | I, II, III, IV, IX, X, XIV | Aggrecan, gelatin, fibronectin |
| Collagenase-4 | MMP-18 | ( Xenopus) | 70/53 | ||
| Gelatinases | |||||
| Gelatinasi A | MMP-2 | 16q13 | 72/66 | I, II, III, IV, VII, X | Gelatin, fibronectin, fibrillin |
| Gelatinasi B | MMP-9 | 20q11.2-q13.1 | 92/86 | IV, V | Gelatin, elastin, fibrillin |
| Stromelysins | |||||
| Stromelysin -1 | MMP-3 | 11q23 | 57/45 | II, III, IV,V,IX, X, XI | Gelatin, plasminogen |
| Stromelysin -2 | MMP-10 | 11q22.3-q23 | 57/44 | IV, | Laminin, fibronectin elastin, |
| Stromelysin -3 | MMP-11 | 22q11.2 | 51/44 | IV | Fibronectin, laminin, aggrecan |
| Matrilysins | |||||
| Matrylisin-1 | MMP-7 | 11q21-q22 | 28/19 | IV | Fibronectin, laminin, gelatin |
| Matrylisin-2 | MMP-26 | 11p-15 | 28/19 | IV | Fibrinogen, fibronectin, gelatin |
| Metalloelastase | MMP-12 | 11q22.2-q22.3 | 54/45 | IV | Elastin, fibronectin, latent TNF |
| MT-MMP | |||||
| Tm-type I | |||||
| MT1-MMP | MMP-14 | 14q11-q12 | 66/56 | I, II, III | Gelatin, fibronectin, laminin |
| MT2-MMP | MMP-15 | 15q13-q21 | 72/60 | Gelatin, fibronectin, laminin | |
| MT3-MMP | MMP-16 | 8q21 | 64/52 | III | Gelatin, fibronectin, laminin |
| MT5-MMP | MMP-24 | 20q11.2 | -/52 | Gelatin, fibronectin, laminin | |
| GPI-anchored | Fibrinogen, fibrin | ||||
| MT4-MMP | MMP-17 | 12q24.3 | 57/63 | Fibrin, gelatin | |
| MT6-MMP | MMP-25 | 16p13.3 | IV | Fibronectin, gelatin, laminin | |
| Other MMPs | |||||
| MMP-19 | 12q14 | 54/45 | IV | ||
| Enamelysin | MMP-20 | 11q22.3 | 54/22 | Aggrecan, elastin, fibrillin Gelatin | |
| MMP-21 | ND | 70/53 | Aggrecan | ||
| CA-MMP | MMP-23 | 1p36.3 | Aggrecan | ||
| MMP-27 | 11q24 | Gelatin, casein, fibronectin | |||
| Epylisin | MMP-28 | 17q21.1 | 56/45 | Casein |
4. Matrix Metalloproteinase Biology in Parasitic Infections of CNS
4.1. MMPs and Protozoan Infections
4.1.1. Malaria
4.1.2. African Trypanosomosis
4.1.3. Toxoplasmosis
4.2. MMPs and Helminth Infections
4.2.1. Neurocysticercosis
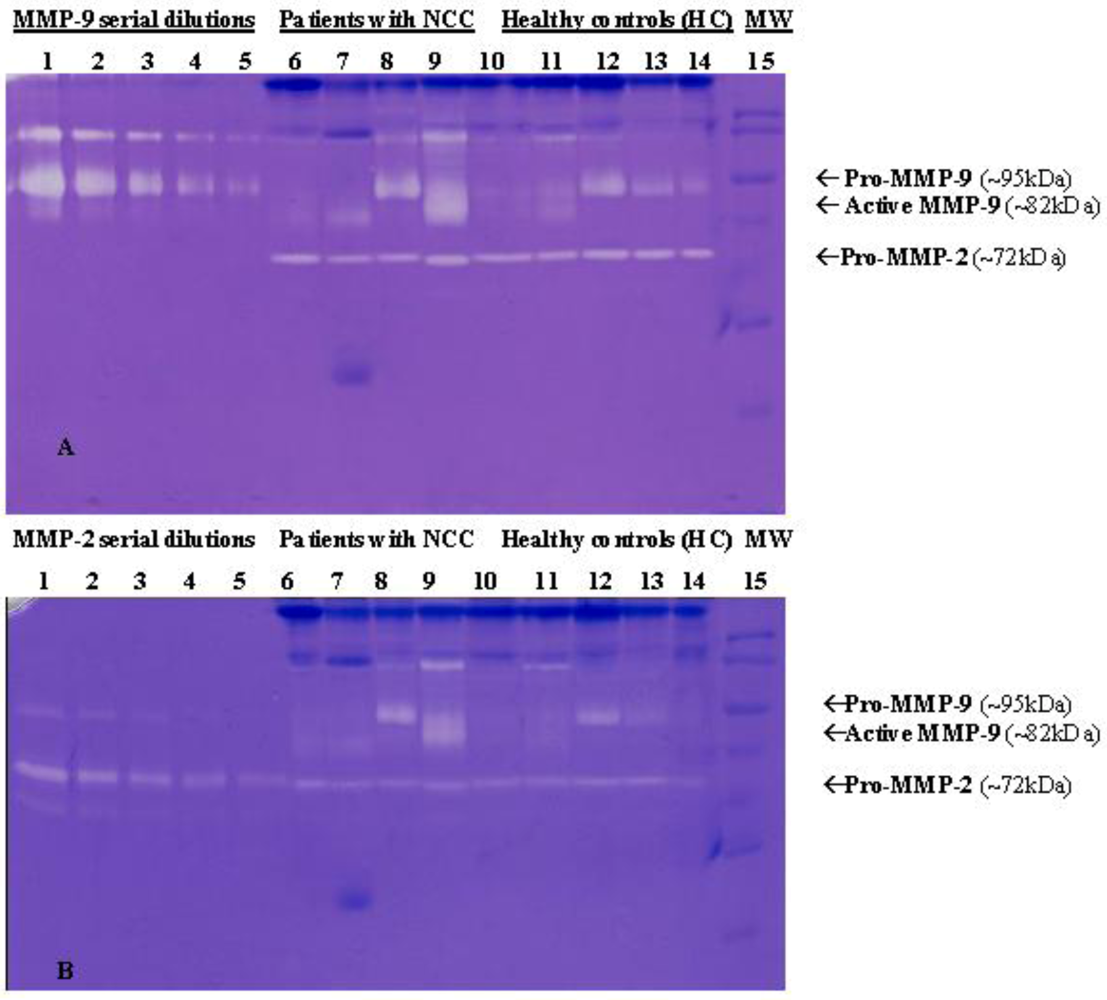
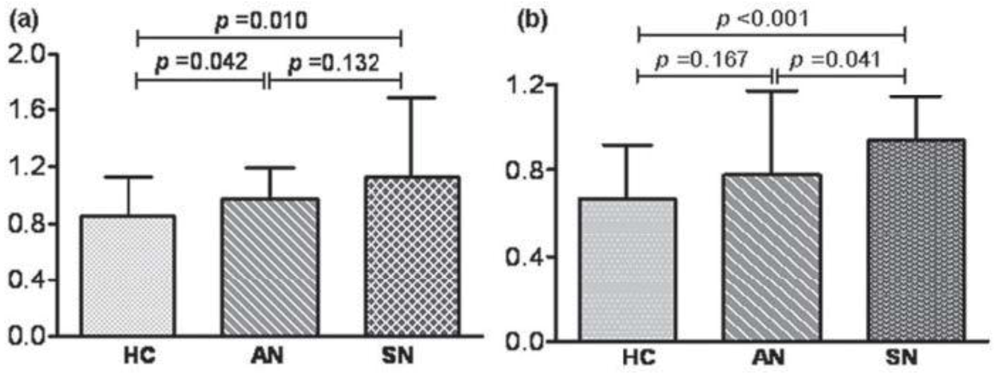
4.2.2. Infections with Nematodes
5. Concluding Remarks
| Parasitic infection | MODIFICATION OF MMP levels | MOFIFICATION OF TIMP levels | Refs. |
|---|---|---|---|
| Cerebral malaria | MMP-9 levels increased or unchanged | TIMP-2 level decreased | [107,108] |
| MMP-8 level increased [108] | TIMP-1 level increased | [108] | |
| MMP-1 accumulation in CNS [113] | [113] | ||
| African trypanosomosis | MMP-2 and MMP-9 levels increased | TIMP-1 and TIMP-2 levels unchanged | [117,120] |
| Cerebral toxoplasmosis | MMP-8 and MMP-10 produced by CD4+ and CD8+ T cells | Expression of TIMP-1 in the CNS | [125] |
| Neurocysticercosis | MMP-2 and MMP-9 levels increate in symptomatic patients [129] | [129] | |
| Angiostrongyloidosis | MMP-9 accumulation in inflammatory cells invading the CNS in experimental infection [139] | [139] | |
| TIMP-1 level increased in patients | |||
| MMP-2 and MMP-9 levels increased in patient | TIMP-4 level decreased | [132] |
Acknowledgments
Conflict of Interest
References
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef]
- Kumar, V.; Fausto, N.; Abbas, A. Robbins and Cotran: Pathologic Basis of Disease, 7th ed.; Elsevier: Philadelphia, PA, USA, 2004. [Google Scholar]
- Ingber, D.E. Mechanical control of tissue morphogenesis during embryological development. Int. J. Dev. Biol. 2006, 50, 255–266. [Google Scholar] [CrossRef]
- Maleski, M.; Hockfield, S. Glial cells assemble hyaluronan-based pericellular matrices in vitro. Glia 1997, 20, 193–202. [Google Scholar] [CrossRef]
- Rauch, U. Modeling an extracellular environment for axonal pathfinding and fasciculation in the central nervous system. Cell Tissue Res. 1997, 290, 349–356. [Google Scholar] [CrossRef]
- Fujita, M.; Spray, D.C.; Choi, H.; Saez, J.; Jefferson, D.M.; Hertzberg, E.; Rosenberg, L.C.; Reid, L.M. Extracellular matrixregulation ofcell-cell communicationand tissue-specific gene expression in primary liver cultures. Prog. Clin. Biol. Res. 1986, 226, 333–360. [Google Scholar]
- Noguera, R.; Nieto, O.A.; Tadeo, I.; Fariñas, F.; Alvaro, T. Extracellular matrix, biotensegrity and tumor microenvironment. An update and overview. Histol. Histopathol. 2012, 27, 693–705. [Google Scholar]
- Mosher, D.F.; Adams, J.B. Adhesion-modulating/matricellular ECM protein families: A structural, functional and evolutionary appraisal. Matrix Biol. 2012, 31, 155–161. [Google Scholar] [CrossRef]
- Lin, C.Q.; Bissell, M.J. Multi-faceted regulation of cell differentiation by extracellular matrix. FASEB J. 1993, 7, 737–743. [Google Scholar]
- Werb, Z.; Sympson, C.J.; Alexander, C.M.; Thomasset, N.; Lund, L.R.; MacAuley, A.; Ashkenas, J.; Bissell, M.J. Extracellular matrix remodeling and the regulation of epithelial-stromal interactions during differentiation and involution. Kidney Int. Suppl. 1996, 54, S68–S74. [Google Scholar]
- Gentili, C.; Cancedda, R. Cartilage and bone extracellular matrix. Curr. Pharm. Des. 2009, 15, 1334–1348. [Google Scholar] [CrossRef]
- Davis, G.E.; Donald, R.; Senger, D.R. Endothelial Extracellular Matrix: Biosynthesis, Remodeling, and Functions During Vascular Morphogenesis and Neovessel Stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef]
- Plopper, G. The extracellular matrix and cell adhesion, in Cells; Lewin, B., Cassimeris, L., Lingappa, V., Plopper, G., Sudbury, M.A., Eds.; Jones and Bartlett: Burlington, MA, USA, 2007. [Google Scholar]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef]
- Nicholson, C.; Syková, E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998, 21, 207–215. [Google Scholar] [CrossRef]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell. Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- Roycik, M.D.; Fang, X.; Sang, Q.X. A fresh prospect of extracellular matrix hydrolytic enzymes and their substrates. Curr. Pharm. Des. 2009, 15, 1295–1308. [Google Scholar] [CrossRef]
- Hijova, E. Matrix metalloproteinases: their biological functions and clinical implication. Bratisl. Lek. Listy. 2005, 106, 127–132. [Google Scholar]
- Ghajar, C.M.; George, S.C.; Putnam, A.J. Matrix metalloproteinase control of capillary morphogenesis. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 251–278. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Ravi, A.; Pallavi, G.; Sitaraman, S.V. matrix metalloproteinases in inflammatory bowel disease: boon or a bane? Inflamm. Bowel Dis. 2007, 13, 97–107. [Google Scholar] [CrossRef]
- Yong, V.W.; Power, C.; Forsyth, P.; Edwards, D.R. Metalloproteinases in biology and pathology of the nervous system. Nature Rev. 2001, 2, 502–511. [Google Scholar] [CrossRef]
- Dzwonek, J.; Rylski, M.; Kaczmarek, L. Matrix metalloproteinases and their endogenous inhibitors in neuronal physiology of the adult brain. FEBS Lett. 2004, 567, 129–135. [Google Scholar] [CrossRef]
- Sedlacek, R.; Mauch, S.; Kolb, B.; Schätzlein, C.; Eibel, H.; Peter, H.H.; Schmitt, J.; Krawinkel, U. Matrix metalloproteinase MMP-19 (RASI-1) is expressed on the surface of activated peripheral blood mononuclear cells and is detected as an autoantigen in rheumatoid arthritis. Immunobiol. 1998, 198, 408–423. [Google Scholar] [CrossRef]
- Tetlow, L.C.; Adlam, D.J.; Woolley, D.E. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001, 44, 585–594. [Google Scholar] [CrossRef]
- Yoshida, W.; Uzuki, M.; Nishida, J.; Shimamura, T.; Sawai, T. Examination of in vivo gelatinolytic activity in rheumatoid arthritis synovial tissue using newly developed in situ zymography and image analyzer. Clin. Exp. Rheumatol. 2009, 27, 587–593. [Google Scholar]
- Creemers, E.E.; Cleutjens, J.P.; Smits, J.F.; Daemen, M.J. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ. Res. 2001, 89, 201–210. [Google Scholar] [CrossRef]
- Siefert, S.A.; Sarkar, R. Matrix metalloproteinases in vascular physiology and disease. Vascular. 2012, 20, 210–216. [Google Scholar] [CrossRef]
- Briasoulis, A.; Tousoulis, D.; Papageorgiou, N.; Kampoli, A.M.; Androulakis, E.; Antoniades, C.; Tsiamis, E.; Latsios, G.; Stefanadis, C. Novel therapeutic approaches targeting matrix metalloproteinases in cardiovascular disease. Curr. Top. Med. Chem. 2012, 12, 1214–1221. [Google Scholar] [CrossRef]
- Srivastava, P.K.; Dastidar, S.G.; Ray, A. Chronic obstructive pulmonary disease: role of matrix metalloproteases and future challenges of drug therapy. Expert. Opin. Investig. Drugs. 2007, 16, 1069–1078. [Google Scholar] [CrossRef]
- Oikonomidi, S.; Kostikas, K.; Tsilioni, I.; Tanou, K.; Gourgoulianis, K.I.; Kiropoulos, T.S. Matrix metalloproteinases in respiratory diseases: from pathogenesis to potential clinical implications. Curr. Med. Chem. 2009, 16, 1214–1228. [Google Scholar] [CrossRef]
- Mocchegiani, E.; Giacconi, R.; Costarelli, L. Metalloproteases/anti-metalloproteases imbalance in chronic obstructive pulmonary disease: genetic factors and treatment implications. Curr. Opin. Pulm. Med. 2011, 17, S11–S19. [Google Scholar] [CrossRef]
- Leppert, D.; Lindberg, R.L.; Kappos, L.; Leib, S.L. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res. Rev. 2001, 36, 249–257. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; Lòpez-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. 2004, 4, 617–628. [Google Scholar]
- Gharagozlian, S.; Svennevig, K.; Bangstad, H.J.; Winberg, J.O.; Kolset, S.O. Matrix metalloproteinases in subjects with type 1 diabetes. BMC Clin. Pathol. 2009, 16, 7. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Matrix metalloproteinases and the development of cancer. Chem. Biol. 1996, 3, 895–904. [Google Scholar] [CrossRef]
- van Kempen, L.C.; Coussens, L.M. MMP-9 potentiates pulmonary metastasis formation. Cancer Cell. 2002, 2, 251–252. [Google Scholar] [CrossRef]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Zucker, S.; Vacirca, J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 101–117. [Google Scholar] [CrossRef]
- Affara, N.I.; Andreu, P.; Coussens, L.M. Delineating protease functions during cancer development. Methods Mol. Biol. 2009, 539, 1–32. [Google Scholar] [CrossRef]
- Rooprai, H.K.; McCormick, D. Proteases and their inhibitors in human brain tumours: a review. Anticancer Res. 1997, 17, 4151–4162. [Google Scholar]
- Avolio, C.; Ruggieri, M.; Giuliani, F.; Liuzzi, G.M.; Leante, R.; Riccio, P.; Livrea, P.; Trojano, M. Serum MMP-2 and MMP-9 are elevated in ifferent multiple sclerosis subtypes. J. Neuroimmunol. 2003, 136, 46–53. [Google Scholar] [CrossRef]
- Backstrom, J.R.; Miller, C.A.; Tokes, Z.A. Characterization of neutral proteinases from Alzheimer-affected and control brain specimens: identification of calcium-dependent metalloproteinases from the hippocampus. J. Neurochem. 1992, 58, 983–992. [Google Scholar] [CrossRef]
- Créange, A.; Sharshar, T.; Planchenault, T.; Christov, C.; Poron, F.; Raphae¨l, J.C.; Gherardi, R.K. Matrix metalloproteinase-9 is increased and correlates with severity in Guillain-Barre´ syndrome. Neurol. 1999, 53, 1683–1691. [Google Scholar] [CrossRef]
- Lim, G.P.; Backstrom, J.R.; Cullen, M.J.; Miller, C.A.; Atkinson, R.D.; Tokes, Z.A. Matrix metalloproteinases in the neocortex and spinal cord of amyotrophic lateral sclerosis patients. J. Neurochem. 1996, 67, 251–259. [Google Scholar]
- Rosenberg, G.A. Matrix metalloproteinases in brain injury. J. Neurotrauma 1995, 12, 833–842. [Google Scholar] [CrossRef]
- Lukes, A.; Mun-Bryce, S.; Lukes, M.; Rosenberg, G.A. Extracellular matrix degradation by metalloproteinases and central nervous system diseases. Mol. Neurobiol. 1999, 19, 267–284. [Google Scholar] [CrossRef]
- Liuzzi, G.M.; Mastroianni, C.M.; Santacroce, M.P.; Fanelli, M.; D’Agostino, C.; Vullo, V.; Riccio, P. Increased activity of matrix metalloproteinases in the cerebrospinal fluid of patients with HIV-associated neurological disease. J. Neurovirol. 2000, 6, 156–163. [Google Scholar] [CrossRef]
- Sterlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behaviour. Ann. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef]
- Welgus, H.G.; Campbell, E.J.; Cury, J.D.; Eisen, A.Z.; Senior, R.M.; Wilhelm, S.M.; Goldberg, G.I. Neutral metalloproteinases produced by human mononuclear phagocytes. Enzyme profile, regulation, and expression during cellular development. J. Clin. Invest. 1990, 86, 1496–1502. [Google Scholar] [CrossRef]
- Leppert, D.; Waubant, E.; Galardy, R.; Bunnett, N.W.; Hauser, S.L. T cell gelatinases mediate basement membrane transmigration in vitro. J. Immunol. 1995, 154, 4379–4389. [Google Scholar]
- Masure, S.; Proost, P.; van Damme, J.; Opdenakker, G. Purification and identification of 91-kDa neutrophil gelatinase: release by the activating peptide interleukin-8. Eur. J. Biochem. 1991, 198, 391–398. [Google Scholar] [CrossRef]
- Wells, G.M.; Catlin, G.; Cossins, J.A.; Mangan, M.; Ward, G.A.; Miller, K.M.; Clements, J.M. Quantitation of matrix metalloproteinases in cultured rat astrocytes using the polymerase chain reaction with a multi-competitor cDNA standard. Glia 1996, 18, 332–340. [Google Scholar] [CrossRef]
- Gottschall, P.E.; Yu, X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J. Neurochem. 1995, 64, 1513–1520. [Google Scholar] [CrossRef]
- Okada, S.; Kita, H.; George, T.J.; Gleich, G.J.; Leiferman, K.M. Migration of eosinophils through basement membrane components in vitro: role of matrix metalloproteinase-9. Am. J. Respir. Cell. Mol. Biol. 1997, 17, 519–528. [Google Scholar]
- Nielsen, B.S.; Timshel, S.; Kjeldsen, L.; Sehested, M.; Pyke, C.; Borregaard, N.; Dano, K. 92 kDa type IV collagenase (MMP-9) is expressed in neutrophils and macrophages but not in malignant epithelial cells in human colon cancer. Int. J. Cancer 1996, 65, 57–62. [Google Scholar] [CrossRef]
- Herron, G.S.; Werb, Z.; Dwyer, K.; Banda, M.J. Secretion of metalloproteinases by stimulated capillary endothelial cells. In Production of procollagenase and prostromelysin exceeds expression of proteolytic activity. J. Biol. Chem. 1986, 261, 2810–2813. [Google Scholar]
- Birkedal-Hansen, H.; Moore, W.G.; Bodden, M.K.; Windsor, L.J.; Birkedal-Hansen, B.; DeCarlo, A.; Engler, J.A. Matrix metalloproteinases: a review. Crit. Rev. Oral Biol. Med. 1993, 4, 197–250. [Google Scholar]
- Birkedal-Hansen, H.; Yamada, S.; Windsor, J.; Pollard, A.H.; Lyons, G.; Stetler-Stevenson, W.; Birkedal-Hansen, B. Matrix metalloproteinases. Curr. Protoc. Cell Biol. 2008, 10, 8. [Google Scholar]
- Uría, J.A.; López-Otín, C. Matrilysin-2, a new matrix metalloproteinase expressed in human tumors and showing the minimal domain organization required for secretion, latency, and activity. Cancer Res. 2000, 60, 4745–4751. [Google Scholar]
- Nagase, H.; Woessner, J.F., Jr. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef]
- Overall, C.M. Molecular determinants of metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol. Biotechnol. 2002, 22, 51–86. [Google Scholar] [CrossRef]
- Stamenkovic, I. Extracellular matrix remodelling: the role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef]
- Steffens, B.; Hakkinen, L.; Larjava, H. Proteolytic events of wound-healing-coordinated interactions among matrix metalloproteinanses (MMPs), integrins, and extracellular matrix molecules. Crit. Rev. Oral Biol. Med. 2001, 12, 373–398. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Kobayashi, D.K.; Ley, T.J. Cloning and characterization of a unique elastolytic metalloproteinase produced by human alveolar macrophages. J. Biol. Chem. 1993, 268, 23824–23829. [Google Scholar]
- Salmela, M.T.; Karjalainen-Lindsberg, M.L.; Puolakkainen, P.; Saarialho-Kere, U. Upregulation and differential expression of matrilysin (MMP-7) and metalloelastase (MMP-12) and their inhibitors TIMP-1 and TIMP-3 in Barrett's oesophageal adenocarcinoma. Br. J. Cancer 2001, 85, 383–392. [Google Scholar] [CrossRef]
- Wilson, C.L.; Matrisian, L.M. Matrilysin: an epithelial matrix metalloproteinase with potentially novel functions. Int. J. Biochem. Cell Biol. 1996, 28, 123–136. [Google Scholar] [CrossRef]
- Park, H.I.; Ni, J.; Gerkema, F.E.; Liu, D.; Belozerow, V.; Sang, Q.X.A. Identification and characterization of human endometase (matrix metalloproteinase-26) from endometrial tumor. J. Biol. Chem. 2000, 27, 20540–20544. [Google Scholar]
- Galewskaa, Z.; Romanowicza, L.; Jaworskib, S.; Bańkowskia, E. Matrix metalloproteinases, MMP-7 and MMP-26, in plasma and serum of control and preeclamptic umbilical cord blood. Eur. J. Obstetrics Gynecol. Repr. Biol. 2010, 150, 152–156. [Google Scholar] [CrossRef]
- Massova, I.; Kotra, L.P.; Fridman, R.; Mobashery, S. Matrix metalloproteinases: structures, evolution, and diversification. FASEB J. 1998, 12, 1075–1095. [Google Scholar]
- Das, S.; Mandal, M.; Chakraborti, T.; Mandal, A.; Chakraborti, S. Structure and evolutionary aspects of matrix metalloproteinases: a brief overview. Mol. Cell Biochem. 2003, 253, 31–40. [Google Scholar] [CrossRef]
- Ii, M.; Yamamoto, H.; Adachi, Y.; Maruyama, Y.; Shinomura, Y. Role of matrix metalloproteinase-7 (matrilysin) in human cancer invasion, apoptosis, growth, and angiogenesis. Exp. Biol. Med. (Maywood) 2006, 231, 20–27. [Google Scholar]
- Jones, C.B.; Sane, D,C.; Herrington, D.M. Matrix metalloproteinases: a review of their structure and role in acute coronary syndrome. Cardiovasc. Res. 2003, 59, 812–823. [Google Scholar] [CrossRef]
- Fillmore, H.L.; VanMeter, T.E.; Broaddus, W.C. Membrane-type matrix metalloproteinases (MT-MMPs): expression and function during glioma invasion. J. Neurooncol. 2001, 53, 187–202. [Google Scholar] [CrossRef]
- Zucker, S.; Pei, D.; Cao, J.; Lopez-Otin, C. Membrane type-matrix metalloproteinases (MT-MMP). Curr. Top. Dev. Biol. 2003, 54, 1–74. [Google Scholar] [CrossRef]
- Stracke, O.J.; Fosang, J.A.; Last, K.; Mercuri, A.F.; Pendas, M.A.; Llano, E.; Perris, R.; Di Cesare, E.P. Matrix metalloproteinases 19 and 20 cleave aggrecan and cartilage oligomeric matrix protein (COMP). FEBS Lett. 2000, 478, 52–56. [Google Scholar] [CrossRef]
- Lohi, J.; Wilson, C.L.; Roby, J.D.; Parks, W.C. Epilysin, a novel human matrix metalloproteinase (MMP-28) expressed in testis and keratinocytes and in response to injury. J. Biol. Chem. 2001, 276, 10134–10144. [Google Scholar]
- Fini, M.E.; Cook, J.R.; Mohan, R. Proteolytic mechanisms in corneal ulceration and repair. Arch. Dermatol. Res. 1998, 290, S12–S23. [Google Scholar] [CrossRef]
- van den Berg, W.B. The role of cytokines and growth factors in cartilage destruction in osteoarthritis and rheumatoid arthritis. J. Rheumatol. 1999, 58, 136–141. [Google Scholar]
- Cawston, T.E.; Wilson, A.J. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract. Res. Clin. Rheumatol. 2006, 20, 983–1002. [Google Scholar] [CrossRef]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell. Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef]
- Clark, I.M.; Swingler, T.E.; Sampieri, C.L.; Edwards, D.R. The regulation of matrix metalloproteinases and their inhibitors. Int. J. Biochem. Cell. Biol. 2008, 40, 1362–1378. [Google Scholar] [CrossRef] [Green Version]
- Springman, E.B.; Angleton, E.L.; Birkedal-Hansen, H.; Van Wart, H.E. Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc. Natl. Acad. Sci. USA 1990, 87, 364–368. [Google Scholar]
- Van Wart, H.E. Birkedal-Hansen, H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef]
- Kotra, L.P.; Zhang, L.; Fridman, R.; Orlando, R.; Mobashery, S. N-Glycosylation pattern of the zymogenic form of human matrix metalloproteinase-9. Bioorg. Chem. 2002, 30, 356–370. [Google Scholar] [CrossRef]
- Yang, Z.; Strickland, D.K.; Bornstein, P. Extracellular MMP-2 levels are regulated by the low-density lipoprotein-related scavenger receptor and thrombospondin 2. J. Biol. Chem. 2001, 276, 8403–8408. [Google Scholar]
- Murphy, G. Tissue inhibitors of metalloproteinases. Murphy Genome Biol. 2011, 12, 1–7. [Google Scholar]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim. Biophys. Acta. 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Bode, W.; Maskos, K. Structural basis of the matrix metalloproteinases and their physiological inhibitors, the tissue inhibitors of metalloproteinases. Biol. Chem. 2003, 384, 863–872. [Google Scholar]
- Gomez, D.E.; Alonso, D.F.; Yoshiji, H.; Thorgeirsson, U.P. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur. J. Cell. Biol. 1997, 7, 111–122. [Google Scholar]
- Jiang, Y.; Goldberg, I.D.; Shi, Y.E. Complex roles of tissue inhibitors of metalloproteinases in cancer. Oncogene 2002, 21, 2245–2252. [Google Scholar] [CrossRef]
- Guedez, L.; Stetler-Stevenson, W.G.; Wolff, L.; Wang, J.; Fukushima, P.; Mansoor, A.; Stetler-Stevenson, M. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J. Clin. Invest. 1998, 102, 2002–2010. [Google Scholar] [CrossRef]
- Fata, J.E.; Leco, K.J.; Voura, E.B.; Yu, H.Y.; Waterhouse, P.; Murphy, G.; Moorehead, R.A.; Khokha, R. Accelerated apoptosis in the Timp-3-deficient mammary gland. J. Clin. Invest. 2001, 108, 831–841. [Google Scholar]
- Hadler-Olsen, E.; Fadnes, B.; Sylte, I.; Uhlin-Hansen, L.; Winberg, J.O. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 2011, 278, 28–45. [Google Scholar] [CrossRef]
- Geurts, N.; Opdenakker, G.; Van den Steen, P.E. Matrix metalloproteinases as therapeutic targets in protozoan parasitic infections. Pharmacol. Ther. 2012, 133, 257–279. [Google Scholar] [CrossRef]
- Snow, R.W.; Guerra, C.A.; Noor, A.M.; Myint, H.Y.; Hay, S.I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 2005, 434, 214–217. [Google Scholar] [CrossRef]
- Kappe, S.H.; Vaughan, A.M.; Boddey, J.A.; Cowman, A.F. Thatwas then but this is now: malaria research in the time of an eradication agenda. Science 2010, 328, 862–866. [Google Scholar] [CrossRef]
- van der Heyde, H.C.; Nolan, J.; Combes, V.; Gramaglia, I.; Grau, G.E. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006, 22, 503–508. [Google Scholar] [CrossRef]
- Miller, L.H.; Dror, I.; Baruch, K.M.; Ogobara, K.D. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- Armah, H.; Dodoo, A.K.; Wiredu, E.K.; Stiles, J.K.; Adjei, A.A.; Gyasi, R.K.; Tettey, Y. High-level cerebellar expression of cytokines and adhesion molecules in fatal, paediatric, cerebral malaria. Ann. Trop. Med. Parasitol. 2005, 99, 629–647. [Google Scholar] [CrossRef]
- Shikani, H.J.; Freeman, B.D.; Lisanti, M.P.; Weiss, L.M.; Tanowitz, H.B.; Desruisseaux, M.S. Cerebral malaria:We Have Come a Long Way. Am. J. Pathol. 2012, 181, 1484–1492. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Yong, V.W. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat. Rev. Neurosci. 2005, 6, 931–944. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Kornfeld, M.; Estrada, E.; Kelley, R.O.; Liotta, L.A.; Stetler-Stevenson, W.G. TIMP-2 reduces proteolytic opening of blood-brain barrier by type IV collagenase. Brain Res. 1992, 576, 203–207. [Google Scholar] [CrossRef]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of immune cells in the central nervous system. J. Clin. Invest. 2010, 120, 1368–1379. [Google Scholar] [CrossRef]
- Griffiths, M.J.; Shafi, M.J.; Popper, S.J.; Hemingway, C.A.; Kortok, M.M.; Wathen, A.; Rockett, K.A.; Mott, R.; Levin, M.; Newton, C.R.; Marsh, K.; Relman, D.A.; Kwiatkowski, D.P. Genomewide analysis of the host response to malaria in Kenyan children. J. Infect. Dis. 2005, 191, 1599–1611. [Google Scholar]
- Dietmann, A.; Helbok, R.; Lackner, P.; Issifou, S.; Lell, B.; Matsiegui, P.B.; Reindl, M.; Schmutzhard, E.; Kremsner, P.G. Matrix metalloproteinases and their tissue inhibitors (TIMPs) in Plasmodium falciparum malaria: serum levels of TIMP-1 are associated with disease severity. J. Infect. Dis. 2008, 197, 1614–1620. [Google Scholar] [CrossRef]
- Mun-Bryce, S.; Rosenberg, G.A. Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am. J. Physiol. 1998, 274, R1203–R1211. [Google Scholar]
- Muroski, M.E.; Roycik, M.D.; Newcomer, R.G.; Van den Steen, P.E.; Opdenakker, G.; Monroe, H.R.; Sahab, Z.J.; Sang, Q.X. Matrix metalloproteinase-9/gelatinase B is a putative therapeutic target of chronic obstructive pulmonary disease and multiple sclerosis. Curr. Pharm. Biotechnol. 2008, 9, 34–46. [Google Scholar] [CrossRef]
- Prato, M.; D'Alessandro, S.; Van den Steen, P.E.; Opdenakker, G.; Arese, P.; Taramelli, D.; Basilico, M. Natural haemozoin modulates matrix metalloproteinases and induces morphological changes in human microvascular endothelium. Cell Microbiol. 2011, 13, 1275–1285. [Google Scholar] [CrossRef]
- Deininger, M.H.; Winkler, S.; Kremsner, P.G.; Meyermann, R.; Schluesener, H.J. Angiogenic proteins in brains of patients who died with cerebral malaria. J. Neuroimmunol. 2003, 142, 101–111. [Google Scholar] [CrossRef]
- Deininger, M.H.; Kremsner, P.G.; Meyermann, R.; Schluesener, H. Macrophages/microglial cells in patients with cerebral malaria. Eur. Cytokine Netw. 2002, 13, 173–185. [Google Scholar]
- Kristensson, K.; Nygård, M.; Bertini, G.; Bentivoglio, M. African trypanosome infections of the nervous system: parasite entry and effects on sleep and synaptic functions. Prog. Neurobiol. 2010, 91, 152–171. [Google Scholar] [CrossRef]
- Matthews, K.R.; Gull, K. Cycles within cycles: the interplay between differentiation and cell division in Trypanosoma brucei. Parasitol. Today. 1994, 10, 473–476. [Google Scholar] [CrossRef]
- Enanga, B.; Burchmore, R. J.; Stewart, M.L.; Barrett, M.P. Sleeping sickness and the brain. Cell. Mol. Life Sci. 2002, 59, 845–858. [Google Scholar] [CrossRef]
- Grab, D. J.; Kennedy, P.G. Traversal of human and animal trypanosomes across the blood-brain barrier. J. Neurovirol. 2008, 14, 344–351. [Google Scholar] [CrossRef]
- Hainard, A.; Tiberti, N.; Robin, X.; Ngoyi, D.M.; Matovu, E.; Enyaru, J.C.; Müller, M.; Turck, N.; Ndung'u, J.M.; Lejon, V.; Sanchez, J.C. Matrix metalloproteinase-9 and intercellular adhesion molecule 1 are powerful staging markers for human African trypanosomiasis. Trop. Med. Int. Health. 2011, 16, 119–126. [Google Scholar] [CrossRef]
- Agrawal, S.; Anderson, P.; Durbeej, M.; van Rooijen, N.; Ivars, F.; Opdenakker, G.; Sorokin, L.M. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1007–1019. [Google Scholar] [CrossRef]
- Masocha, W.; Rottenberg, M.E.; Kristensson, K. Minocycline impedes African trypanosome invasion of the brain in a murine model. Antimicrob. Agents Chemother. 2006, 50, 1798–1804. [Google Scholar] [CrossRef]
- Darcy, F.; Santoro, F. Toxoplasmosis. In Parasitic Infections and the Immune System; Kierszenbaum, F., Ed.; Academic Press: Waltham, MA, USA, 1994; pp. 163–201. [Google Scholar]
- Wong, S.Y.; Remington, J.S. Biology of Toxoplasma gondii. AIDS 1993, 7, 299–316. [Google Scholar] [CrossRef]
- Gazzinelli, R.; Xu, Y.; Hieny, S.; Cheever, A.; Sher, A. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J. Immunol. 1992, 149, 175–180. [Google Scholar]
- Strack, A.; Asensio, V.C.; Campbell, I.L.; Schluter, D.; Deckert, M. Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in Toxoplasma encephalitis and critically regulated by interferon-gamma. Acta Neuropathol. 2002, 103, 458–468. [Google Scholar] [CrossRef]
- Clark, R.T.; Nance, J.P.; Noor, S.; Wilson, E.H. T cell production of matrix metalloproteases and inhibition of parasite clearance by TIMP-1 during chronic toxoplasma infection in the brain. ASN Neurol. 2010, 3, 1–12. [Google Scholar]
- García, H.H.; Gonzalez, A.E.; Evans, C.A.; Gilman, R.H. Cysticercosis Working Group in Peru. Taenia solium cysticercosis. Lancet 2003, 362, 547–556. [Google Scholar]
- Sciutto, E.; Fragoso, G.; Fleury, A.; Laclette, J.P.; Sotelo, J.; Aluja, A.; Vargas, L.; Larralde, C. Taenia solium disease in humans and pigs:an ancient parasitosis disease rooted in developing countries and emerging as a major health problem of global dimensions. Microbes Infect. 2000, 2, 1875–1890. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Teale, J.M. Multiple expression of matrix metalloproteinases in murine neurocysticercosis: implications for leukocyte migration through multiple central nervous system barriers. Brain Res. 2008, 1214, 145–158. [Google Scholar] [CrossRef]
- Verma, A.; Prasad, K.N.; Nyati, K.K.; Singh, S.K.; Singh, A.K.; Paliwal, V.K.; Gupta, R.K. Association of MMP-2 and MMP-9 with clinical outcome of neurocysticercosis. Parasitol. 2011, 138, 1423–1428. [Google Scholar] [CrossRef]
- Heuser, K.; Hoddevik, E.H.; Taubøll, E.; Gjerstad, L.; Indahl, U.; Kaczmarek, L.; Berg, P.R.; Lien, S.; Nagelhus, E.A.; Ottersen, O.P. Temporal lobe epilepsy and matrix metalloproteinase 9: a tempting relation but negative genetic association. Seizure 2010, 19, 335–338. [Google Scholar] [CrossRef]
- Yin, P.; Yang, L.; Zhou, H.Y.; Sun, R.P. Matrix metalloproteinase-9 may be a potential therapeutic target in epilepsy. Med. Hypotheses 2011, 76, 184–186. [Google Scholar] [CrossRef]
- Tsai, H.C.; Chung, L.Y.; Chen, E.R.; Liu, Y.C.; Lee, S.S.J.; Chen, Y.S.; Sy, C.L.; Wann, S.R.; Yen, C.M. Association of Matrix Metalloproteinase-9 and Tissue Inhibitors of Metalloproteinase-4 in Cerebrospinal Fluid with Blood-Brain Barrier Dysfunction in Patients with Eosinophilic Meningitis Caused by Angiostrongylus cantonensis. Am. J. Trop. Med. Hyg. 2008, 78, 20–27. [Google Scholar]
- Wang, Q.P.; Wu, Z.D.; Wei, J.; Owen, R.L.; Lun, Z.R. Human Angiostrongylus cantonensis: an update. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 389–395. [Google Scholar] [CrossRef]
- Nishimura, K.; Hung, T. Current views on geographic distribution and modes of infection of neurohelminthic diseases. J. Neurol Sci. 1997, 145, 5–14. [Google Scholar] [CrossRef]
- Hsu, W.Y.; Chen, J.Y.; Chien, C.T.; Chi, C.S.; Han, N.T. Eosinophilic meningitis caused by Angiostrongylus cantonensis. Pediatr. Infect. Dis. J. 1990, 9, 443–445. [Google Scholar] [CrossRef]
- Sasaki, O.; Sugaya, H.; Ishida, K.; Yoshimura, K. Ablation of eosinophils with anti-IL-5 antibody enhances the survival of intracranial worms of Angiostrongylus cantonensis in the mouse. Parasite Immunol. 1993, 15, 349–354. [Google Scholar] [CrossRef]
- Sugaya, H.; Yoshimura, K. T-cell-dependent eosinophilia in the cerebrospinal fluid of the mouse infected with Angiostrongylus cantonensis. Parasite Immunol. 1998, 10, 127–138. [Google Scholar] [CrossRef]
- Lai, S.C.; Jiang, S.T.; Chen, K.M.; Lee, H.H. Matrix metalloproteinases activity demonstrated in the infective stage of the nematodes, Angiostrongylus cantonensis. Parasitol. Res. 2005, 97, 466–471. [Google Scholar]
- Lee, H.H.; Chou, H.L.; Chen, K.M.; Lai, S.C. Association of matrix metalloproteinase-9 in eosinophilic meningitis of BALB/c mice caused by Angiostrongylus cantonensis. Parasitol. Res. 2004, 94, 321–328. [Google Scholar] [CrossRef]
- Tseng, Y.K.; Tu, W.C.; Lee, H.H.; Chen, K.M.; Chou, H.L.; Lai, S.C. Ultrastructural localization of matrix metalloproteinase-9 in eosinophils from the cerebrospinal fluid of mice with eosinophilic meningitis caused by Angiostrongylus cantonensis. Ann. Trop. Med. Parasitol. 2004, 98, 831–841. [Google Scholar] [CrossRef]
- Lai, S.C.; Twu, J.J.; Jiang, S.T.; Hsu, J.D.; Chen, K.M.; Chiaing, H.C.; Wang, C.J.; Tseng, C.K.; Shyu, L.Y.; Lee, H.H. Induction of matrix metalloproteinase-9 in murine eosinophilic meningitis caused by Angiostrongylus cantonensis. Ann. Trop. Med. Parasitol. 2004, 98, 715–724. [Google Scholar] [CrossRef]
- Chen, K.M.; Lee, H.H.; Lu, K.H.; Tseng, Y.K.; Hsu, L.S.; Chou, H.L.; Lai, S.C. Association of matrix metalloproteinase-9 and Purkinje cell degeneration in mouse cerebellum caused by Angiostrongylus cantonensis. Int. J. Parasitol. 2004, 34, 1147–1156. [Google Scholar] [CrossRef]
- Chen, K.M.; Lee, H.H.; Chou, H.L.; Liu, J.Y.; Tsai, B.; Lai, S.C. Upregulation of MMP-9/TIMP-1 enzymatic system in eosinophilic meningitis caused by Angiostrongylus cantonensis. Int. J. Exp. Pathol. 2005, 86, 81–89. [Google Scholar] [CrossRef]
- Chen, K.M.; Liu, J.Y.; Lai, S.C.; Hsu, L.S.; Lee, H.H. Association of plasminogen activators and matrix metalloproteinase-9 proteolytic cascade with blood-CNS barrier damage of angiostrongyliasis. Int. J. Exp. Path. 2006, 87, 113–119. [Google Scholar] [CrossRef]
- Wei, P.C.; Tsai, C.H.; Chiu, P.S.; Lai, S.C. Matrix metalloproteinase-12 leads to elastin degradation in BALB/c mice with eosinophilic meningitis caused by Angiostrongylus cantonensis. Int. J. Parasitol. 2011, 41, 1175–1183. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bruschi, F.; Pinto, B. The Significance of Matrix Metalloproteinases in Parasitic Infections Involving the Central Nervous System. Pathogens 2013, 2, 105-129. https://doi.org/10.3390/pathogens2010105
Bruschi F, Pinto B. The Significance of Matrix Metalloproteinases in Parasitic Infections Involving the Central Nervous System. Pathogens. 2013; 2(1):105-129. https://doi.org/10.3390/pathogens2010105
Chicago/Turabian StyleBruschi, Fabrizio, and Barbara Pinto. 2013. "The Significance of Matrix Metalloproteinases in Parasitic Infections Involving the Central Nervous System" Pathogens 2, no. 1: 105-129. https://doi.org/10.3390/pathogens2010105
APA StyleBruschi, F., & Pinto, B. (2013). The Significance of Matrix Metalloproteinases in Parasitic Infections Involving the Central Nervous System. Pathogens, 2(1), 105-129. https://doi.org/10.3390/pathogens2010105