
Iron, Aging, and Neurodegeneration

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Iron in the Brain
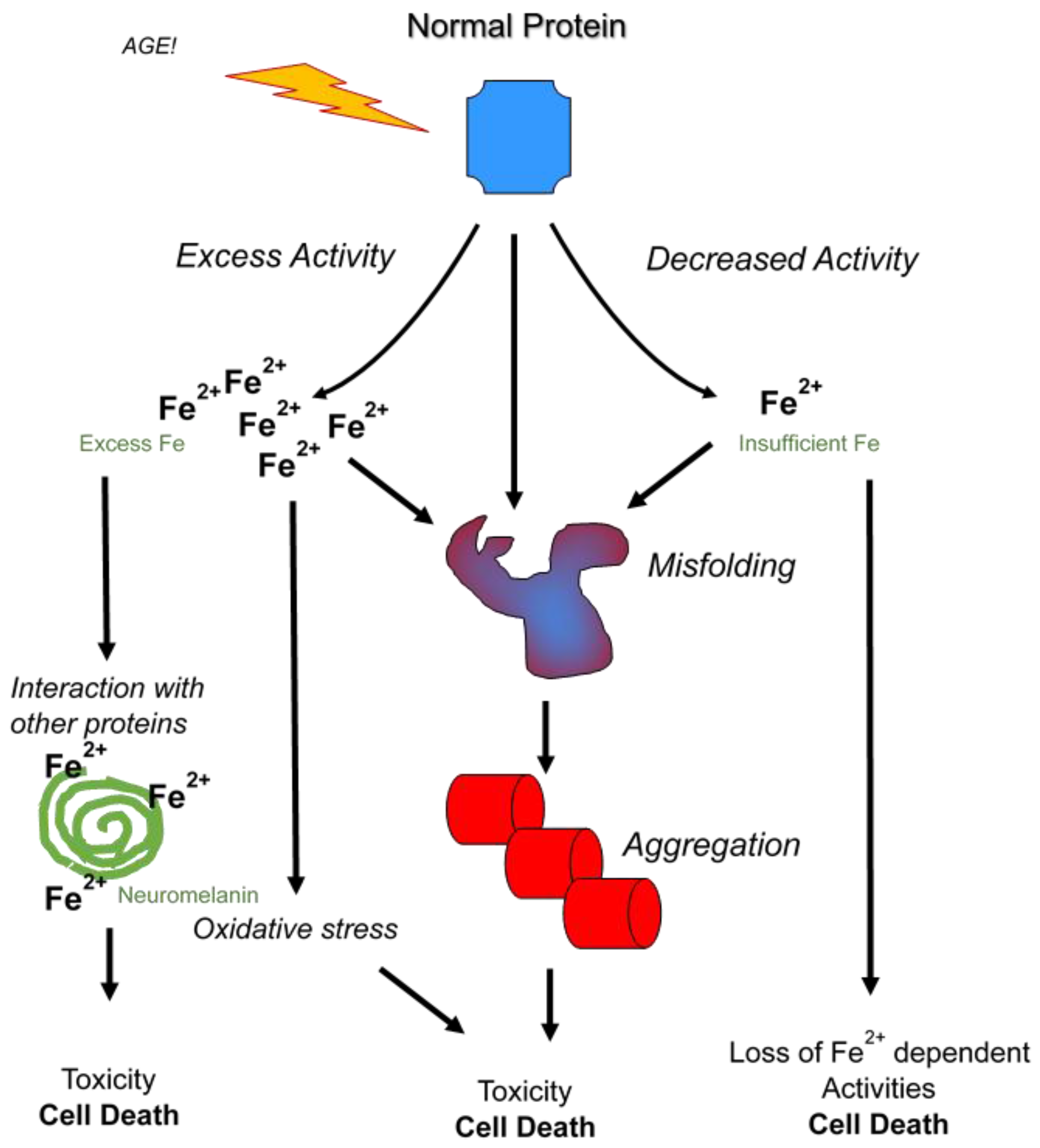
3. Neurodegenerative Diseases Linked to Iron
4. Iron in Alzheimer’s Disease
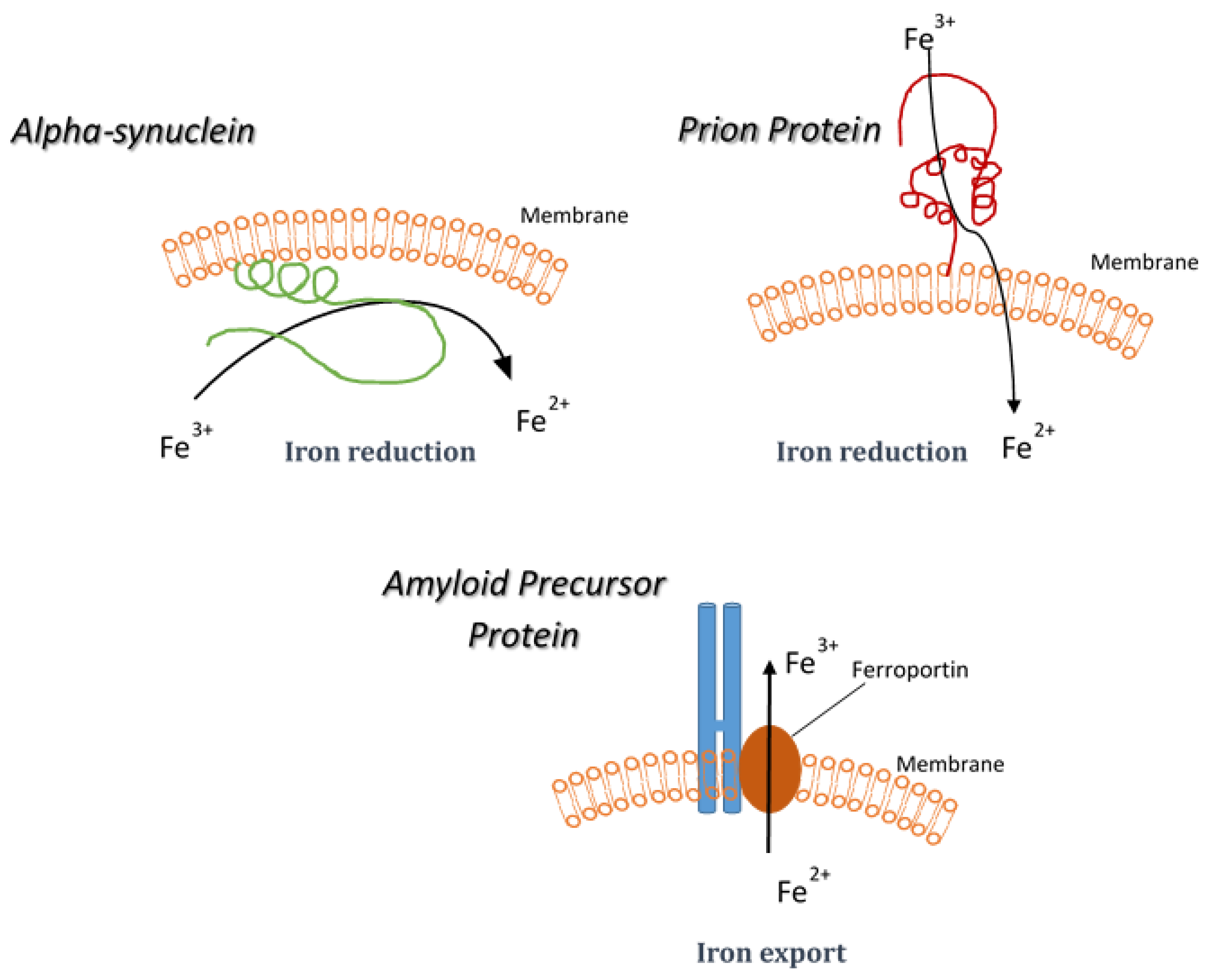
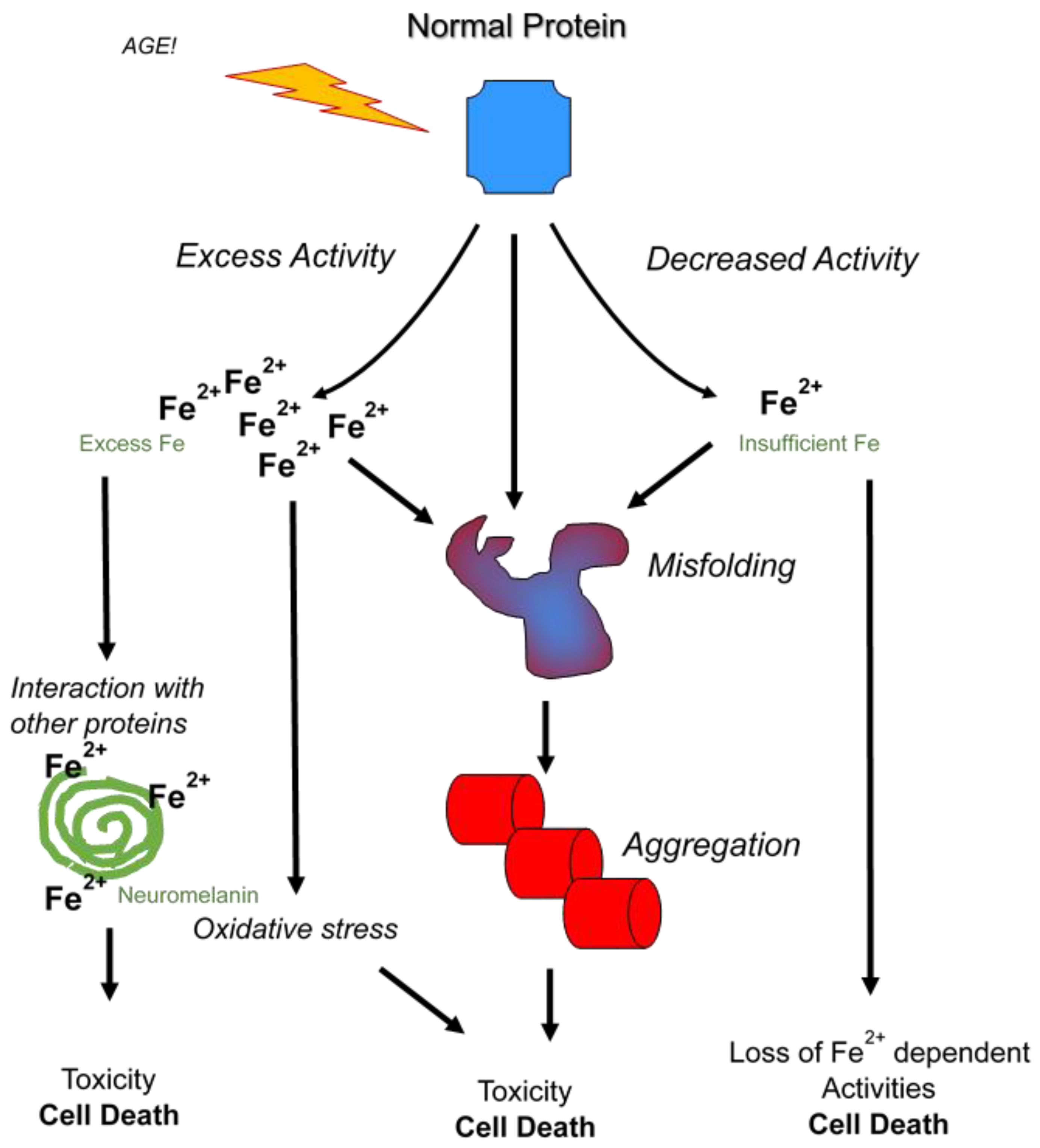
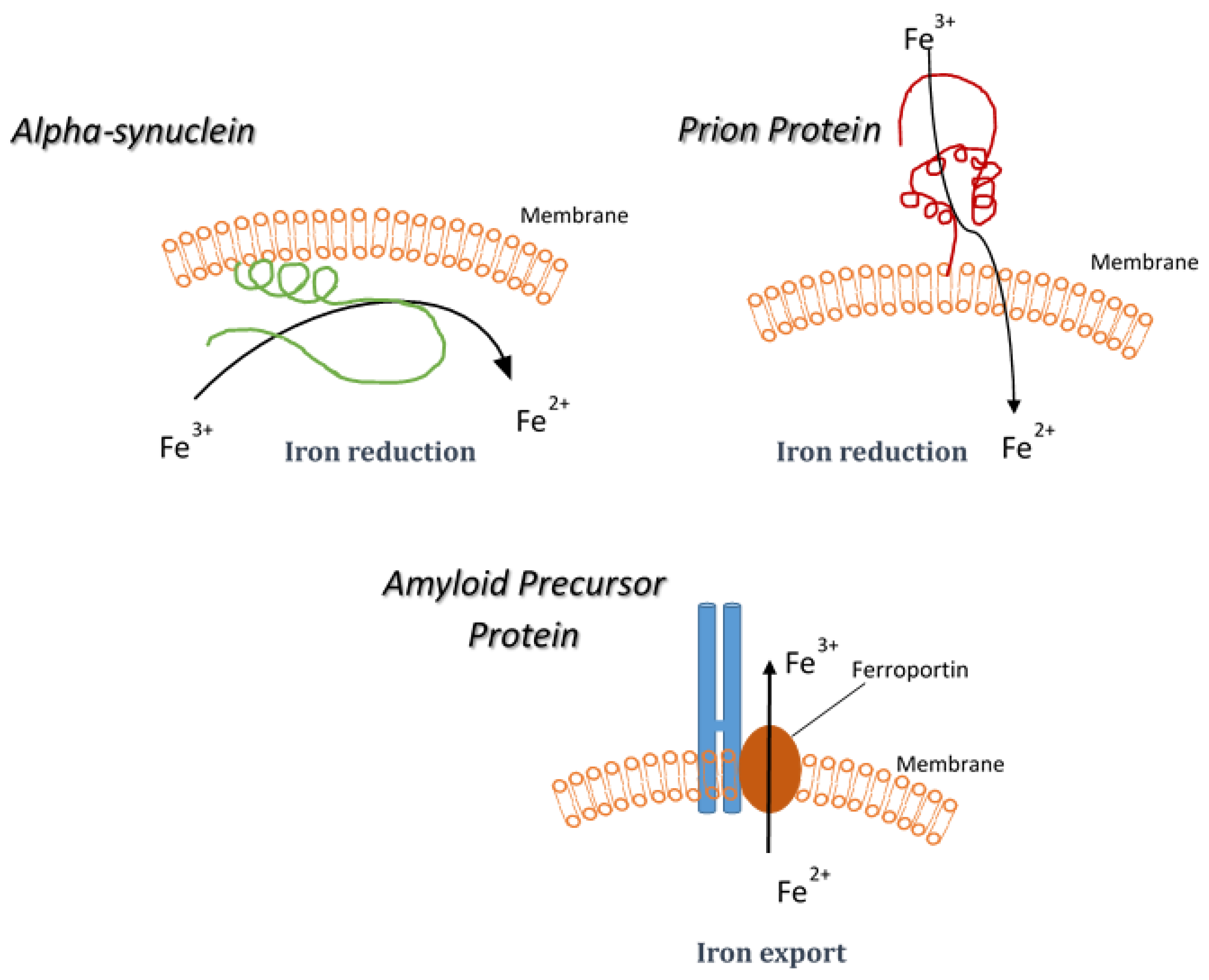
5. Synucleinopathies and Iron Reduction
6. Prion Diseases
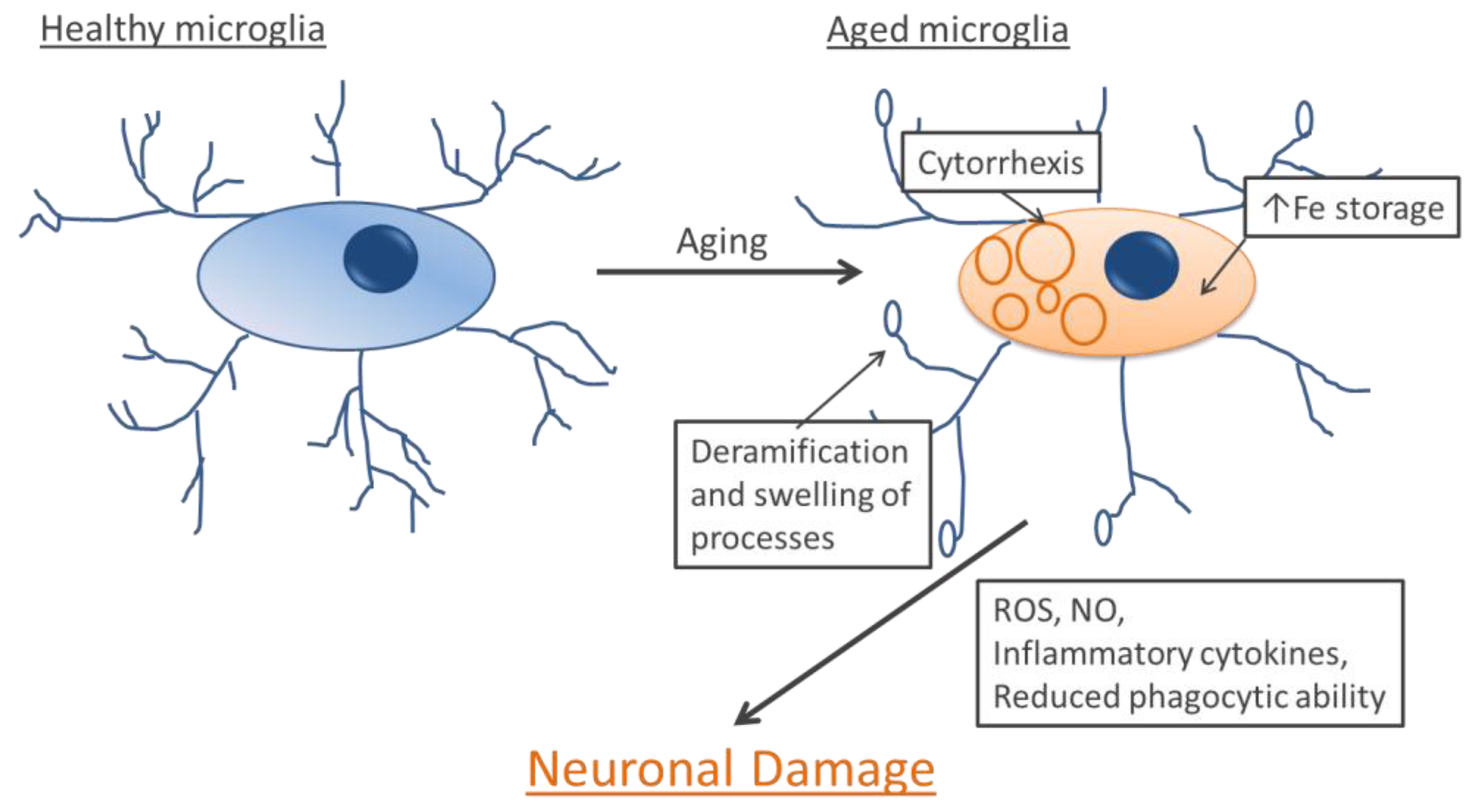
7. Microglia and Aging
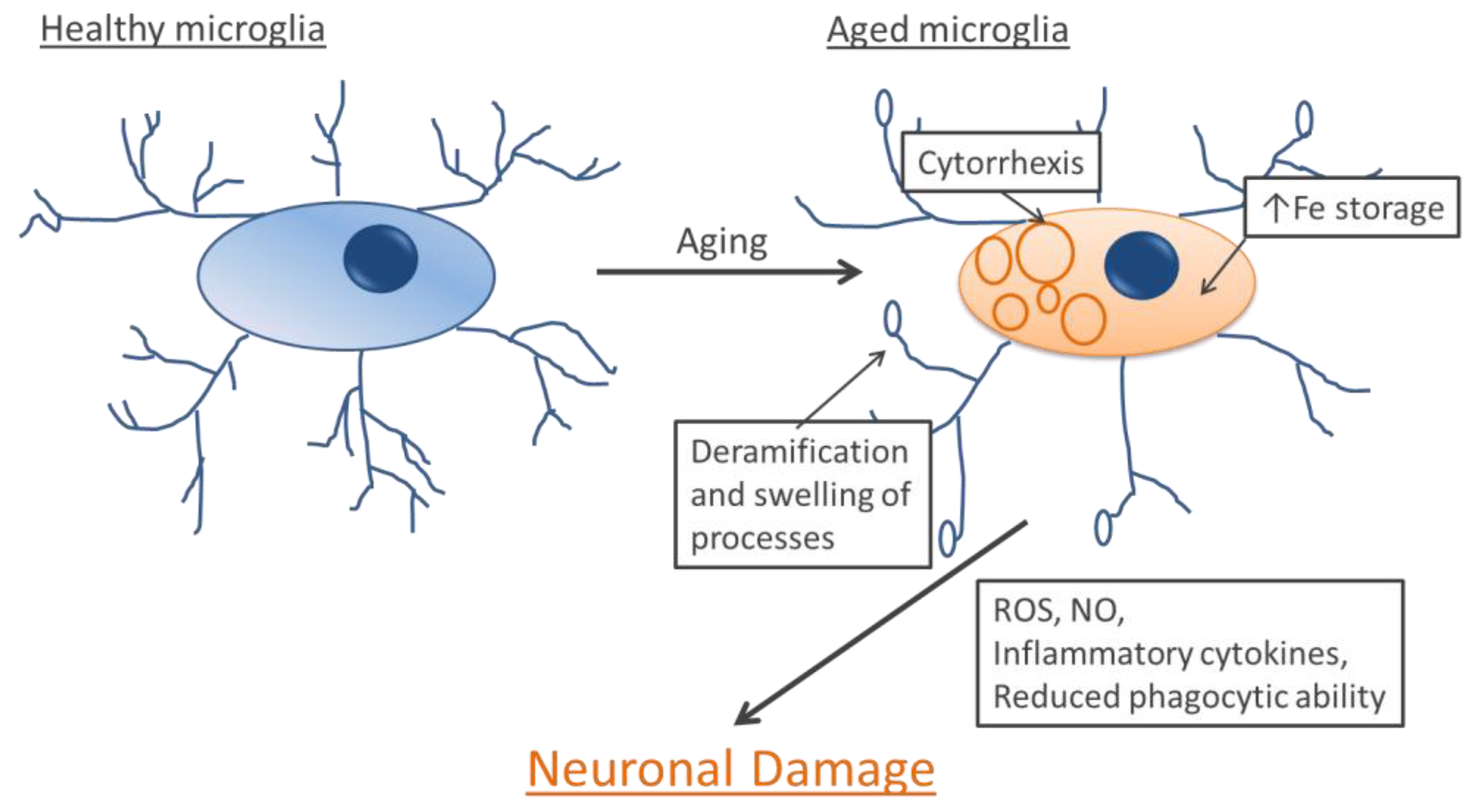
8. Microglia and Neurodegenerative Diseases
9. Conclusions
Author Contributions
Conflicts of Interest
References
- Oliveira, B.F.; Nogueira-Machado, J.A.; Chaves, M.M. The role of oxidative stress in the aging process. Sci. World J. 2010, 10, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, O.T. Management of the aging risk factor for Parkinson’s disease. Neurobiol. Aging 2014, 35, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Crichton, R.R.; Ward, R.J. Iron homeostasis. Met. Ions Biol. Syst. 1998, 35, 633–665. [Google Scholar] [PubMed]
- Drennan, C.L.; Peters, J.W. Surprising cofactors in metalloenzymes. Curr. Opin. Struct. Biol. 2003, 13, 220–226. [Google Scholar] [CrossRef]
- Dunn, L.L.; Rahmanto, Y.S.; Richardson, D.R. Iron uptake and metabolism in the new millennium. Trends Cell Biol. 2007, 17, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sargent, P.J.; Farnaud, S.; Evans, R.W. Structure/function overview of proteins involved in iron storage and transport. Curr. Med. Chem. 2005, 12, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Koorts, A.M.; Viljoen, M. Ferritin and ferritin isoforms II: Protection against uncontrolled cellular proliferation, oxidative damage and inflammatory processes. Arch. Physiol. Biochem. 2007, 113, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Winter, W.E.; Bazydlo, L.A.; Harris, N.S. The molecular biology of human iron metabolism. Lab. Med. 2014, 45, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Kong, X. Iron speciation in the cytosol: An overview. Dalton Trans. 2013, 42, 3220–3229. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. A brief history of brain iron research. J. Neurol. Sci. 2003, 207, 95–97. [Google Scholar] [CrossRef]
- Koeppen, A.H. The history of iron in the brain. J. Neurol. Sci. 1995, 134, 1–9. [Google Scholar] [CrossRef]
- Gotz, M.E.; Double, K.; Gerlach, M.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Burdo, J.R.; Martin, J.; Menzies, S.L.; Dolan, K.G.; Romano, M.A.; Fletcher, R.J.; Garrick, M.D.; Garrick, L.M.; Connor, J.R. Cellular distribution of iron in the brain of the belgrade rat. Neuroscience 1999, 93, 1189–1196. [Google Scholar] [CrossRef]
- Connor, J.R.; Boeshore, K.L.; Benkovic, S.A.; Menzies, S.L. Isoforms of ferritin have a specific cellular distribution in the brain. J. Neurosci. Res. 1994, 37, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Morgan, E.H. The significance of the mutated divalent metal transporter (DMT1) on iron transport into the belgrade rat brain. J. Neurochem. 2004, 88, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Casella, L.; Albertini, A.; Bellei, C.; Zucca, F.A.; Engelen, M.; Zadlo, A.; Szewczyk, G.; Zareba, M.; Sarna, T. Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson’s disease. J. Neurochem. 2008, 106, 1866–1875. [Google Scholar] [PubMed]
- Zucca, F.A.; Giaveri, G.; Gallorini, M.; Albertini, A.; Toscani, M.; Pezzoli, G.; Lucius, R.; Wilms, H.; Sulzer, D.; Ito, S.; et al. The neuromelanin of human substantia nigra: Physiological and pathogenic aspects. Pigment Cell Res. 2004, 17, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Gallorini, M.; Schunemann, V.; Trautwein, A.X.; Gerlach, M.; Riederer, P.; Vezzoni, P.; Tampellini, D. Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: Consequences for iron storage and neurodegenerative processes. J. Neurochem. 2001, 76, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, B.; Sourander, P. The effect of age on the non-haemin iron in the human brain. J. Neurochem. 1958, 3, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Bonnet, A.M.; Agid, Y.; Hirsch, E.C. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef]
- Brun, A.; Englund, E. Brain changes in dementia of Alzheimer’s type relevant to new imaging diagnostic methods. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1986, 10, 297–308. [Google Scholar] [CrossRef]
- Pandolfo, M. Friedreich’s ataxia: Clinical aspects and pathogenesis. Semin. Neurol. 1999, 19, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Napier, I.; Ponka, P.; Richardson, D.R. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Liu, Y.; Montermini, L.; Stifani, S.; Pandolfo, M. Frataxin shows developmentally regulated tissue-specific expression in the mouse embryo. Neurobiol. Dis. 1997, 4, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Boddaert, N.; Sang, K.H.L.Q.; Rotig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Rovida, E. Neuroferritinopathy: From ferritin structure modification to pathogenetic mechanism. Neurobiol. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.R.; Fey, C.; Morris, C.M.; Bindoff, L.A.; Ince, P.G.; Chinnery, P.F.; Coulthard, A.; Jackson, M.J.; Jackson, A.P.; McHale, D.P.; et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 2001, 28, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Muhoberac, B.B.; Garringer, H.J.; Hurley, T.D.; Vidal, R. Unraveling of the e-helices and disruption of 4-fold pores are associated with iron mishandling in a mutant ferritin causing neurodegeneration. J. Biol. Chem. 2010, 285, 1950–1956. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Davidzon, G.; Kurlan, R.M.; Tawil, R.; Bonilla, E.; Mauro, S.D.; Powers, J.M. Hereditary ferritinopathy: A novel mutation, its cellular pathology, and pathogenetic insights. J. Neuropathol. Exp. Neurol. 2005, 64, 280–294. [Google Scholar] [PubMed]
- Barbeito, A.G.; Garringer, H.J.; Baraibar, M.A.; Gao, X.; Arredondo, M.; Nunez, M.T.; Smith, M.A.; Ghetti, B.; Vidal, R. Abnormal iron metabolism and oxidative stress in mice expressing a mutant form of the ferritin light polypeptide gene. J. Neurochem. 2009, 109, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Chinnery, P.F. Neuroferritinopathy. Semin. Pediatr. Neurol. 2006, 13, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J. Unraveling the hallervorden-spatz syndrome: Pantothenate kinase-associated neurodegeneration is the name. Curr. Opin. Pediatr. 2003, 15, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A novel pantothenate kinase gene (pank2) is defective in hallervorden-spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Zeidman, L.A.; Pandey, D.K. Declining use of the hallervorden-spatz disease eponym in the last two decades. J. Child Neurol. 2012, 27, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, P. Neurodegeneration with brain iron accumulation: Diagnosis and management. J. Mov. Disord. 2015, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Szumowski, J.; Bas, E.; Gaarder, K.; Schwarz, E.; Erdogmus, D.; Hayflick, S. Measurement of brain iron distribution in hallevorden-spatz syndrome. J. Magn. Reson. Imaging 2010, 31, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Pantothenate kinase-associated neurodegeneration (hallervorden-spatz syndrome). Eur. J. Paediatr. Neurol. 2002, 6, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Hogarth, P. As iron goes, so goes disease? Haematologica 2011, 96, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Zhang, L.; Rouault, T.A. Iron misregulation and neurodegenerative disease in mouse models that lack iron regulatory proteins. Neurobiol. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Mammalian iron-sulphur proteins: Novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Hardy, J. Genetics of Alzheimer’s disease. Neurotherapeutics 2014, 11, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Barbato, C.; Canu, N.; Zambrano, N.; Serafino, A.; Minopoli, G.; Ciotti, M.T.; Amadoro, G.; Russo, T.; Calissano, P. Interaction of Tau with Fe65 links tau to APP. Neurobiol. Dis. 2005, 18, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Hesse, L.; Beher, D.; Masters, C.L.; Multhaup, G. The beta A4 amyloid precursor protein binding to copper. FEBS Lett. 1994, 349, 109–116. [Google Scholar] [CrossRef]
- Spoerri, L.; Vella, L.J.; Pham, C.L.; Barnham, K.J.; Cappai, R. The amyloid precursor protein copper binding domain histidine residues 149 and 151 mediate App stability and metabolism. J. Biol. Chem. 2012, 287, 26840–26853. [Google Scholar] [CrossRef] [PubMed]
- Dahms, S.O.; Konnig, I.; Roeser, D.; Guhrs, K.H.; Mayer, M.C.; Kaden, D.; Multhaup, G.; Than, M.E. Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J. Mol. Biol. 2012, 416, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, K.H.; Dienemann, C.; Hoefgen, S.; Than, M.E.; Hagedoorn, P.L.; Hagen, W.R. The amyloid precursor protein (APP) does not have a ferroxidase site in its E2 domain. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. β-amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, T.; Camakaris, J.; Cappai, R.; Masters, C.L.; Beyreuther, K.; Multhaup, G. Copper inhibits β-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion. Biochem. J. 1999, 344, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Baumkotter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J. Neurosci. 2014, 34, 11159–11172. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Rodriguez, W.; Yatsimirsky, A.K.; Glabe, C.G. Binding of Zn(II), Cu(II), and Fe(II) ions to Alzheimer’s A beta peptide studied by fluorescence. Bioorg. Med. Chem. Lett. 1999, 9, 2243–2248. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H., Jr.; Paradis, M.D.; Tanzi, R.E. Modulation of a beta adhesiveness and secretase site cleavage by zinc. J. Biol. Chem. 1994, 269, 12152–12158. [Google Scholar] [PubMed]
- Streltsov, V.A.; Titmuss, S.J.; Epa, V.C.; Barnham, K.J.; Masters, C.L.; Varghese, J.N. The structure of the amyloid-beta peptide high-affinity copper II binding site in alzheimer disease. Biophys. J. 2008, 95, 3447–3456. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [PubMed]
- Ayton, S.; Lei, P.; Bush, A.I. Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 2015, 12, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Dedman, D.J.; Treffry, A.; Candy, J.M.; Taylor, G.A.; Morris, C.M.; Bloxham, C.A.; Perry, R.H.; Edwardson, J.A.; Harrison, P.M. Iron and aluminium in relation to brain ferritin in normal individuals and Alzheimer’s-disease and chronic renal-dialysis patients. Biochem. J. 1992, 287, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Sultzer, D.; Mintz, J.; Holt, L.E.; Marx, P.; Phelan, C.K.; Marder, S.R. In vivo evaluation of brain iron in Alzheimer’s disease and normal subjects using mri. Biol. Psychiatry 1994, 35, 480–487. [Google Scholar] [CrossRef]
- Castellani, R.J.; Honda, K.; Zhu, X.; Cash, A.D.; Nunomura, A.; Perry, G.; Smith, M.A. Contribution of redox-active iron and copper to oxidative damage in alzheimer disease. Ageing Res. Rev. 2004, 3, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Lee, H.G.; Blair, J.A.; Zhu, X.; Perry, G.; Smith, M.A. Role of metal dyshomeostasis in Alzheimer’s disease. Metallomics 2011, 3, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Faux, N.G.; Bush, A.I. Alzheimer’s Disease Neuroimaging Initiative. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rottkamp, C.A.; Raina, A.K.; Zhu, X.; Gaier, E.; Bush, A.I.; Atwood, C.S.; Chevion, M.; Perry, G.; Smith, M.A. Redox-active iron mediates amyloid-beta toxicity. Free Radic. Biol. Med. 2001, 30, 447–450. [Google Scholar] [CrossRef]
- Stephenson, D.T.; Clemens, J.A. In vivo effects of beta-amyloid implants in rodents: Lack of potentiation of damage associated with transient global forebrain ischemia. Brain Res. 1992, 586, 235–246. [Google Scholar] [CrossRef]
- Friedland-Leuner, K.; Stockburger, C.; Denzer, I.; Eckert, G.P.; Muller, W.E. Mitochondrial dysfunction: Cause and consequence of Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2014, 127, 183–210. [Google Scholar] [PubMed]
- Atamna, H. Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 2004, 3, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [PubMed]
- Perry, G.; Taddeo, M.A.; Petersen, R.B.; Castellani, R.J.; Harris, P.L.; Siedlak, S.L.; Cash, A.D.; Liu, Q.; Nunomura, A.; Atwood, C.S.; et al. Adventiously-bound redox active iron and copper are at the center of oxidative damage in alzheimer disease. Biometals 2003, 16, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Ruberu, N.N.; Sawabe, M.; Arai, T.; Kazama, H.; Hosoi, T.; Yamanouchi, H.; Murayama, S. Lewy body-related α-synucleinopathy in aging. J. Neuropathol. Exp. Neurol. 2004, 63, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. α-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987, 2, 1219–1220. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.J.; Bonifati, V. The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov. Disord. 2013, 28, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Leenders, K.L.; Oertel, W.H. Parkinson’s disease: Clinical signs and symptoms, neural mechanisms, positron emission tomography, and therapeutic interventions. Neural Plast. 2001, 8, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A. Presynaptic mechanisms of L-DOPA-induced dyskinesia: The findings, the debate, and the therapeutic implications. Front Neurol 2014. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Brandel, J.P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Revesz, T.; Jacques, T.S.; Papacostas, S.; Chataway, J. Adult-onset neurodegeneration with brain iron accumulation and cortical α-synuclein and tau pathology: A distinct clinicopathological entity. Arch. Neurol. 2007, 64, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241. [Google Scholar] [CrossRef]
- Gorrell, J.M.; DiMonte, D.; Graham, D. The role of the environment in Parkinson’s disease. Environ. Health Perspect. 1996, 104, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Cunningham, S.; Kotamraju, S.; Joseph, J.; Hillard, C.J.; Kalyanaraman, B. α-synuclein up-regulation and aggregation during mpp+-induced apoptosis in neuroblastoma cells: Intermediacy of transferrin receptor iron and hydrogen peroxide. J. Biol. Chem. 2004, 279, 15240–15247. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Maor, G.; Youdim, M.B. Iron and α-synuclein in the substantia nigra of mptp-treated mice: Effect of neuroprotective drugs R-apomorphine and green tea polyphenol (−)-epigallocatechin-3-gallate. J. Mol. Neurosci. 2004, 24, 401–416. [Google Scholar] [CrossRef]
- Sengstock, G.J.; Olanow, C.W.; Menzies, R.A.; Dunn, A.J.; Arendash, G.W. Infusion of iron into the rat substantia nigra: Nigral pathology and dose-dependent loss of striatal dopaminergic markers. J. Neurosci. Res. 1993, 35, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L.; Halliday, G.M. New face of neuromelanin. J. Neural Transm. Suppl. 2006, 119–123. [Google Scholar]
- Li, J.; Yang, J.; Zhao, P.; Li, S.; Zhang, R.; Zhang, X.; Liu, D.; Zhang, B. Neuromelanin enhances the toxicity of alpha-synuclein in SK-N-SH cells. J. Neural Transm. 2012, 119, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Riederer, P.; Double, K.L. Neuromelanin-bound ferric iron as an experimental model of dopaminergic neurodegeneration in Parkinson’s disease. Parkinsonism Relat. Disord. 2008, 14, S185–S188. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Bergamasco, B.; Lopiano, L. Is neuromelanin changed in Parkinson’s disease? Investigations by magnetic spectroscopies. J. Neural Transm. 2006, 113, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.S.; Leibold, E.A. Regulation of the iron regulatory proteins by reactive nitrogen and oxygen species. Gene Expr. 1999, 7, 367–376. [Google Scholar] [PubMed]
- Connor, J.R.; Snyder, B.S.; Arosio, P.; Loeffler, D.A.; LeWitt, P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer’s diseased brains. J. Neurochem. 1995, 65, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Jinsmaa, Y.; Sullivan, P.; Gross, D.; Cooney, A.; Sharabi, Y.; Goldstein, D.S. Divalent metal ions enhance dopal-induced oligomerization of alpha-synuclein. Neurosci. Lett. 2014, 569, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Lin, T.S.; Minteer, S.; Burke, W.J. 3,4-dihydroxyphenylacetaldehyde and hydrogen peroxide generate a hydroxyl radical: Possible role in Parkinson’s disease pathogenesis. Brain Res. Mol. Brain Res. 2001, 93, 1–7. [Google Scholar] [CrossRef]
- Davies, P.; Wang, X.; Sarell, C.J.; Drewett, A.; Marken, F.; Viles, J.H.; Brown, D.R. The synucleins are a family of redox-active copper binding proteins. Biochemistry 2010, 50, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Structural characterization of copper(II) binding to α-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef] [PubMed]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Interaction of α-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Moualla, D.; Brown, D.R. α-synuclein is a cellular ferrireductase. PLoS ONE 2011. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular nk between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.A.; Wang, X.; Brown, D.R. Unique copper-induced oligomers mediate α-synuclein toxicity. FASEB J. 2009, 23, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Moualla, D.; Wright, J.A.; Brown, D.R. Copper binding regulates intracellular α-synuclein localisation, aggregation and toxicity. J. Neurochem. 2010, 113, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Hogen, T.; Hillmer, A.S.; Bader, B.; Schmidt, F.; Kamp, F.; Kretzschmar, H.A.; Botzel, K.; Giese, A. Generation of ferric iron links oxidative stress to α-synuclein oligomer formation. J. Parkinson’s Dis. 2011, 1, 205–216. [Google Scholar] [CrossRef]
- Ortega, R.; Carmona, A.; Roudeau, S.; Perrin, L.; Ducic, T.; Carboni, E.; Bohic, S.; Cloetens, P.; Lingor, P. α-synuclein over-expression induces increased iron accumulation and redistribution in iron-exposed neurons. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Ironside, J.W.; Sutherland, K.; Bell, J.E.; McCardle, L.; Barrie, C.; Estebeiro, K.; Zeidler, M.; Will, R.G. A new variant of Creutzfeldt-Jakob disease: Neuropathological and clinical features. Cold Spring Harb. Symp. Quant. Biol. 1996, 61, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Hope, J.; Reekie, L.J.; Hunter, N.; Multhaup, G.; Beyreuther, K.; White, H.; Scott, A.C.; Stack, M.J.; Dawson, M.; Wells, G.A. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature 1988, 336, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Knight, R. Creutzfeldt-Jakob disease: A rare cause of dementia in elderly persons. Clin. Infect. Dis. 2006, 43, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Groth, D.; Prusiner, S.B.; Kobata, A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 1989, 28, 8380–8388. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Wong, B.S.; Hafiz, F.; Clive, C.; Haswell, S.J.; Jones, I.M. Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 1999, 344, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Prion and prejudice: Normal protein and the synapse. Trends Neurosci. 2001, 24, 85–90. [Google Scholar] [CrossRef]
- Davies, P.; Brown, D.R. The chemistry of copper binding to PrP: Is there sufficient evidence to elucidate a role for copper in protein function? Biochem. J. 2008, 410, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Metalloproteins and neuronal death. Metallomics 2010, 2, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Prions and manganese: A maddening beast. Metallomics 2011, 3, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Hafiz, F.; Glasssmith, L.L.; Wong, B.S.; Jones, I.M.; Clive, C.; Haswell, S.J. Consequences of manganese replacement of copper for prion protein function and proteinase resistance. EMBO J. 2000, 19, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Brazier, M.W.; Davies, P.; Player, E.; Marken, F.; Viles, J.H.; Brown, D.R. Manganese binding to the prion protein. J. Biol. Chem. 2008, 283, 12831–12839. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Brown, D.R. Manganese enhances prion protein survival in model soils and increases prion infectivity to cells. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, S.; Sassoon, J.; Knight, R.; Hopkins, J.; Brown, D.R. Elevated manganese levels in blood and central nervous system occur before onset of clinical signs in scrapie and bovine spongiform encephalopathy. J. Anim. Sci. 2007, 85, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, S.; Sassoon, J.; Knight, R.; Brown, D.R. Elevated manganese levels in blood and CNS in human prion disease. Mol. Cell. Neurosci. 2008, 37, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Thackray, A.M.; Knight, R.; Haswell, S.J.; Bujdoso, R.; Brown, D.R. Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem. J. 2002, 362, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.J.; Gilbert, P.U.; Abrecht, M.; Baldwin, K.L.; Russell, R.E.; Pedersen, J.A.; Aiken, J.M.; McKenzie, D. Low copper and high manganese levels in prion protein plaques. Viruses 2013, 5, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Haldar, S.; Tripathi, A.K.; McElwee, M.K.; Horback, K.; Beserra, A. Iron in neurodegenerative disorders of protein misfolding: A case of prion disorders and Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Jun, Y.C.; Jin, J.K.; Kim, J.I.; Kim, N.H.; Leibold, E.A.; Connor, J.R.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Alteration of iron regulatory proteins (IRP1 and IRP2) and ferritin in the brains of scrapie-infected mice. Neurosci. Lett. 2007, 422, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Mohan, M.L.; Isaac, A.O.; Luo, X.; Petrak, J.; Vyoral, D.; Singh, N. Prion protein modulates cellular iron uptake: A novel function with implications for prion disease pathogenesis. PLoS ONE 2009. [Google Scholar] [CrossRef]
- Singh, A.; Isaac, A.O.; Luo, X.; Mohan, M.L.; Cohen, M.L.; Chen, F.; Kong, Q.; Bartz, J.; Singh, N. Abnormal brain iron homeostasis in human and animal prion disorders. PLoS Pathog. 2009. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kong, Q.; Luo, X.; Petersen, R.B.; Meyerson, H.; Singh, N. Prion protein (PrP) knock-out mice show altered iron metabolism: A functional role for PrP in iron uptake and transport. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]
- Kralovicova, S.; Fontaine, S.N.; Alderton, A.; Alderman, J.; Ragnarsdottir, K.V.; Collins, S.J.; Brown, D.R. The effects of prion protein expression on metal metabolism. Mol. Cell. Neurosci. 2009, 41, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552. [Google Scholar] [PubMed]
- Brown, D.R. Role of microglia in age-related changes to the nervous system. Sci. World J. 2009, 9, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Njie, E.G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2012, 33, 195.e1–195.e12. [Google Scholar] [CrossRef] [PubMed]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Eder, C. Microglial K+ channel expression in young adult and aged mice. Glia 2015, 63, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Lopes, K.O.; Sparks, D.L.; Streit, W.J. Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia 2008, 56, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; Casale, M.; Alcon, B.; Pham, N.; Narayan, N.; Lynch, G. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 2007, 55, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.S.; Andersen, M.V.; Christoffersen, P.R.; Jensen, M.D.; Lichota, J.; Moos, T. Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with β-amyloid deposition in mice with alzheimer-like pathology. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Ye, Z.; Kholodenko, D.; Seubert, P.; Selkoe, D.J. Degradation of amyloid β-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J. Biol. Chem. 1997, 272, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, G.; Wilcock, D.; Henderson, D.; Gordon, M.; Morgan, D. Intrahippocampal LPS injections reduce Aβ load in APP + PS1 transgenic mice. Neurobiol. Aging 2001, 22, 1007–1012. [Google Scholar] [CrossRef]
- Li, M.; Pisalyaput, K.; Galvan, M.; Tenner, A.J. Macrophage colony stimulatory factor and interferon-γ trigger distinct mechanisms for augmentation of β-amyloid-induced microglia-mediated neurotoxicity. J. Neurochem. 2004, 91, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 2001, 22, 915–922. [Google Scholar] [CrossRef]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Lewis, C.A.; Zhang, R.; Rossi, F.M. The role of microglia in human disease: Therapeutic tool or target? Acta Neuropathol. 2014, 128, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelova, D.M.; Brown, D.R. Iron, Aging, and Neurodegeneration. Metals 2015, 5, 2070-2092. https://doi.org/10.3390/met5042070
Angelova DM, Brown DR. Iron, Aging, and Neurodegeneration. Metals. 2015; 5(4):2070-2092. https://doi.org/10.3390/met5042070
Chicago/Turabian StyleAngelova, Dafina M., and David R. Brown. 2015. "Iron, Aging, and Neurodegeneration" Metals 5, no. 4: 2070-2092. https://doi.org/10.3390/met5042070
APA StyleAngelova, D. M., & Brown, D. R. (2015). Iron, Aging, and Neurodegeneration. Metals, 5(4), 2070-2092. https://doi.org/10.3390/met5042070