CD20 mAb-Mediated Complement Dependent Cytotoxicity of Tumor Cells is Enhanced by Blocking the Action of Factor I

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
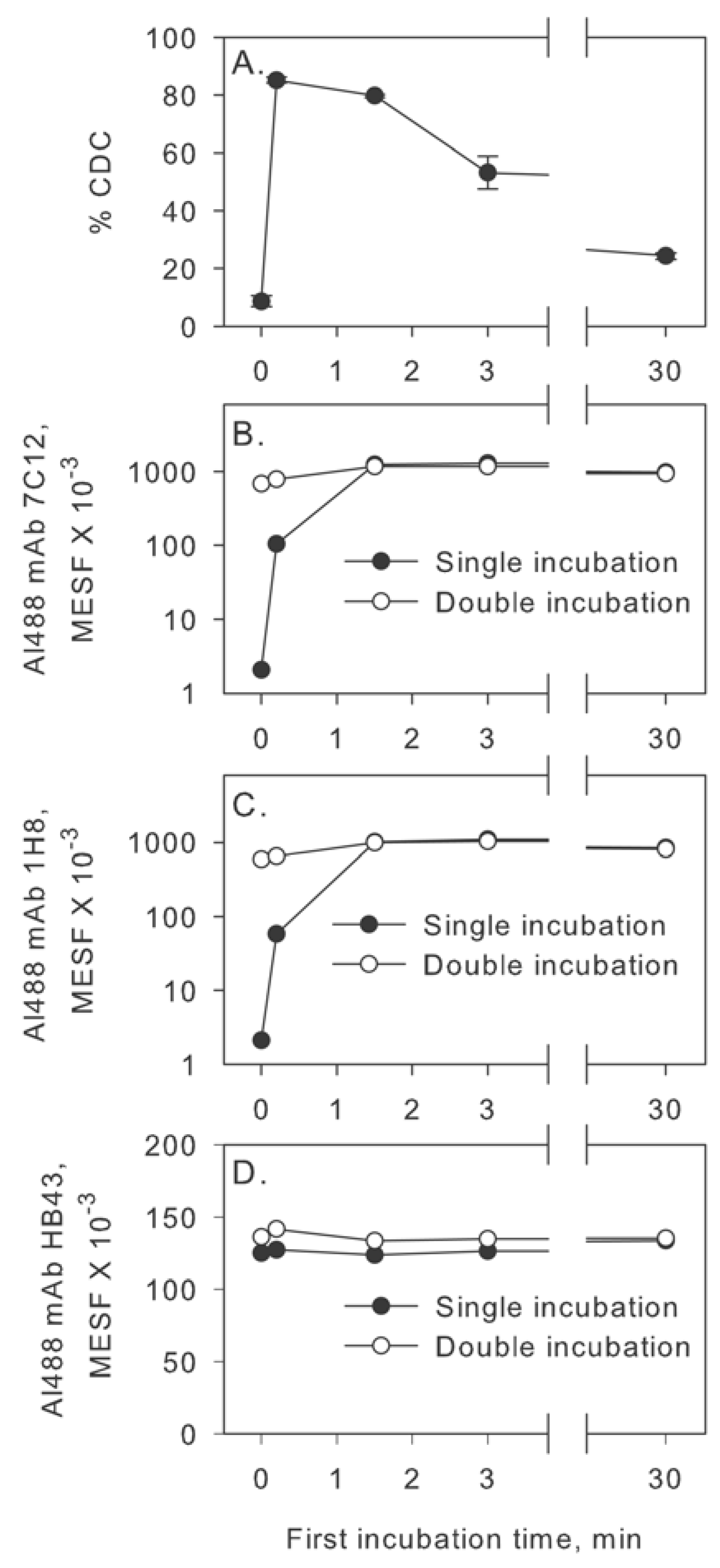
2.1. Complement Activation in Two Steps
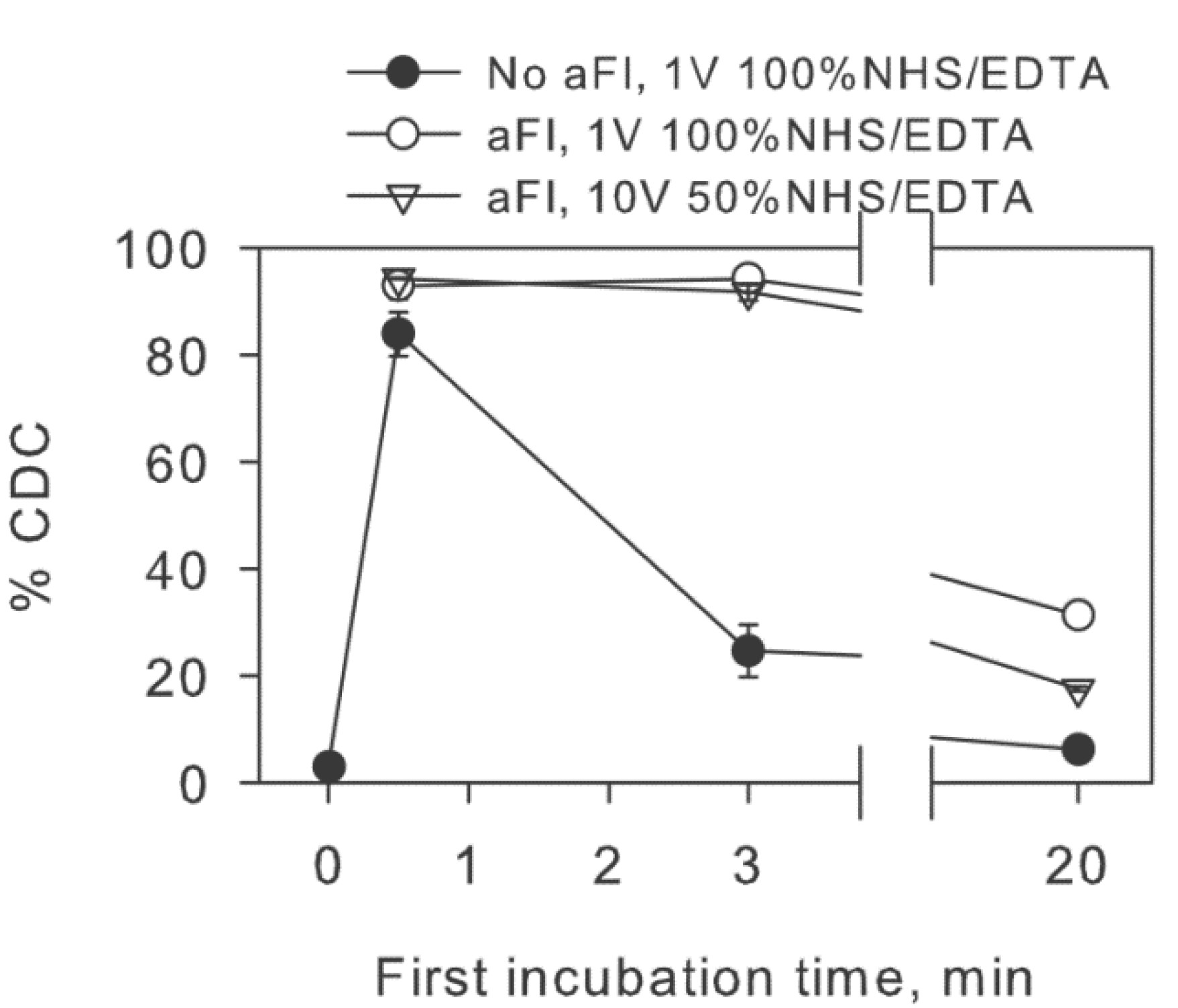
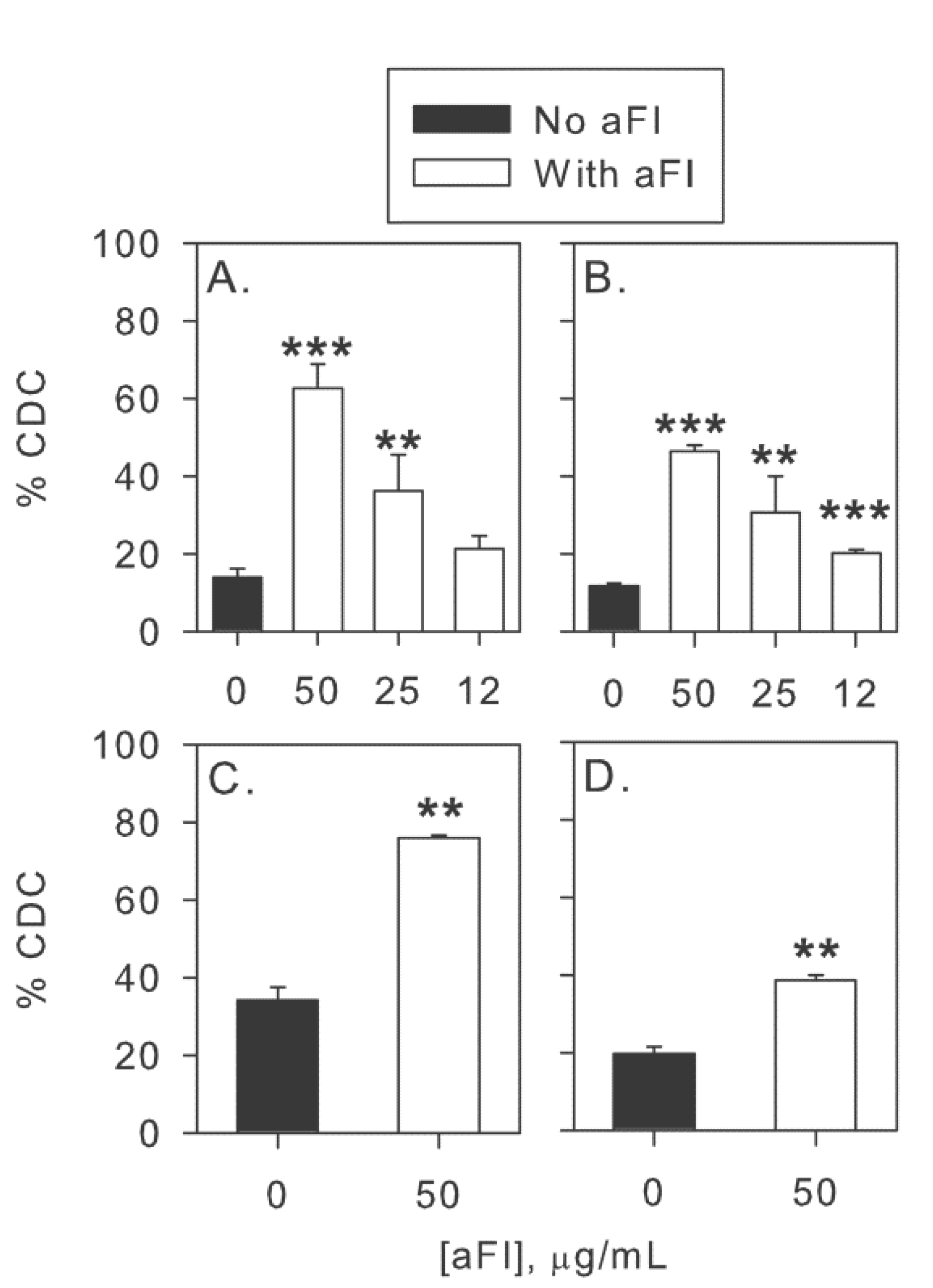
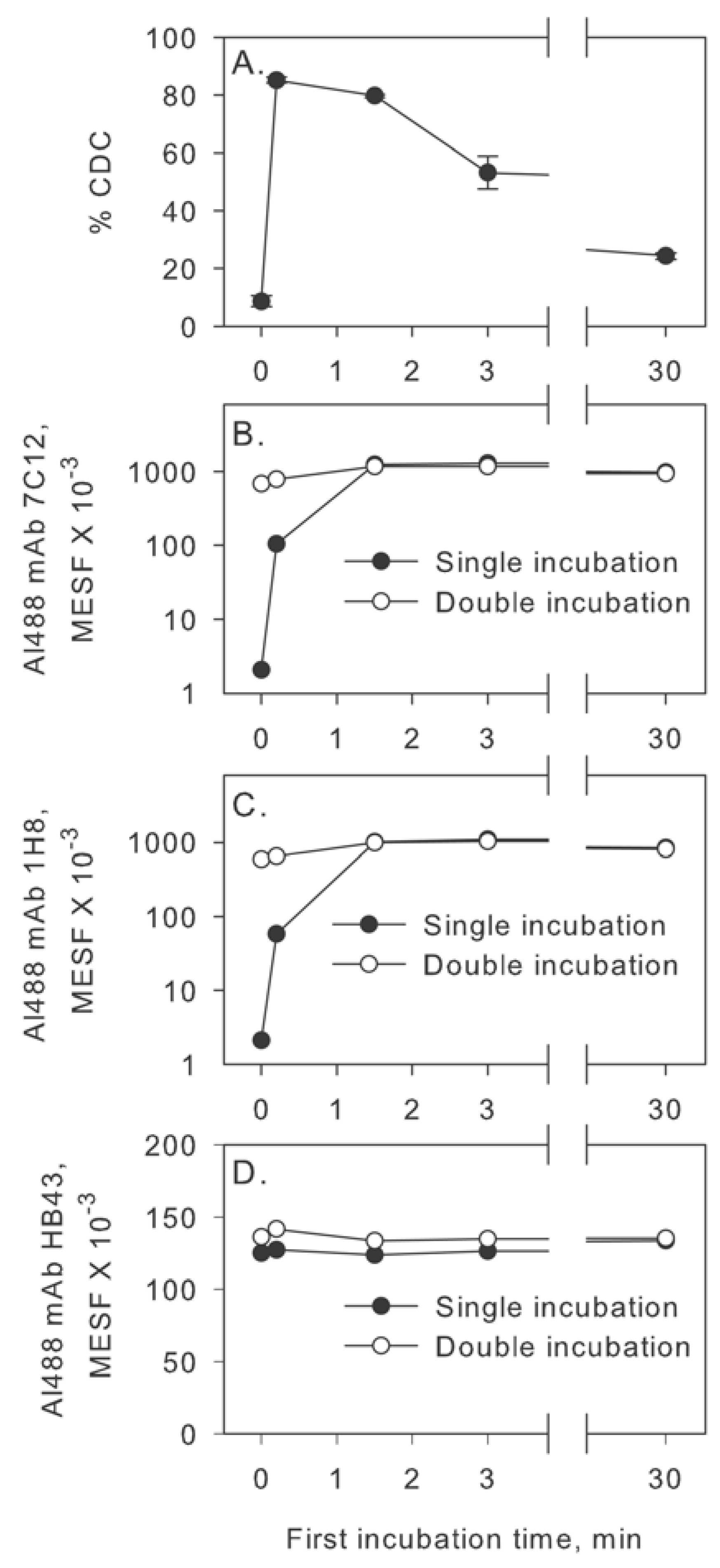
2.2. Inhibition of Factor I Increases CDC in the Two-Step Assay
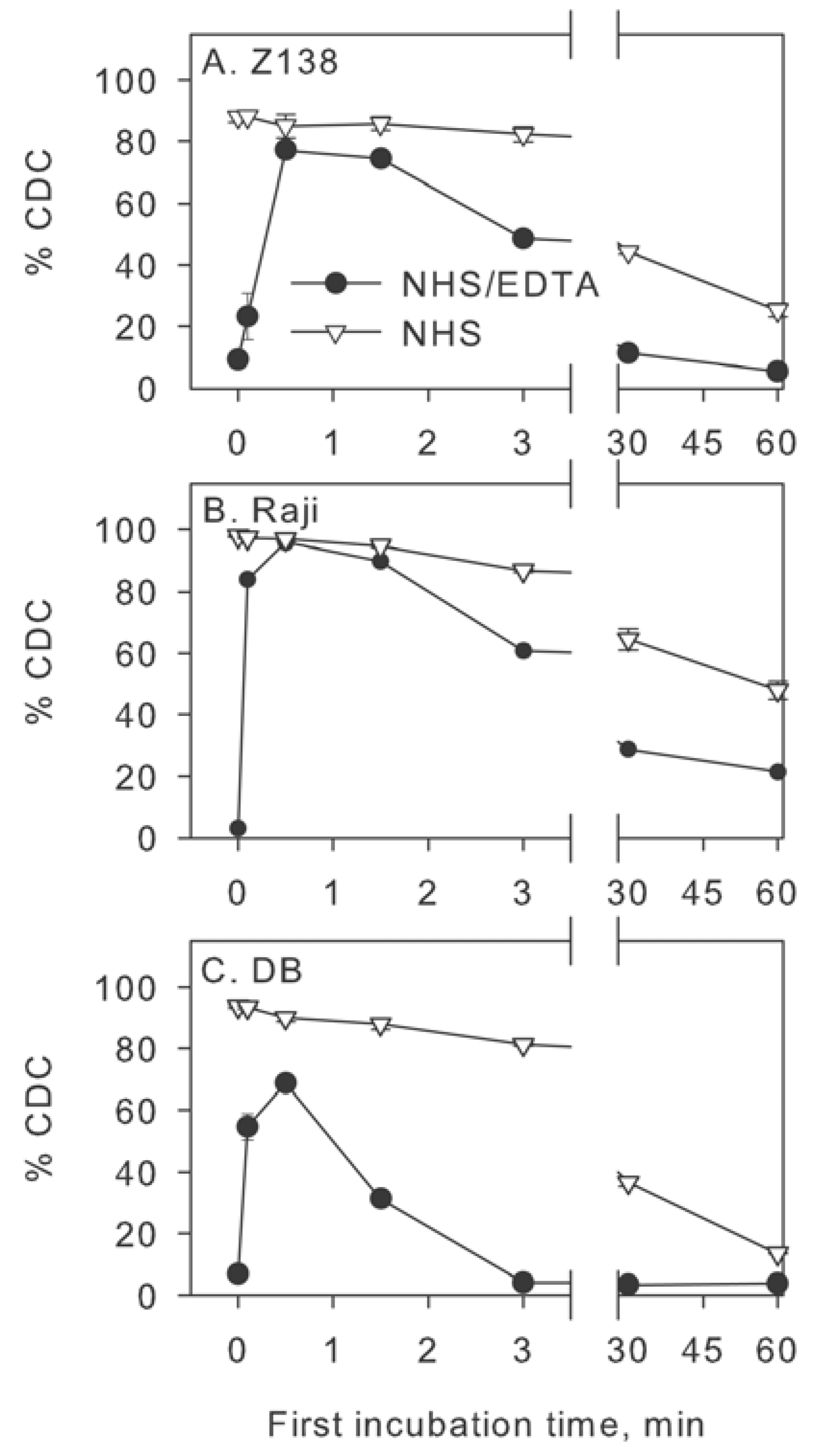
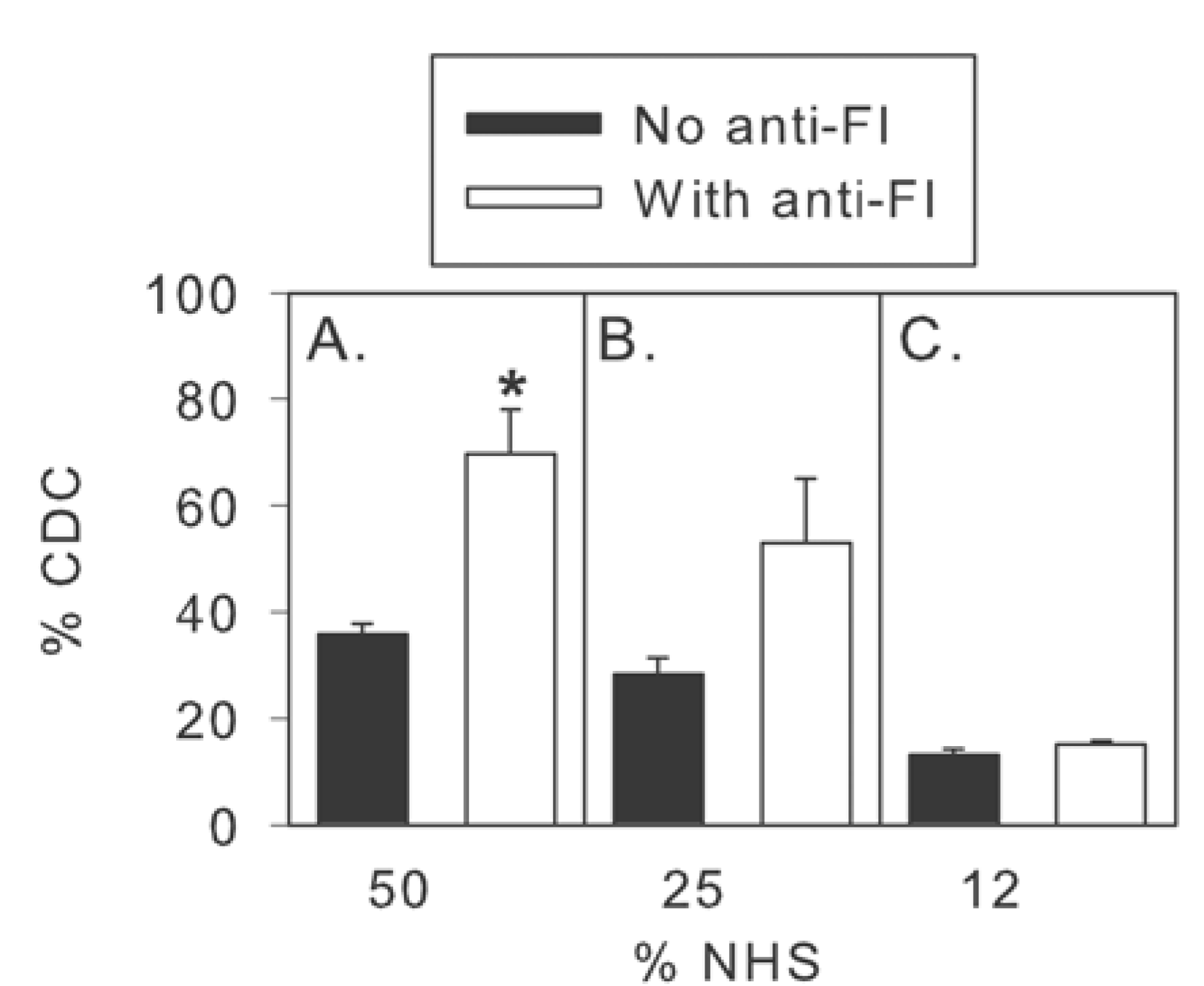
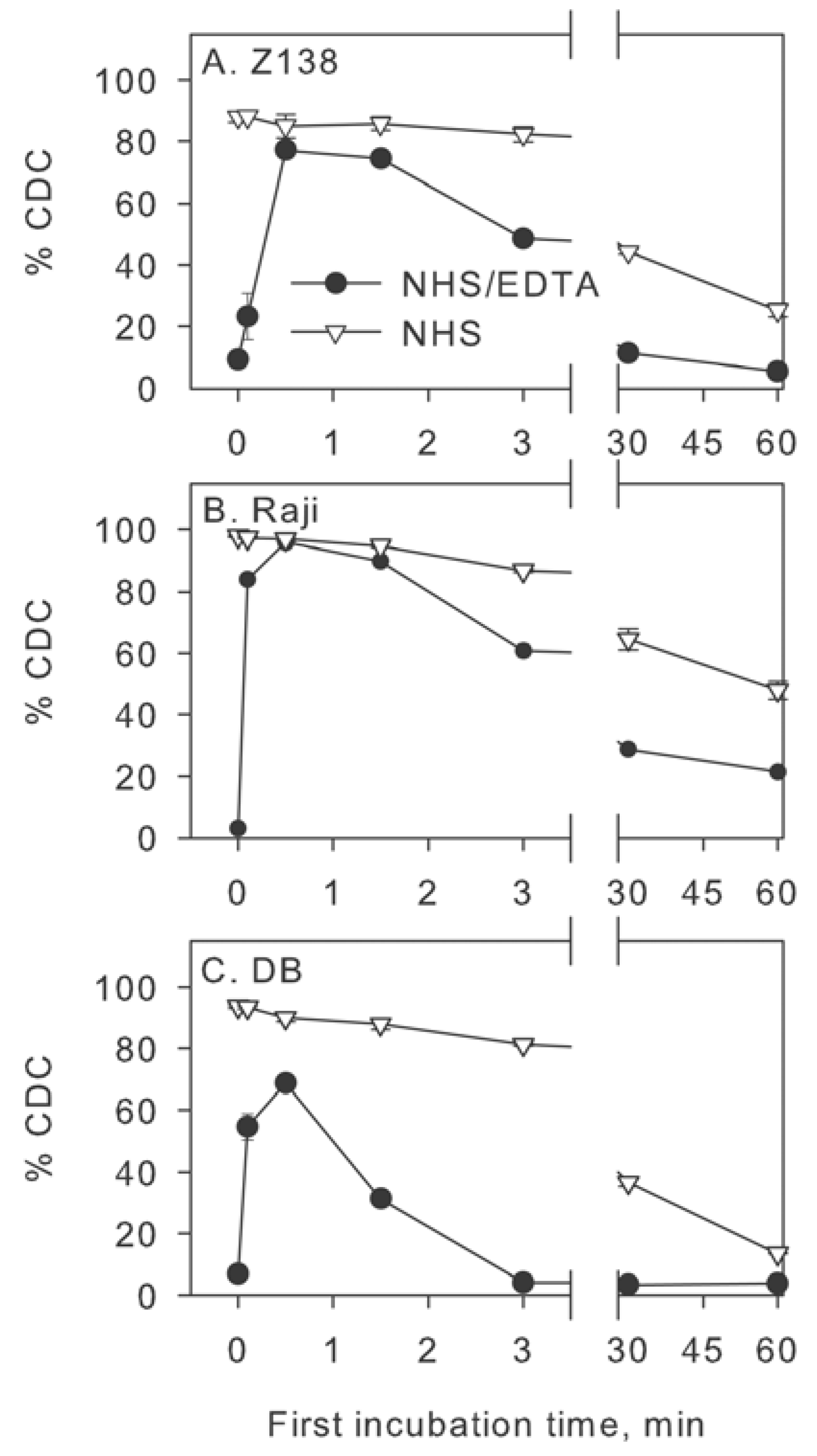
2.3. Inhibition of Factor I Increases CDC in Intact NHS



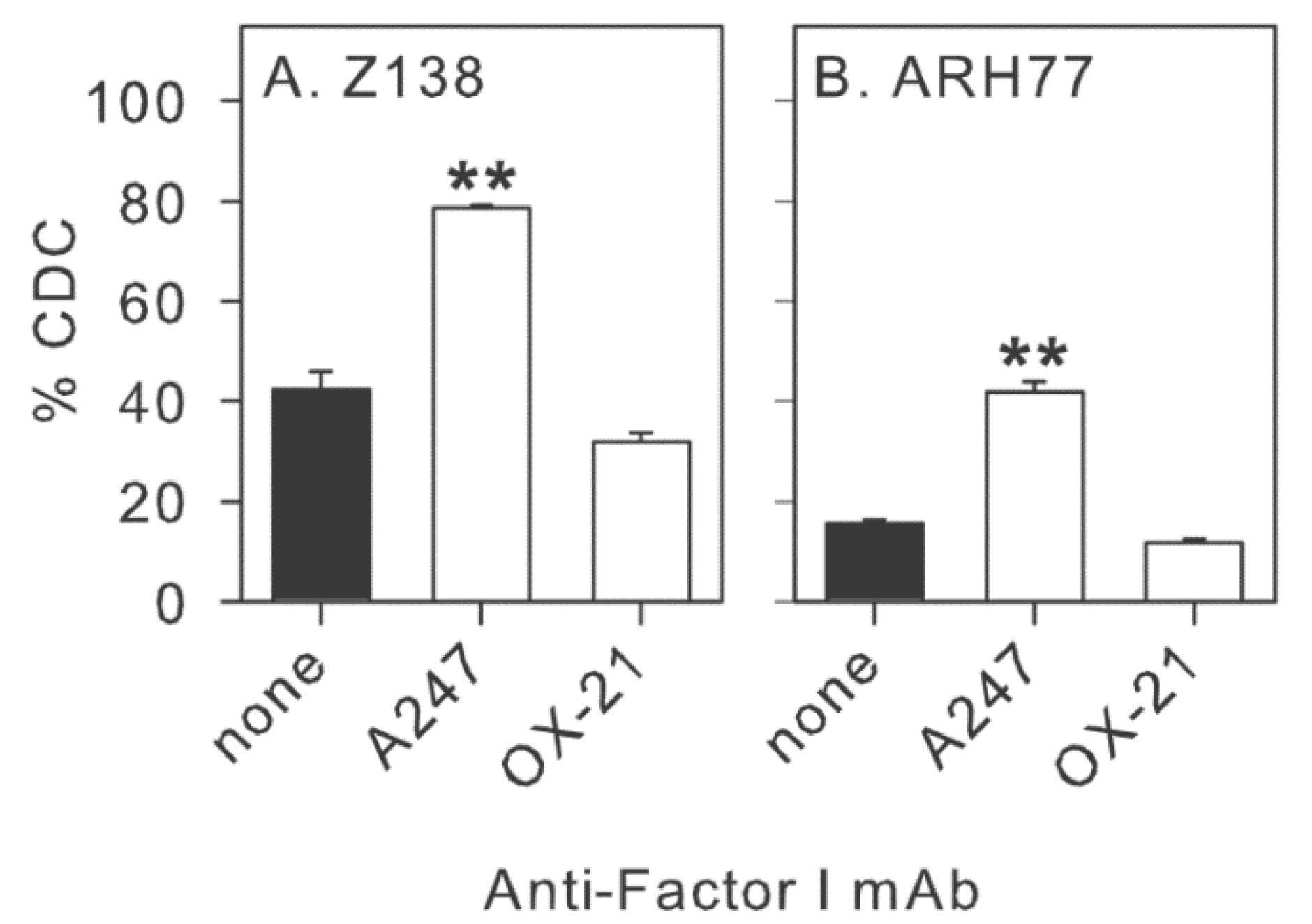
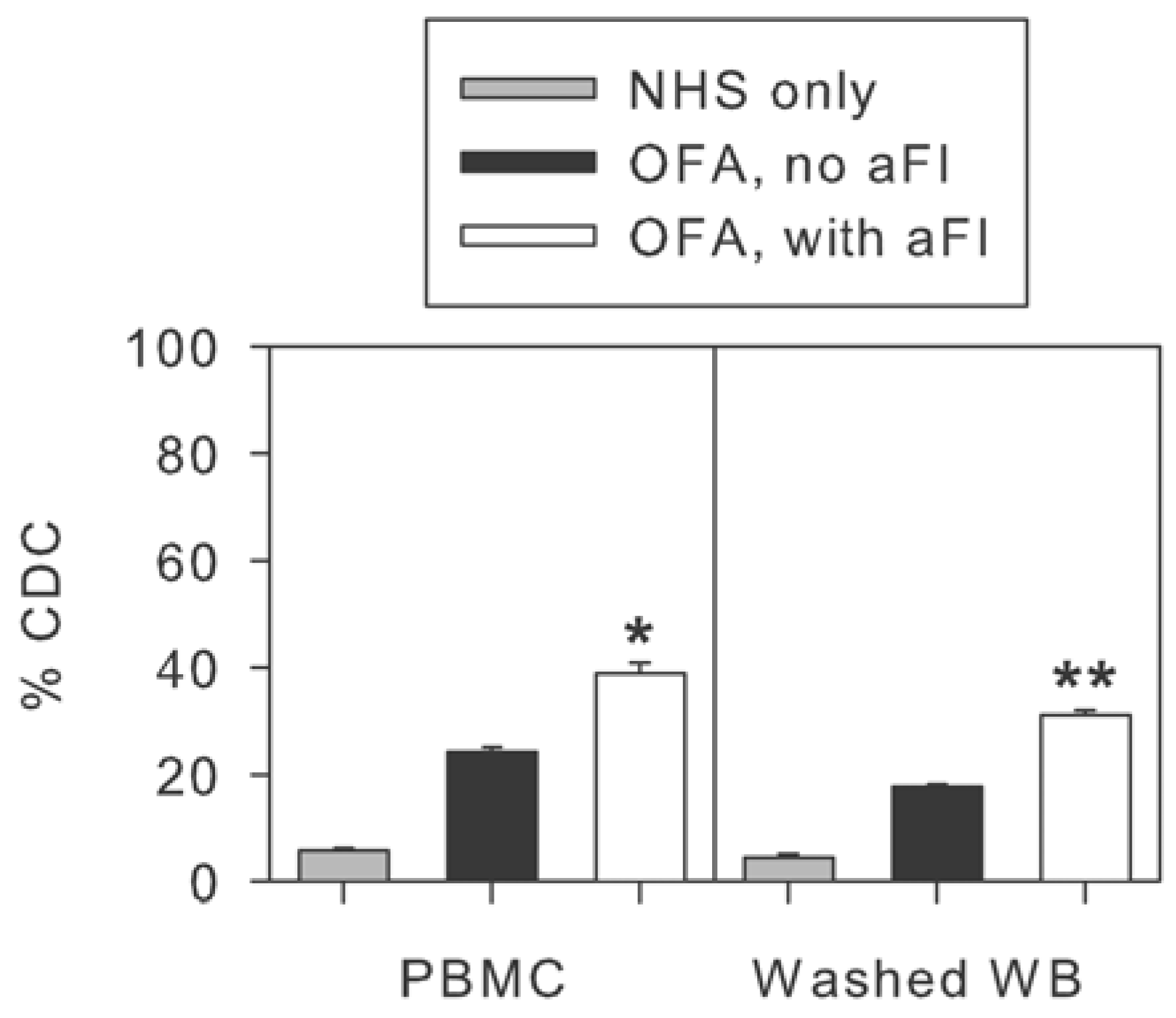
2.4. Inhibition of Factor I Increases CDC of CLL Cells in Intact NHS
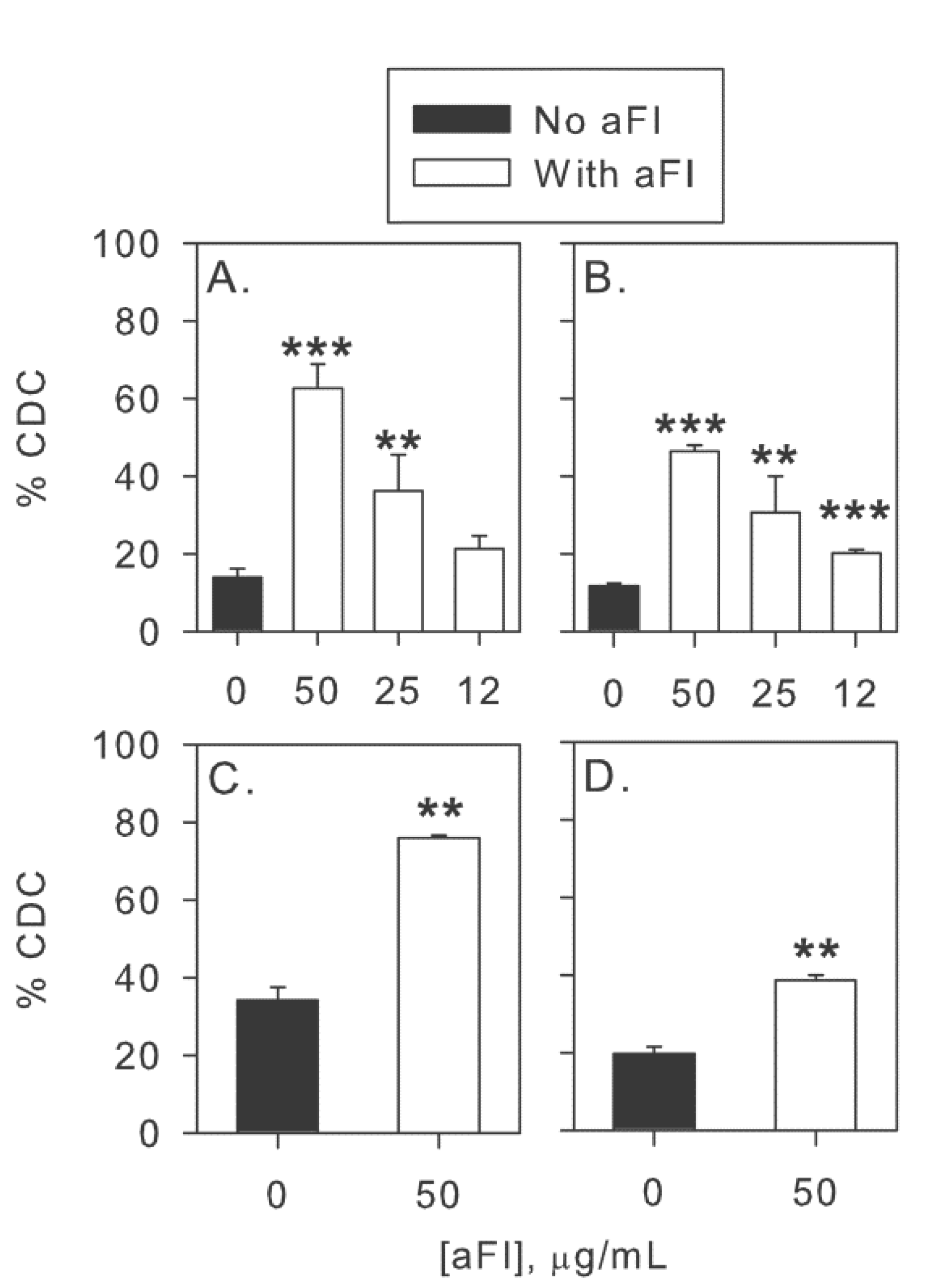
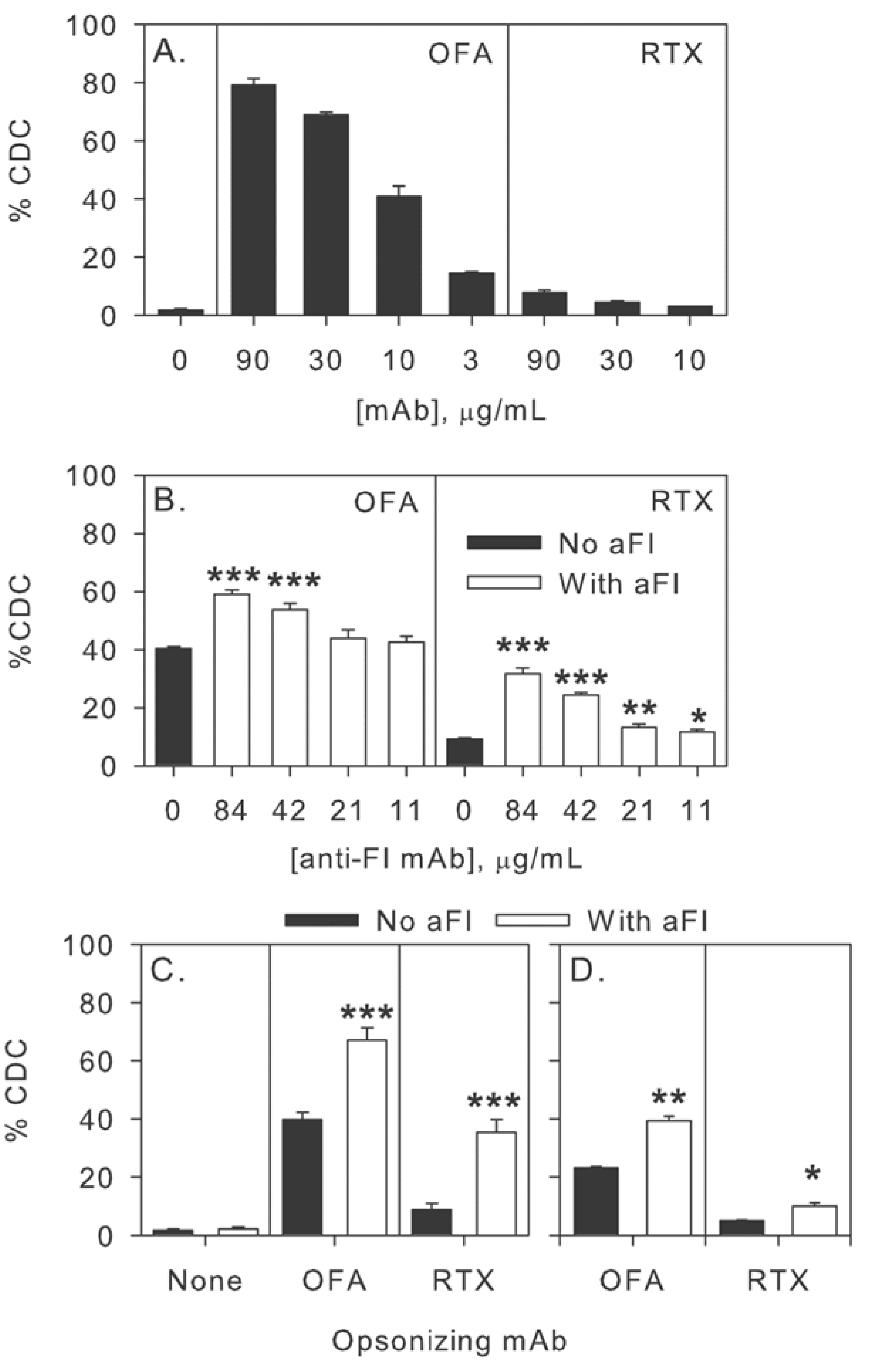
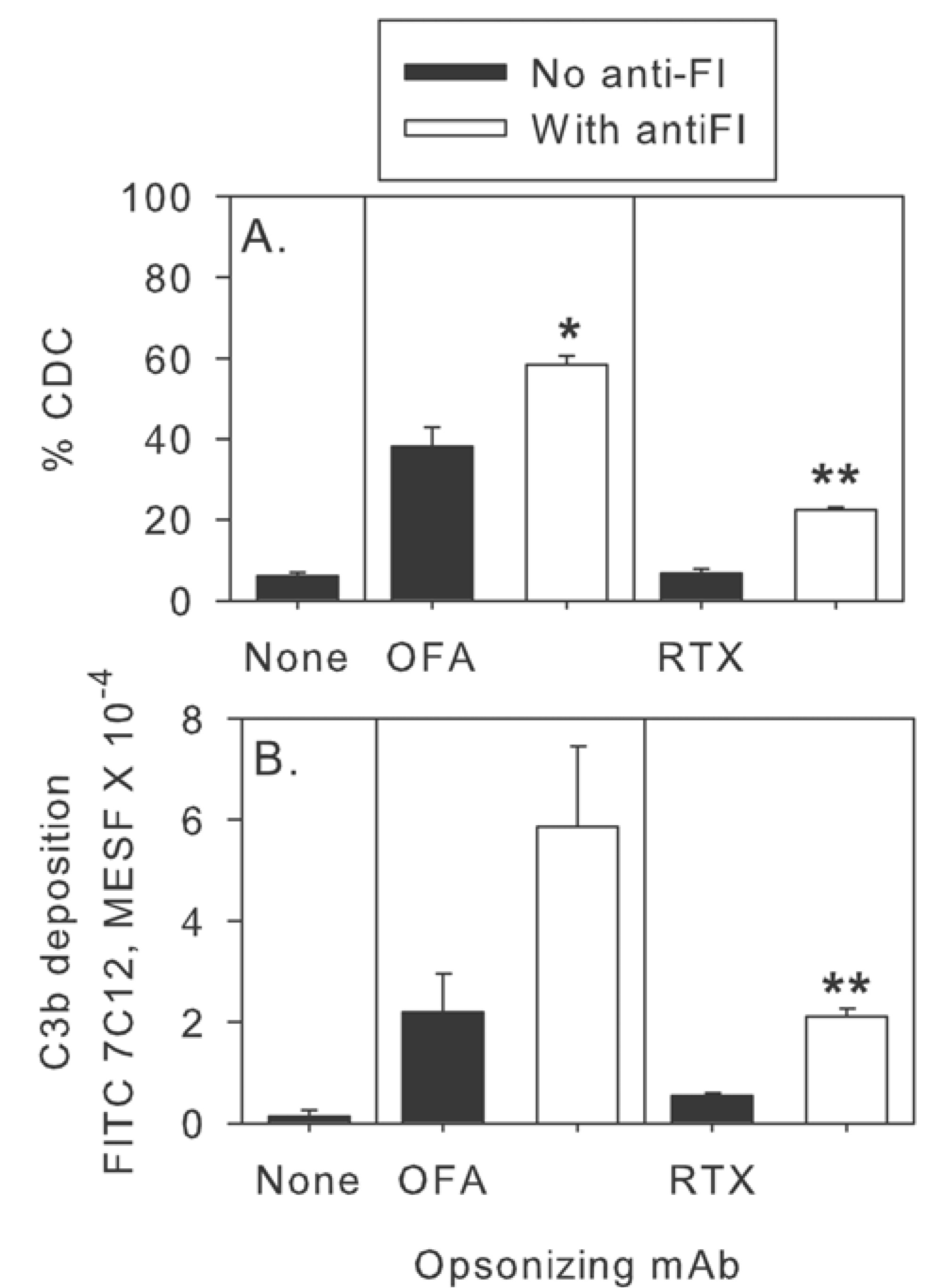

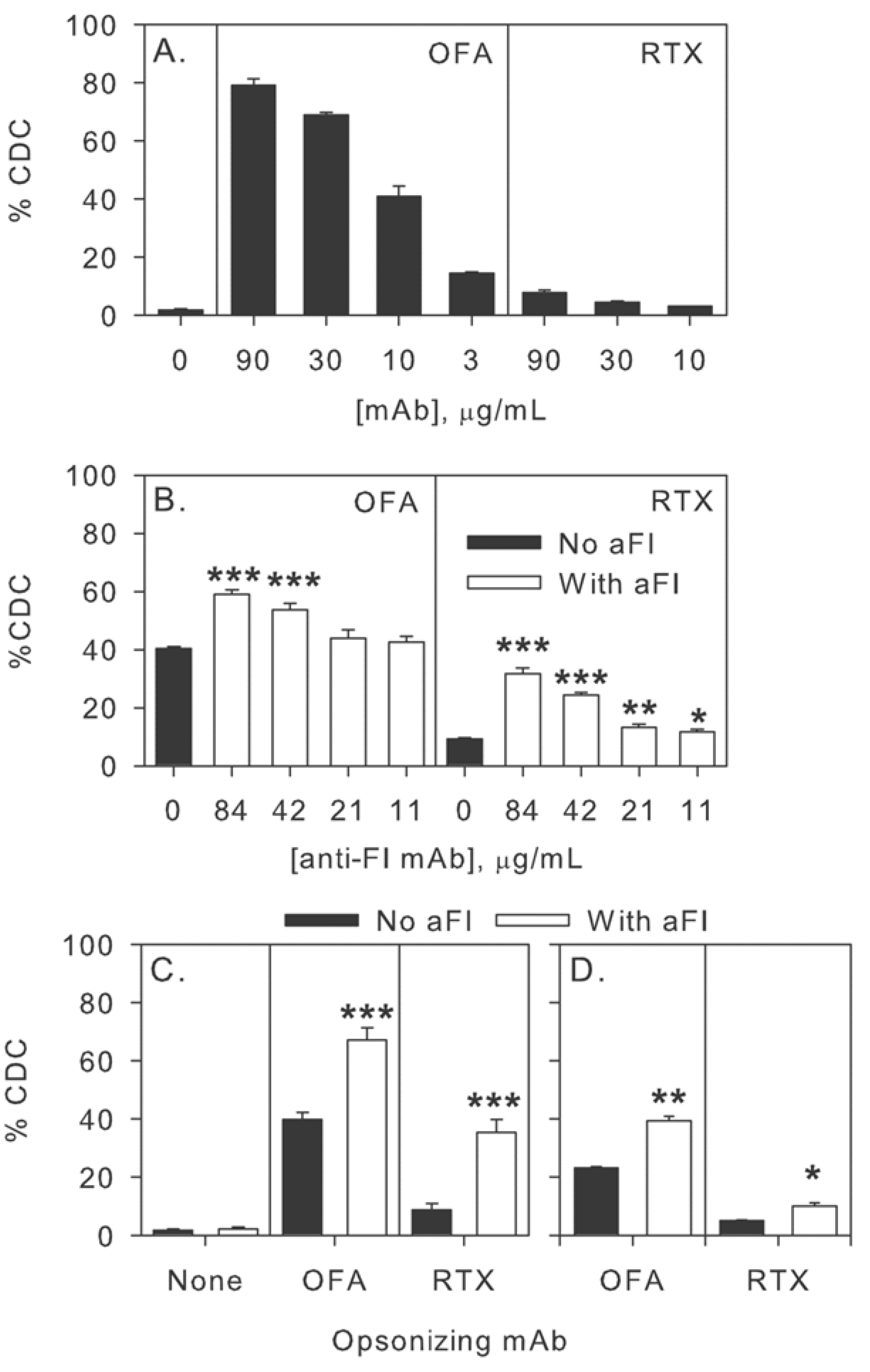
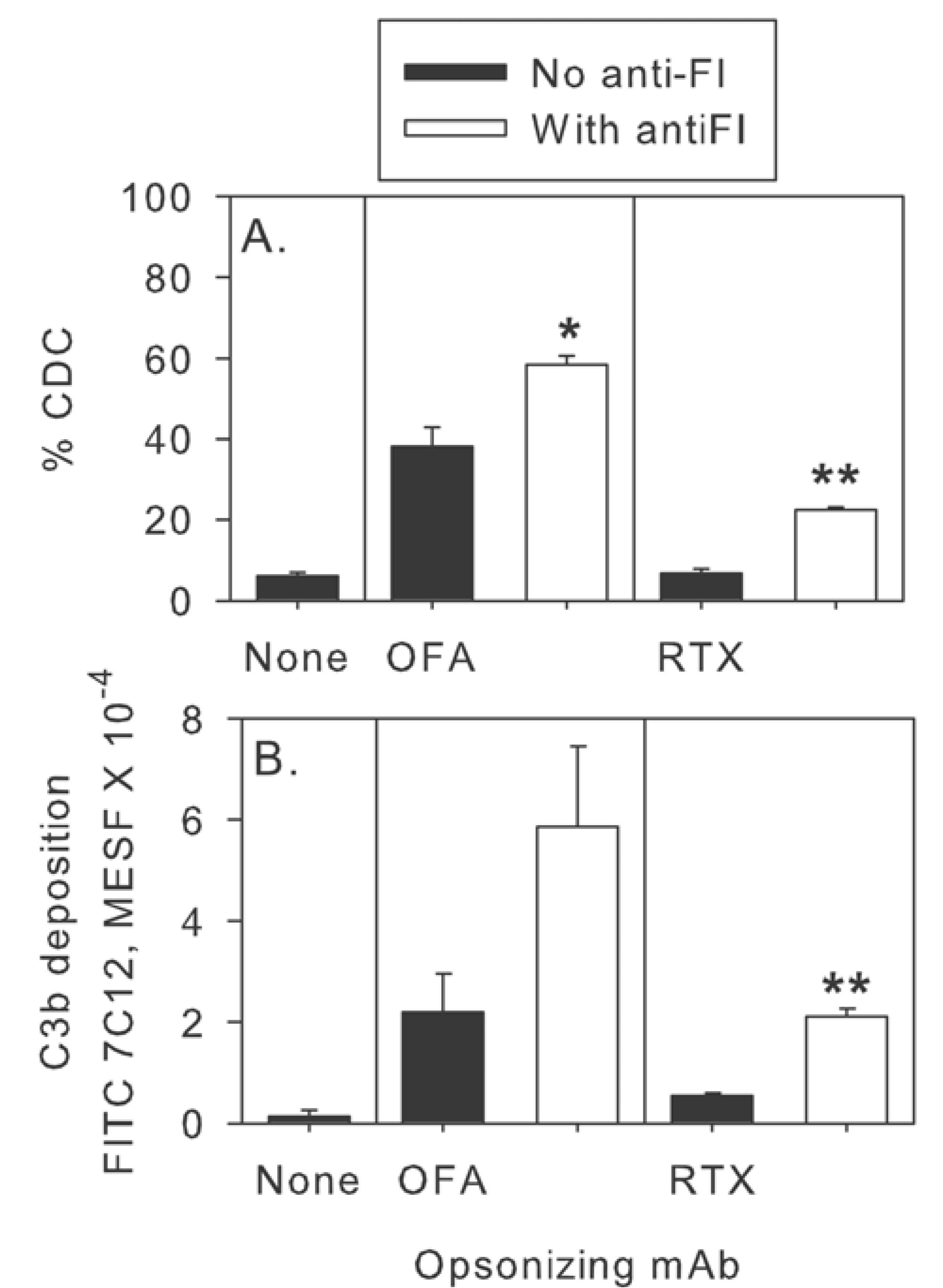
2.5. Modulation of Effector Functions to Increase mAb-mediated Killing of Tumor Cells
2.6. Future Directions
3. Experimental Section
3.1. Cell Lines and Primary Cells
3.2. Antibodies and Reagents
3.3. mAb Opsonization and Complement Activation
3.4. Statistical Analyses
4. Conclusions
Conflicts of Interest
References
- Glennie, M.J.; French, R.; Cragg, M.S.; Taylor, R.P. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol. Immunol. 2007, 44, 3823–3837. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of action. Sem. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef]
- Lim, S.H.; Beers, S.A.; French, R.R.; Johnson, P.W.M.; Glennie, M.J.; Cragg, M.S. Anti-CD20 monoclonal antibodies--historical and future perspectives. Haematologica 2010, 95, 135–143. [Google Scholar] [CrossRef]
- Boross, P.; Leusen, J.H.W. Mechanisms of action of CD20 antibodies. Am. J. Cancer Res. 2012, 2, 676–690. [Google Scholar]
- Golay, J.; Introna, M. Mechanism of action of therapeutic monoclonal antibodies: Promises and pitfalls of in vitro and invivo assays. Arch. Biochem. Biophys. 2012, 526, 146–153. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nature 2012, 12, 278–287. [Google Scholar]
- Goswami, S.; Wang, W.; Arakawa, T.; Ohtake, S. Developments and challenges for mAb-based therapeutics. Antibodies 2013, 2, 452–500. [Google Scholar] [CrossRef]
- Bologna, L.; Gotti, E.; da Roit, F.; Intermesoli, T.; Rambaldi, A.; Introna, M.; Golay, J. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J. Immunol. 2013, 190, 231–239. [Google Scholar]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc g receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef]
- Taylor, R.P.; Lindorfer, M.A. The role of complement in mAb-based therapies of cancer. Methods 2013. [Google Scholar] [CrossRef]
- Okroj, M.; Osterborg, A.; Blom, A.M. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat. Rev. 2013, 39, 632–639. [Google Scholar] [CrossRef]
- Pawluczkowycz, A.W.; Beurskens, F.J.; Beum, P.V.; Lindorfer, M.A.; van de Winkel, J.G.J.; Parren, P.W.H.I.; Taylor, R.P. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): Considerably higher levels of CDC are induced by OFA than by RTX. J. Immunol. 2009, 183, 749–758. [Google Scholar] [CrossRef]
- Gong, Q.; Ou, Q.; Ye, S.; Lee, W.P.; Cornelius, J.; Diehl, L.; Lin, W.Y.; Hu, Z.; Lu, Y.; Chen, Y.; et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 2005, 174, 817–826. [Google Scholar]
- Boross, P.; Jansen, J.H.M.; de Haij, S.; Beurskens, F.J.; van der Poel, C.E.; Bevaart, L.; Nederend, M.; Golay, J.; van de Winkel, J.G.J.; Parren, P.W.H.I.; et al. The in vivo mechanism of action of CD20 monoclonal antibodies depends on local tumor burden. Haematologica 2011, 96, 1822–1830. [Google Scholar] [CrossRef]
- Beurskens, F.J.; Lindorfer, M.A.; Farooqui, M.; Beum, P.V.; Engelberts, P.; Mackus, W.J.M.; Parren, P.W.H.I.; Wiestner, A.; Taylor, R.P. Exhaustion of cytotoxic effector systems may limit monoclonal antibody-based immunotherapy in cancer patients. J. Immunol. 2012, 188, 3532–3541. [Google Scholar]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement activation determines the therapeutic activity of rituximab in vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar]
- Tedder, T.F.; Baras, A.; Xiu, Y. Fcg receptor-dependent effector mechanisms regulate CD19 and CD20 antibody immunotherapies for B lymphocyte malignancies and autoimmunity. Springer Semin. Immun. 2006, 28, 351–364. [Google Scholar] [CrossRef]
- Macor, P.; Tripodo, C.; Zorzet, S.; Piovan, E.; Bossi, F.; Marzari, R.; Amadori, A.; Tedesco, F. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res. 2007, 67, 10556–10563. [Google Scholar]
- Masuda, K.; Kubota, T.; Kaneko, E.; Iida, S.; Wakitani, M.; Kobayashi-Natsume, Y.; Kubota, A.; Shitara, K.; Nakamura, K. Enhanced binding affinity for FcgRIIIa of fucose-negative antibody is sufficient to induce maximal antibody-dependent cellular cytotoxicity. Mol. Immunol. 2007, 44, 3122–3131. [Google Scholar] [CrossRef]
- Imai, M.; Ohta, R.; Varela, J.C.; Song, H.; Tomlinson, S. Enhancement of antibody-dependent mechanisms of tumor cell lysis by a targeted activator of complement. Cancer Res. 2007, 67, 9535–9541. [Google Scholar] [CrossRef]
- Li, B.; Shi, S.; Qian, W.; Zhao, L.; Zhang, D.; Hou, S.; Zheng, L.; Dai, J.; Zhao, J.; Wang, H.; et al. Development of novel tetravalent anti-CD20 antibodies with potent antitumor activity. Cancer Res. 2008, 68, 2400–2408. [Google Scholar] [CrossRef]
- de Romeuf, C.; Dutertre, C.A.; Le Garff-Tavernier, M.; Fournier, N.; Gaucher, C.; Glacet, A.; Jorieux, S.; Bihoreau, N.; Behrens, C.K.; Beliard, R.; et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgRIIIA/CD16. Br. J. Haematol. 2008, 140, 635–643. [Google Scholar] [CrossRef]
- Riaz, W.; Hernandez-Ilizaliturri, F.J.; Czuczman, M.S. Strategies to enhance rituximab anti-tumor activity in the treatment of CD20-positive B-cell neoplasms. Immunol. Res. 2009, 46, 192–205. [Google Scholar]
- Li, B.; Zhao, L.; Guo, H.; Wang, C.; Zhang, X.; Wu, L.; Chen, L.T.Q.; Qian, W.; Wang, H.; Guo, Y. Characterization of a rituximab variant with potent antitumor activity against rituximab-resistant B-cell lymphoma. Blood 2009, 114, 5007–5015. [Google Scholar]
- van Meerten, T.; Hagenbeek, A. Novel antibodies against follicular non-Hodgkin's lymphoma. Best Practice Res. Clin. Haematol. 2011, 24, 231–256. [Google Scholar] [CrossRef]
- Peipp, M.; van de Winkel, J.G.J.; Valerius, T. Molecular engineering to improve antibodies' anti-lymphoma activity. Best Practice Res. Clin. Haematol. 2011, 24, 217–229. [Google Scholar] [CrossRef]
- Lindorfer, M.A.; Wiestner, A.; Zent, C.S.; Taylor, R.P. Monoclonal antibody (mAb)-based cancer therapy: Is it time to reevaluate dosing strategies? Oncoimmunology 2012, 1, 959–961. [Google Scholar] [CrossRef]
- Elvington, M.; Huang, Y.; Morgan, B.P.; Ziao, F.; van Rooijen, N.; Atkinson, C.; Tomlinson, S. A targeted complement-dependent strategy to improve the outcome of mAb therapy, and characterization in a murine model of metastatic cancer. Blood 2012, 119, 6043–6051. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immunity 2012, 12, 13–20. [Google Scholar]
- Zent, C.S.; Wu, W.; Bowen, D.A.; Hanson, C.A.; Pettinger, A.M.; Shanafelt, T.D.; Kay, N.E.; Leis, J.F.; Call, T.G. Addition of granulocyte macrophage colony stimulating factor does not improve response to early treatment of high-risk chronic lymphocytic leukemia with alemtuzumab and rituximab. Leuk. Lymph. 2013, 54, 476–482. [Google Scholar] [CrossRef]
- Mamidi, S.; Cinci, M.; Hasmann, M.; Fehring, V.; Kirschfink, M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol. Oncol. 2013, 7, 580–594. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.M.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Ozkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPa variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Koski, C.; Ramm, L.; Hammer, C.; Mayer, M.; Shin, M. Cytolysis of nucleated cells by complement: Cell death displays multi-hit characteristics. Proc. Natl. Acad. Sci. USA 1983, 80, 3816–3820. [Google Scholar]
- Carney, D.F.; Hammer, C.H.; Shin, M.L. Elimination of terminal complement complexes in the plasma membrane of nucleated cells: influence of extracellular Ca2+ and association with cellular Ca2+. J. Immunol. 1986, 137, 263–270. [Google Scholar]
- Kim, S.; Carney, D.F.; Hammer, C.H.; Shin, M.L. Nucleated cell killing by complement: Effects of C5b-9 channel size and extracellular Ca-2+ on the lytic process. J. Immunol. 1987, 138, 1530–1536. [Google Scholar]
- Reiter, Y.; Ciobotariu, A.; Jones, J.; Morgan, B.P.; Fishelson, Z. Complement membrane attack complex, perforin, and bacterial exotoxins induce in K562 cells calcium-dependent cross-protection from lysis. J. Immunol. 1995, 155, 2203–2210. [Google Scholar]
- Helmy, K.Y.; Katschke, K.J.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 2006, 124, 915–927. [Google Scholar]
- He, J.Q.; Wiesmann, C.; van Lookeren Campagne, M. A role of macrophage complement receptor CR1g in immune clearance and inflammation. Mol. Immunol. 2008, 45, 4041–4047. [Google Scholar] [CrossRef]
- Lindorfer, M.A.; Kohl, J.; Taylor, R.P. Interactions between the complement system and Fcg receptors. In Antibody Fc: Linking Adaptive and Innate Immunity; Ackerman, M.E., Nimmerjahn, F., Eds.; Elsevier: Philadelphia, PA, USA, 2014. [Google Scholar]
- Morgan, B.P.; Harris, C.L. Complement Regulatory Proteins; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Fishelson, Z.; Donin, N.; Zell, S.; Schultz, S.; Kirschfink, M. Obstacles to cancer immunotherapy: Expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol. Immunol. 2003, 40, 109–123. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar]
- Dunkelberger, J.R.; Song, W.C. Role and mechanism of action of complement in regulating T cell immunity. Mol. Immunol. 2010, 47, 2176–2186. [Google Scholar] [CrossRef]
- Ehrnthaller, C.; Ignatius, A.; Gebhard, F.; Huber-Lang, M. New insights of an old defense system: Structure, function and clinical relevance of the complement system. Mol. Med. 2011, 17, 317–329. [Google Scholar]
- Nilsson, S.C.; Sim, R.B.; Lea, S.M.; Fremeaux-Bacchi, V.; Blom, A.M. Complement factor I in health and disease. Mol. Immunol. 2011, 48, 1611–1620. [Google Scholar] [CrossRef]
- Kolev, M.; Towner, L.; Donev, R. Complement in cancer and cancer immunotherapy. Arch. Immunol. Ther. Exp. 2011, 59, 407–419. [Google Scholar] [CrossRef]
- Nilsson, S.C.; Nita, I.; Mansson, L.; Groeneveld, T.W.L.; Trouw, L.A.; Villoutreix, B.O.; Blom, A.M. Analysis of binding sites on complement Factor I that are required for its activity. J. Biol. Chem. 2010, 285, 6235–6245. [Google Scholar]
- Roversi, P.; Johnson, S.; Caesar, J.J.; McLean, F.; Leath, K.J.; Tsiftsoglou, S.A.; Morgan, B.P.; Harris, C.L.; Sim, R.B.; Lea, S.M. Structural basis for complement factor I control and its disease-associated sequence polymorphisms. Proc. Natl. Acad. Sci. USA 2011, 108, 12839–12844. [Google Scholar] [CrossRef]
- Beum, P.V.; Lindorfer, M.A.; Peek, E.M.; Stukenberg, P.T.; de Weers, M.; Beurskens, F.J.; Parren, P.W.H.I.; van de Winkel, J.G.J.; Taylor, R.P. Penetration of antibody-opsonized cells by the membrane attack complex of complement promotes Ca2+ influx and induces streamers. Eur. J. Immunol. 2011, 41, 2436–2446. [Google Scholar] [CrossRef]
- Okroj, M.; Holmquist, E.; King, B.C.; Blom, A.M. Functional analyses of complement convertases using C3 and C5-depleted sera. PLoS One 2012, 7, e47245. [Google Scholar]
- Horl, S.; Banki, Z.; Huber, G.; Ejaz, A.; Windisch, D.; Muellauer, B.; Willenbacher, E.; Steurer, M.; Stoiber, H. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia 2013, 27, 2200–2208. [Google Scholar] [CrossRef]
- Horl, S.; Banki, Z.; Huber, G.; Ejaz, A.; Mullauer, B.; Willenbacher, E.; Steurer, M.; Stoiber, H. Complement factor H-derived short consensus repeat 18-20 enhanced complement-dependent cytotoxicity of Ofatumumab on chronic lymphocytic leukemia cells. Haematologica 2013. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Solga, M.D.; Schuman, T.A.; Chi, A.W.; Lindorfer, M.A.; Sutherland, W.M.; Foley, P.L.; Taylor, R.P. An anti-C3b(i) mAb enhances complement activation, C3b(i) deposition, and killing of CD20+ cells by Rituximab. Blood 2003, 101, 1071–1079. [Google Scholar] [CrossRef]
- Kennedy, A.D.; Beum, P.V.; Solga, M.D.; DiLillo, D.J.; Lindorfer, M.A.; Hess, C.E.; Densmore, J.J.; Williams, M.E.; Taylor, R.P. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J. Immunol. 2004, 172, 3280–3288. [Google Scholar]
- Teeling, J.L.; French, R.R.; Cragg, M.S.; van den Brakel, J.; Pluyter, M.; Huang, H.; Chan, C.; Parren, P.W.; Hack, C.E.; Dechant, M.; et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin's lymphomas. Blood 2004, 104, 1793–1800. [Google Scholar] [CrossRef]
- Beum, P.V.; Peek, E.M.; Lindorfer, M.A.; Beurskens, F.J.; Engelberts, P.J.; Parren, W.H.I.; van de Winkel, J.G.J.; Taylor, R.P. Loss of CD20 and bound CD20 antibody from opsonized B cells occurs more rapidly because of trogocytosis mediated by Fc receptor-expressing effector cells than direct internalization by the B cells. J. Immunol. 2011, 187, 3438–3447. [Google Scholar] [CrossRef]
- Bhat, R.; Watzl, C. Serial killing of tumor cells by human natural killer cells—Enhancement by therapeutic antibodies. PLoS One 2007, 2, e326. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Hess, A.; Lucas, D.M.; O'Donnell, P.; Ambinder, R.F.; Diehl, L.F.; Carter-Brookins, D.; Newton, S.; Flinn, I.W. Systemic interleukin-2 and adoptive transfer of lymphokine-activated killer cells improves antibody-dependent cellular cytotoxicity in patients with relapsed B-cell lymphoma treated with rituximab. Clin. Cancer Res. 2007, 13, 2392–2399. [Google Scholar] [CrossRef]
- Ge, X.; Wu, L.H.W.; Fernandes, S.; Wang, C.; Li, X.; Brown, J.R.; Zin, X. rILYd4, a human CD59 inhibitor, enhances complement-dependent cytotoxicity of ofatumumab against rituximab-resistant B-cell lymphoma cells and chronic lymphocytic leukemia. Clin. Cancer Res. 2011, 17, 6702–6711. [Google Scholar] [CrossRef]
- Teeling, J.L.; Mackus, W.J.M.; Wiegman, L.J.J.M.; van den Brakel, J.H.N.; Bees, S.A.; French, R.R.; van Meerten, T.; Ebeling, S.; Vink, T.; Slootstra, J.W.; et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J. Immunol. 2006, 177, 362–371. [Google Scholar]
- Baig, N.A.; Taylor, R.P.; Lindorfer, M.A.; Church, A.K.; LaPlant, B.R.; Pavey, E.S.; Nowakowski, G.S.; Zent, C.S. Complement dependent cytotoxicity (CDC) in chronic lymphocytic leukemia (CLL): Ofatumumab enhances alemtuzumab CDC and reveals cells resistant to activated complement. Leuk. Lymph. 2012, 53, 2218–2227. [Google Scholar] [CrossRef]
- Wierda, W.; Kipps, T.; Durig, J.; Griskevicius, L.; Stilgenbauer, S.; Mayer, J.S.L.; Hess, G.; Griniute, R.; Hernandez-Ilizaliturri, F.J.; Padmanabhan, S.; et al. Chemoimmunotherapy with O-FC in previously untreated patients with chronic lymphocytic leukemia. Blood 2011, 117, 6450–6458. [Google Scholar] [CrossRef]
- Beum, P.V.; Kennedy, A.D.; Williams, M.E.; Lindorfer, M.A.; Taylor, R.P. The shaving reaction: Rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP-1 monocytes. J. Immunol. 2006, 176, 2600–2609. [Google Scholar]
- Estrov, Z.; Talpaz, M.; Ku, S.; Harris, D.; Van, Q.; Beran, M.; Hirsch-Ginsberg, C.; Huh, Y.; Yee, G.; Kurzrock, R. Z-138: A new mature B-cell acute lymphoblastic leukemia cell line from a patient with transformed chronic lymphocytic leukemia. Leuk. Res. 1998, 22, 341–353. [Google Scholar] [CrossRef]
- Lindorfer, M.A.; Jinivizian, H.B.; Foley, P.L.; Kennedy, A.D.; Solga, M.D.; Taylor, R.P. The B cell complement receptor 2 transfer reaction. J. Immunol. 2003, 170, 3671–3678. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lindorfer, M.A.; Beum, P.V.; Taylor, R.P. CD20 mAb-Mediated Complement Dependent Cytotoxicity of Tumor Cells is Enhanced by Blocking the Action of Factor I. Antibodies 2013, 2, 598-616. https://doi.org/10.3390/antib2040598
Lindorfer MA, Beum PV, Taylor RP. CD20 mAb-Mediated Complement Dependent Cytotoxicity of Tumor Cells is Enhanced by Blocking the Action of Factor I. Antibodies. 2013; 2(4):598-616. https://doi.org/10.3390/antib2040598
Chicago/Turabian StyleLindorfer, Margaret A., Paul V. Beum, and Ronald P. Taylor. 2013. "CD20 mAb-Mediated Complement Dependent Cytotoxicity of Tumor Cells is Enhanced by Blocking the Action of Factor I" Antibodies 2, no. 4: 598-616. https://doi.org/10.3390/antib2040598