Developments and Challenges for mAb-Based Therapeutics

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Molecule name | Brand name | Target antigen | Primary Indication | Antibody type | Comment |
|---|---|---|---|---|---|---|
| 1986 | Muromonab | Orthoclone® | CD3 | Acute transplant rejection | Murine, IgG2a | First ever Ab approved by FDA; later withdrawn [6] |
| 1991 | Nebacumab | Centoxin® | Endotoxin (HA-1A) | Sepsis and Gm-ve bacteraemia | Human hybridoma derived IgM | First human mAb to be approved by regulatory agency (Europe); later withdrawn from the market [7,8] |
| 1993 | Abciximab | ReoPro® | GPIIb/IIIa | Cardiac ischemia, stroke, myocardial infarction | Chimeric Fab of IgG1 | First approved Fab-based therapeutic [9] |
| 1997 | Rituximab | Rituxan® | CD20 | Non-Hodgkin’s lymphoma | Chimeric IgG1 | First approved full length chimeric mAb [10,11] |
| 1997 | Daclizumab | Zenapax® | CD25 | Prevention of kidney transplant rejection | Humanized IgG1 | First approved full length humanized mAb [12] |
| 1998 | Etanercept | Enbrel® | TNF-receptor | RA | TNFR2-Fc fusion | First approved Fc fusion protein [9] |
| 2000 | Gemtuzumab Ozogamicin | Mylotarg® | CD33 | Acute myelogenous leukaemia | Human IgG4 | First ADC to be approved in US; withdrawn from the market [13,14] |
| 2002 | Adalimumab | Humira® | TNFα | RA | Human IgG1 | First human mAb to be approved in USA [12] |
| 2009 | Catumaxomab | Removab® | EpCAM on tumor cells and CD3 on T-cells | Malignant ascites | CD3- and EPCAM-specific mouse–rat hybrid | First bispecific to be approved by EMA [12,15] |
2. mAb
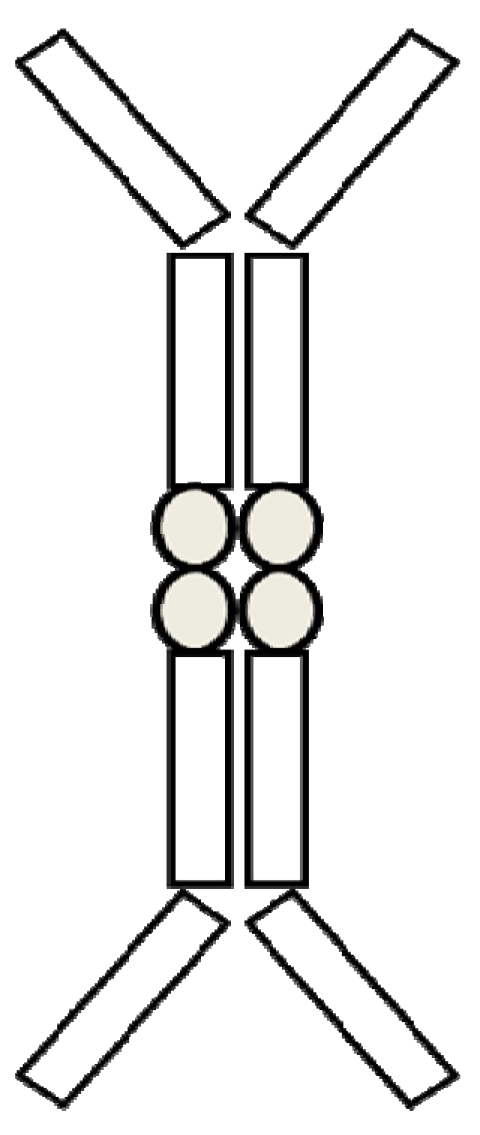
2.1. Structure, Type, and Function
2.2. Glycosylation and Its Importance
2.3. Development of mAb Therapeutics and Associated Challenges
2.3.1. Physical Degradation
2.3.2. Chemical Degradation
2.3.4. Importance of Controlling Degradation
2.3.5. Stabilization Approaches
3. Development of High Concentration Formulation
3.1. Subcutaneous Delivery and Its Limitation
3.2. Challenges of High Concentration Formulation Development
3.3. Analytical Assessment of High Concentration Formulation
4. ADC
4.1. Design and Selection of Different Linkers
4.2. Optimization of Antibody Drug Ratio
4.3. Formulation Challenges
| ADC | Linker | Drug-antibody ratio (DAR) | Dosage form | Excipients | Package system | Product storage | |
|---|---|---|---|---|---|---|---|
| Mylotarg® (withdrawn in 2010) | N-4-(4-acetylphenoxy) butanoic acid and N-acetyl-γ-calicheamicin dimethl hydrazide | 2–3 | Lyo | Dextran 40, sucrose, NaCl and phosphate buffer | Single-use amber glass | 2–8 °C | |
| Adcetris® (approved in 2011) | Maleimidocaproyl-valine-citrulline | ~4 | Lyo | Trehalose, polysorbate 80, and citrate buffer | Single-use glass vials | 2–8 °C | |
| Kadcyla® (approved in 2013) | 4-[N-maleimidomethyl] cyclohexane-1-carboxylate (MCC) | ~3.5 | Lyo | Sucrose, polysorbate 20, and sodium succinate | Single-use glass vial | 2–8 °C (4 h limit after recon at 2–8 °C) |
4.4. Challenges during Product Manufacturing and Stability Monitoring
5. Antibody Fusion Construct
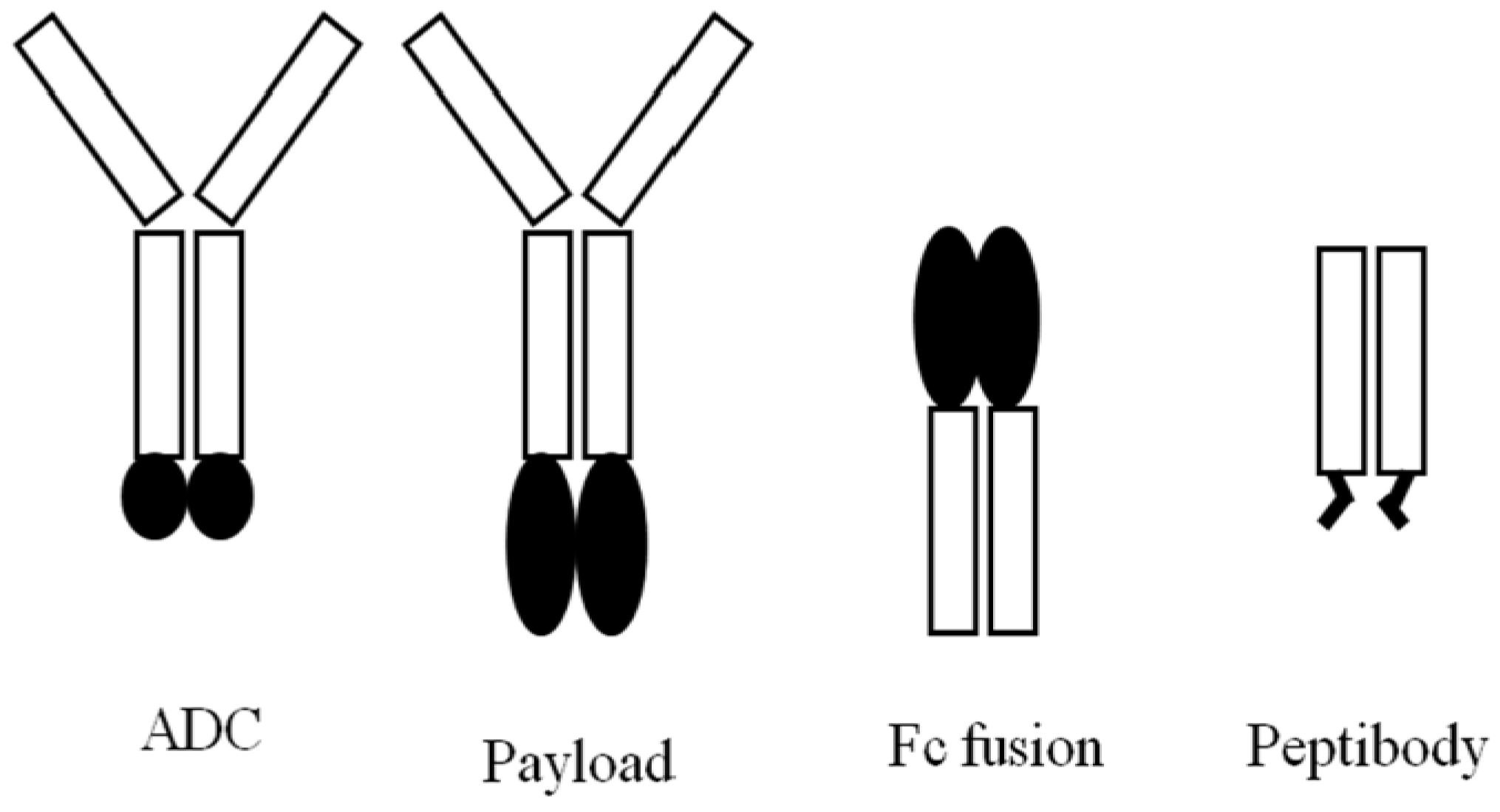
5.1. mAb-Effector Fusion Protein (Payload)
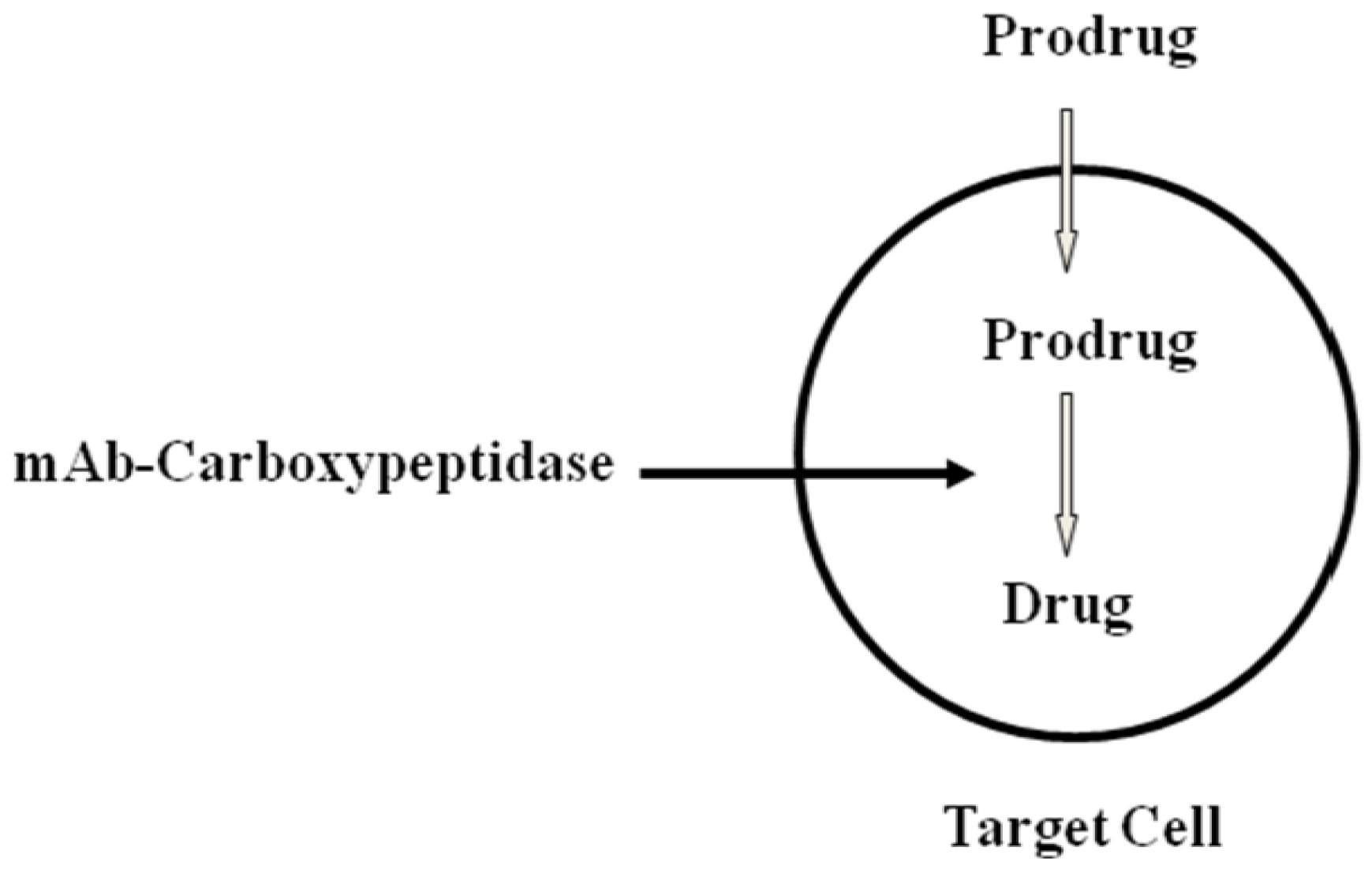
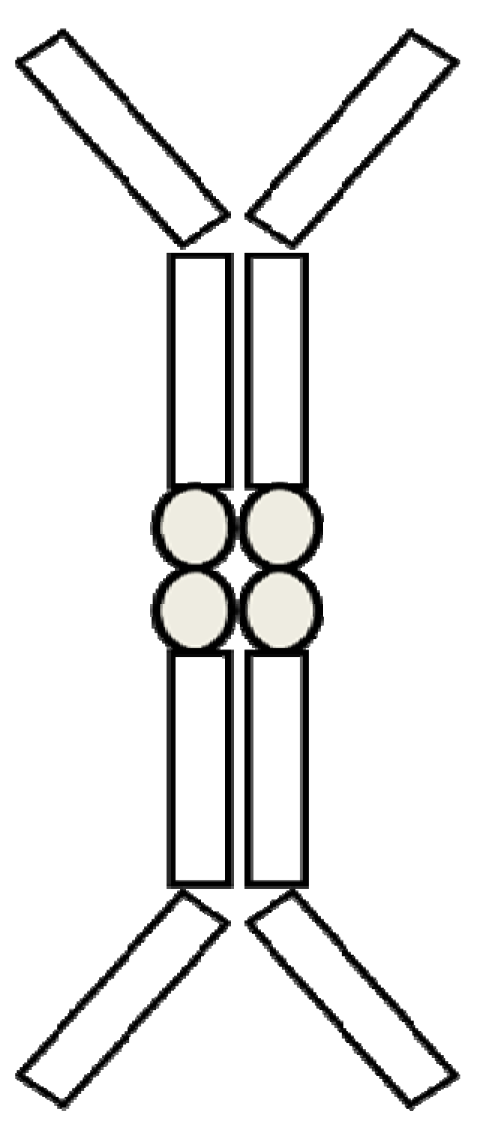
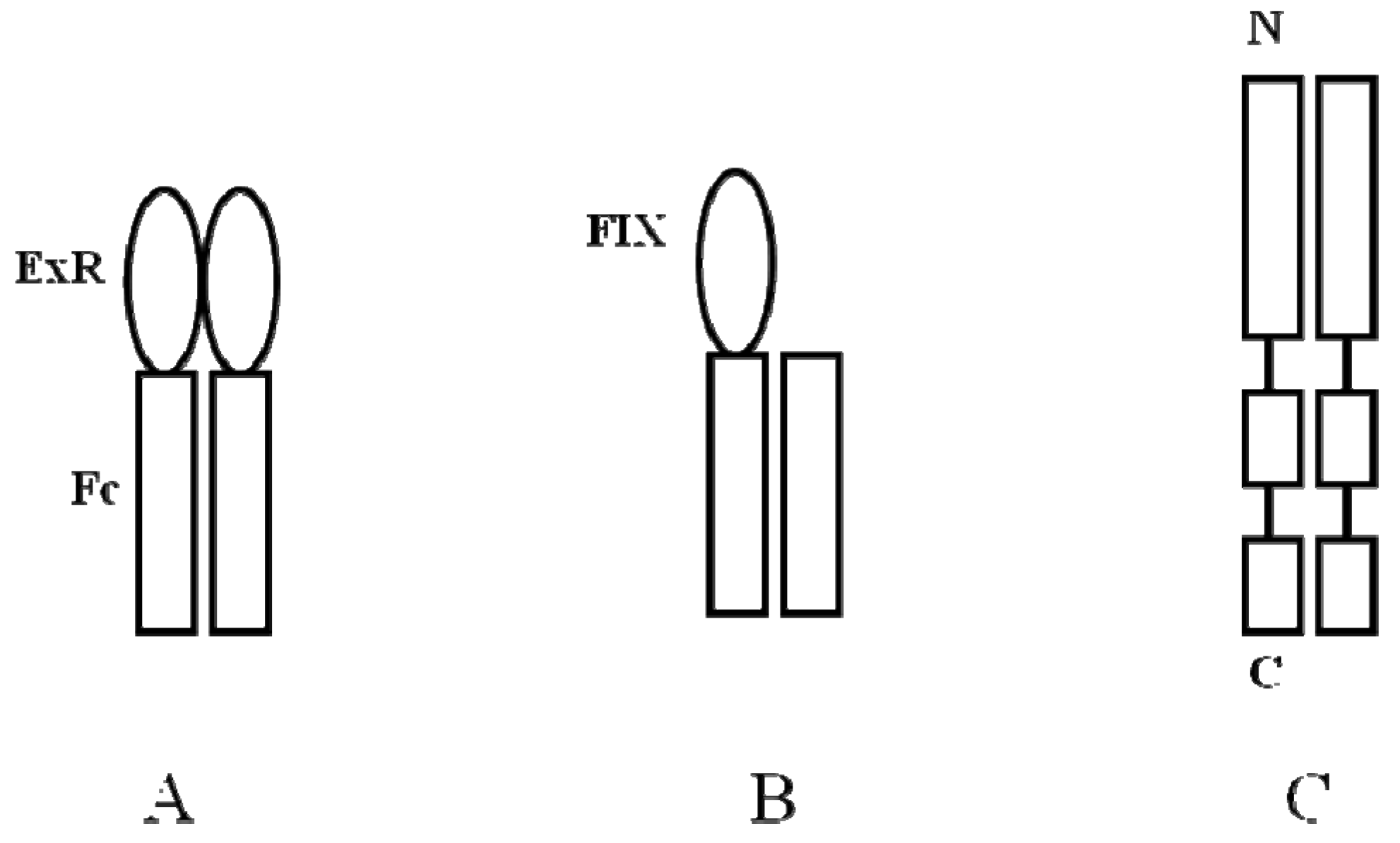
5.2. Protein-Fc Fusion
5.3. Peptibody
6. Other mAb-based Therapeutics
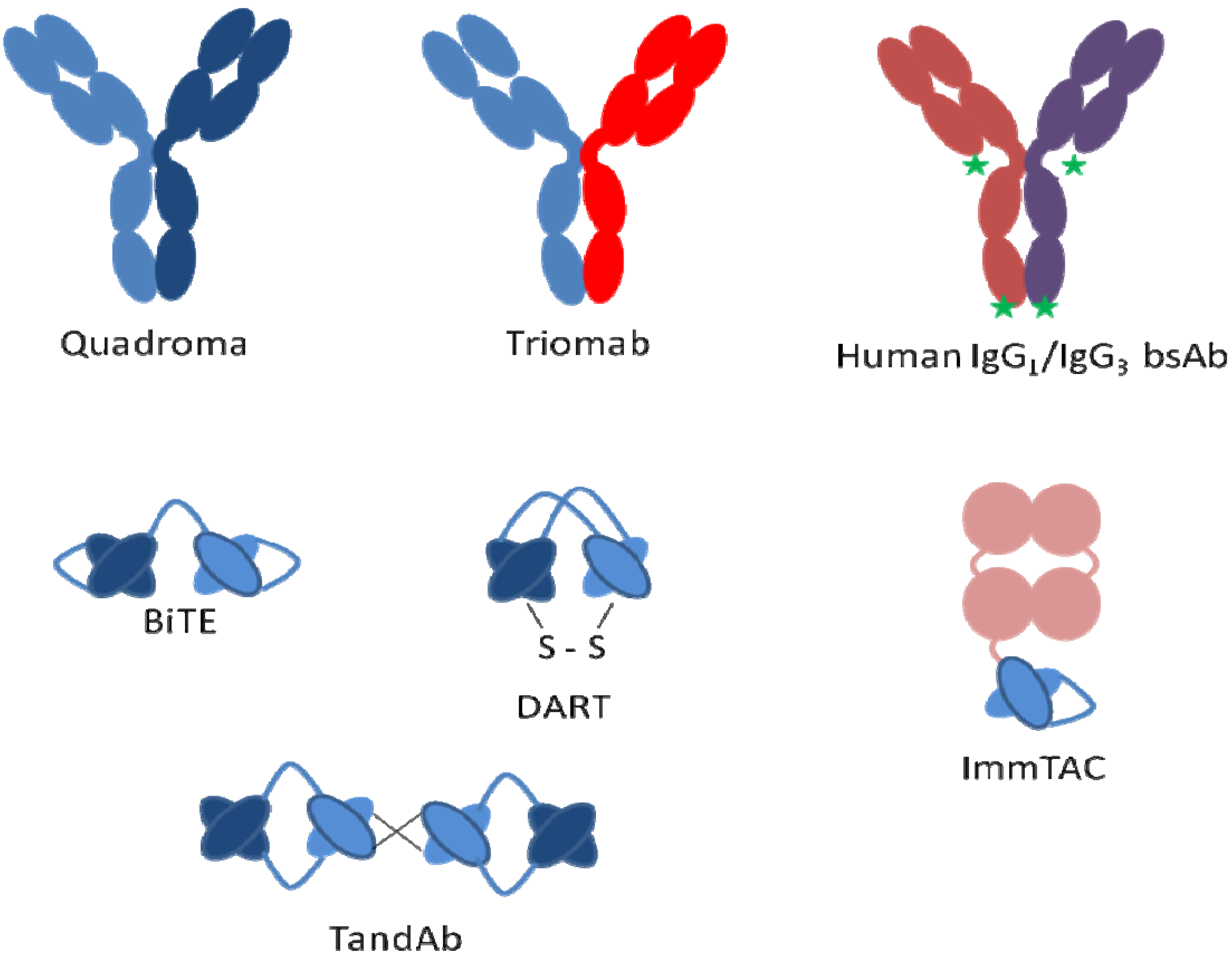
Bispecific
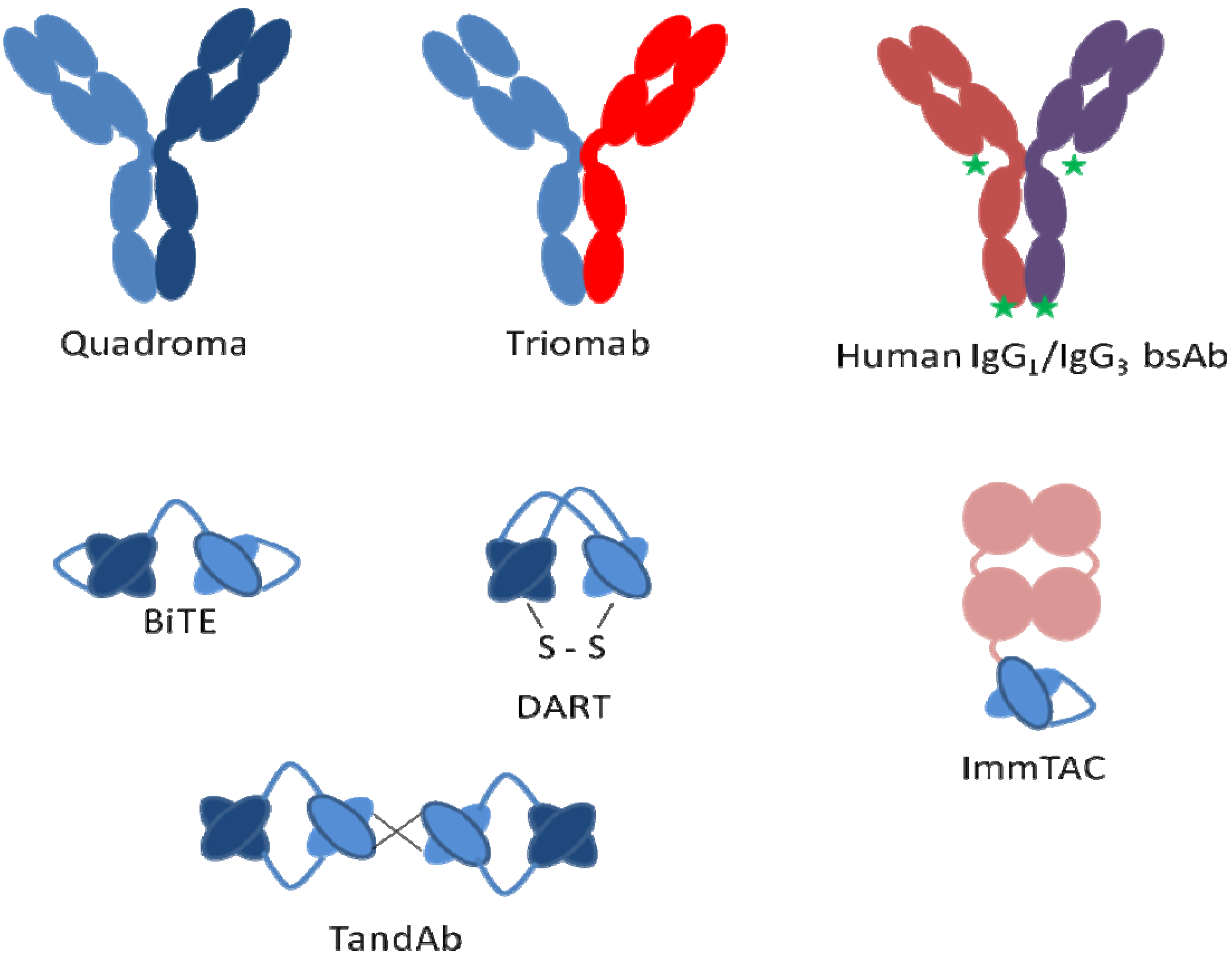
7. mAb Fragments
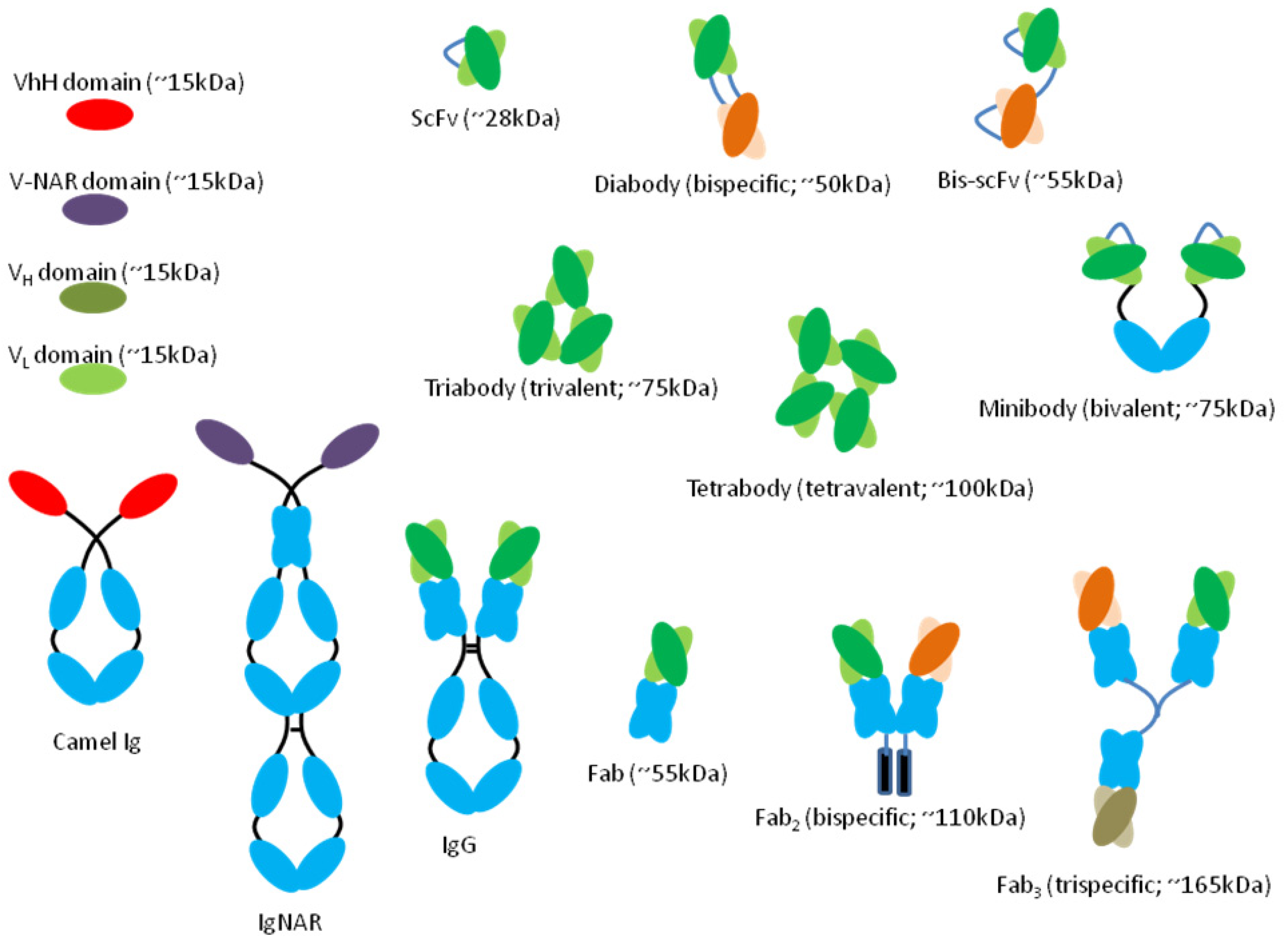
7.1. mAb Cocktail
8. Summary
Conflict of Interest
References
- Casadevall, A.; Scharff, M.D. Return to the past: The case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 1995, 21, 150–161. [Google Scholar] [CrossRef]
- Bruton, O.C. Agammaglobulinemia. Pediatrics 1952, 9, 722–728. [Google Scholar]
- Nadler, L.M.; Stashenko, P.; Hardy, R.; Kaplan, W.D.; Button, L.N.; Kufe, D.W.; Antman, K.H.; Schlossman, S.F. Serotherapy of a patient with a monoclonal antibody directed against a human lymphoma-associated antigen. Cancer Res. 1980, 40, 3147–3154. [Google Scholar]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Oldham, R.K. Monoclonal antibodies as anticancer agents. Adv. Exp. Med. Biol. 1983, 166, 45–57. [Google Scholar] [CrossRef]
- Reichert, J.M. Metrics for antibody therapeutics development. MAbs 2010, 2, 695–700. [Google Scholar] [CrossRef]
- Marks, L. The birth pangs of monoclonal antibody therapeutics: The failure and legacy of Centoxin. MAbs 2012, 4, 403–412. [Google Scholar] [CrossRef]
- Hurley, J.C. Sepsis management and antiendotoxin therapy after nebacumab. A reappraisal. Drugs 1994, 47, 855–861. [Google Scholar] [CrossRef]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef]
- Coiffier, B.; Lepage, E.; Brière, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOPalone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef]
- McLaughlin, P.; Grillo-López, A.J.; Link, B.K.; Levy, R.; Czuczman, M.S.; Williams, M.E.; Heyman, M.R.; Bence-Bruckler, I.; White, C.A.; Cabanillas, F.; et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: Half of patients respond to a four-dose treatment program. J. Clin. Oncol. 1998, 16, 2825–2833. [Google Scholar]
- Reichert, J.M. Antibody-based therapeutics to watch in 2011. MAbs 2011, 3, 76–99. [Google Scholar] [CrossRef]
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: A phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef]
- Oldham, R.K.; Dillman, R.O. Monoclonal antibodies in cancer therapy: 25 years of progress. J. Clin. Oncol. 2008, 26, 1774–1777. [Google Scholar] [CrossRef]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef]
- Lonberg, N. Human antibodies from transgenic animals. Nat. Biotechnol. 2005, 23, 1117–1125. [Google Scholar] [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Graziano, R.F.; Fanger, M.W. Fc gamma RI and Fc gamma RII on monocytes and granulocytes are cytotoxic trigger molecules for tumor cells. J. Immunol. 1987, 139, 3536–3541. [Google Scholar]
- Kimura, H.; Sakai, K.; Arao, T.; Shimoyama, T.; Tamura, T.; Nishio, K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007, 98, 1275–1280. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef]
- Chan, A.C.; Carter, P.J. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 2010, 10, 301–316. [Google Scholar] [CrossRef]
- Ducancel, F.; Muller, B.H. Molecular engineering of antibodies for therapeutic and diagnostic purposes. MAbs 2012, 4, 445–457. [Google Scholar] [CrossRef]
- Dillman, R.O. Magic bullets at last! Finally—Approval of a monoclonal antibody for the treatment of cancer!!! Cancer Biother. Radiopharm. 1997, 12, 223–225. [Google Scholar]
- Huhn, D.; von Schilling, C.; Wilhelm, M.; Ho, A.D.; Hallek, M.; Kuse, R.; Knauf, W.; Riedel, U.; Hinke, A.; Srock, S.; et al. Rituximab therapy of patients with B-cell chronic lymphocytic leukemia. Blood 2001, 98, 1326–1331. [Google Scholar] [CrossRef]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef]
- Wormald, M.R.; Dwek, R.A. Glycoproteins: Glycan presentation and protein-fold stability. Structure 1999, 7, R155–R160. [Google Scholar] [CrossRef]
- Yamane-Ohnuki, N.; Satoh, M. Production of therapeutic antibodies with controlled fucosylation. MAbs 2009, 1, 230–236. [Google Scholar] [CrossRef]
- Lis, H.; Sharon, N. Protein glycosylation. Structural and functional aspects. Eur. J. Biochem. 1993, 218, 1–27. [Google Scholar] [CrossRef]
- Mimura, Y.; Churcha, S.; Ghirlandob, R.; Ashtonc, S.; Donga, S.; Goodalla, M.; Lunda, J.; Jefferisa, R. The influence of glycosylation on the thermal stability and effector function expression of human IgG1-Fc: Properties of a series of truncated glycoforms. Mol. Immunol. 2000, 37, 697–706. [Google Scholar] [CrossRef]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Forrer, K.; Helk, B.; Trout, B.L. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol. J. 2011, 6, 38–44. [Google Scholar]
- Runkel, L.; Meier, W.; Blake Pepinsky, R.; Karpusas, M.; Whitty, A.; Kimball, K.; Brickelmaier, M.; Muldowney, C.; Jones, W.; Goelz, S.E. Structural and functional differences between glycosylated and non-glycosylated forms of human interferon-beta (IFN-beta). Pharm. Res. 1998, 15, 641–649. [Google Scholar] [CrossRef]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef]
- Burton, D.R.; Woof, J.M. Human antibody effector function. Adv. Immunol. 1992, 51, 1–84. [Google Scholar] [CrossRef]
- Anegon, I.; Cuturi, M.C.; Trinchieri, G.; Perussia, B. Interaction of Fc receptor (CD16) ligands induces transcription of interleukin 2 receptor (CD25) and lymphokine genes and expression of their products in human natural killer cells. J. Exp. Med. 1988, 167, 452–472. [Google Scholar] [CrossRef]
- Fanger, M.W.; Graziano, R.F.; Li, S.; Guyre, P.M. Fc gamma R in cytotoxicity exerted by mononuclear cells. Chem. Immunol. 1989, 47, 214–253. [Google Scholar] [CrossRef]
- Anderson, C.L.; Shen, L.; Eicher, D.M.; Wewers, M.D.; Gill, J.K. Phagocytosis mediated by three distinct Fc gamma receptor classes on human leukocytes. J. Exp. Med. 1990, 171, 1333–1345. [Google Scholar] [CrossRef]
- Jefferis, R.; Lund, J.; Pound, J.D. IgG-Fc-mediated effector functions: Molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol. Rev. 1998, 163, 59–76. [Google Scholar] [CrossRef]
- Vugmeyster, Y.; Howell, K. Rituximab-mediated depletion of cynomolgus monkey B cells in vitro in different matrices: Possible inhibitory effect of IgG. Int. Immunopharmacol. 2004, 4, 1117–1124. [Google Scholar] [CrossRef]
- Matsumiya, S.; Yamaguchi, Y.; Saito, J.; Nagano, M.; Sasakawa, H.; Otaki, S.; Satoh, M.; Shitara, K.; Kato, K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 2007, 368, 767–779. [Google Scholar] [CrossRef]
- Ferrara, C.; Stuart, F.; Sondermann, P.; Brünker, P.; Umaña, P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J. Biol. Chem. 2006, 281, 5032–5036. [Google Scholar]
- Iida, S.; Misaka, H.; Inoue, M.; Shibata, M.; Nakano, R.; Yamane-Ohnuki, N.; Wakitani, M.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated therapeutic IgG1 antibody can evade the inhibitory effect of serum immunoglobulin G on antibody-dependent cellular cytotoxicity through its high binding to FcgammaRIIIa. Clin. Cancer Res. 2006, 12, 2879–2887. [Google Scholar] [CrossRef]
- Shitara, K. Potelligent antibodies as next generation therapeutic antibodies. Yakugaku Zasshi 2009, 129, 3–9. [Google Scholar] [CrossRef]
- Wang, W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 1999, 185, 29–88. [Google Scholar] [CrossRef]
- Mahler, H.C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–3934. [Google Scholar] [CrossRef]
- Daugherty, A.L.; Mrsny, R.J. Formulation and delivery issues for monoclonal antibody therapeutics. Adv. Drug Deliv. Rev. 2006, 58, 686–706. [Google Scholar] [CrossRef]
- Parkins, D.A.; Lashmar, U.T. The formulation of biopharmaceutical products. Pharm. Sci. Technol. Today 2000, 3, 129–137. [Google Scholar] [CrossRef]
- Narhi, L.O.; Jiang, Y.; Cao, S.; Benedek, K.; Shnek, D. A critical review of analytical methods for subvisible and visible particles. Curr. Pharm. Biotechnol. 2009, 10, 373–381. [Google Scholar] [CrossRef]
- Das, T.K. Protein particulate detection issues in biotherapeutics development—Current status. AAPS PharmSciTech. 2012, 13, 732–746. [Google Scholar] [CrossRef]
- Karshikoff, A. Non-Covalent Interactions in Proteins, 1st ed.; Imperial College Press: London, UK, 2006. [Google Scholar]
- Andya, J.D.; Hsu, C.C.; Shire, S.J. Mechanisms of aggregate formation and carbohydrate excipient stabilization of lyophilized humanized monoclonal antibody formulations. AAPS PharmSci. 2003, 5, E10. [Google Scholar] [CrossRef]
- Malencik, D.A.; Anderson, S.R. Dityrosine as a product of oxidative stress and fluorescent probe. Amino Acids 2003, 25, 233–247. [Google Scholar] [CrossRef]
- Hawe, A.; Kasperb, J.C.; Friessb, W.; Jiskoota, W. Structural properties of monoclonal antibody aggregates induced by freeze-thawing and thermal stress. Eur. J. Pharm. Sci. 2009, 38, 79–87. [Google Scholar] [CrossRef]
- Kreilgaard, L.; Jones, L.S.; Randolph, T.W.; Frokjaer, S.; Flink, J.M.; Manning, M.C.; Carpenter, J.F. Effect of Tween 20 on freeze-thawing- and agitation-induced aggregation of recombinant human factor XIII. J. Pharm. Sci. 1998, 87, 1597–1603. [Google Scholar]
- Strambini, G.B.; Gonnelli, M. Protein stability in ice. Biophys. J. 2007, 92, 2131–2138. [Google Scholar] [CrossRef]
- Pikal-Cleland, K.A.; Cleland, J.L.; floatdoquy, T.J.; Carpenter, J.F. Effect of glycine on pH changes and protein stability during freeze-thawing in phosphate buffer systems. J. Pharm. Sci. 2002, 91, 1969–1979. [Google Scholar] [CrossRef]
- Kueltzo, L.A.; Wang, W.; Randolph, T.W.; Carpenter, J.F. Effects of solution conditions, processing parameters, and container materials on aggregation of a monoclonal antibody during freeze-thawing. J. Pharm. Sci. 2008, 97, 1801–1812. [Google Scholar] [CrossRef]
- Schreiber, G. Kinetic studies of protein-protein interactions. Curr. Opin. Struct. Biol. 2002, 12, 41–47. [Google Scholar] [CrossRef]
- Cleland, J.L.; Powell, M.F.; Shire, S.J. The development of stable protein formulations: A close look at protein aggregation, deamidation, and oxidation. Crit. Rev. Ther. Drug Carrier. Syst. 1993, 10, 307–377. [Google Scholar]
- Tyler-Cross, R.; Schirch, V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J. Biol. Chem. 1991, 266, 22549–22556. [Google Scholar]
- Son, K.; Kwon, C. Stabilization of human epidermal growth factor (hEGF) in aqueous formulation. Pharm. Res. 1995, 12, 451–454. [Google Scholar] [CrossRef]
- Daniel, R.M.; Dines, M.; Petach, H.H. The denaturation and degradation of stable enzymes at high temperatures. Biochem. J. 1996, 317, 1–11. [Google Scholar]
- Strickley, R.G.; Anderson, B.D. Solid-state stability of human insulin. I. Mechanism and the effect of water on the kinetics of degradation in lyophiles from pH 2-5 solutions. Pharm. Res. 1996, 13, 1142–1153. [Google Scholar] [CrossRef]
- Cacia, J.; Keck, R.; Presta, L.G.; Frenz, J. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: Identification and effect on binding affinity. Biochemistry 1996, 35, 1897–1903. [Google Scholar] [CrossRef]
- Li, S.; Schoneich, C.; Borchardt, R.T. Chemical pathways of peptide degradation. VIII. Oxidation of methionine in small model peptides by prooxidant/transition metal ion systems: Influence of selective scavengers for reactive oxygen intermediates. Pharm. Res. 1995, 12, 348–355. [Google Scholar] [CrossRef]
- Powell, M.F. A compendium and hydropathy/flexibility analysis of common reactive sites in proteins: Reactivity at Asn, Asp, Gln, and Met motifs in neutral pH solution. In Formulation, Characterization, and Stability of Protein Drugs; Pearlman, R., Wang, Y.J., Eds.; Plenum Press: New York, NY, USA, 1996; pp. 1–140. [Google Scholar]
- Brange, J.; Langkj, L.; Havelund, S.; Vølund, A. Chemical stability of insulin. 1. Hydrolytic degradation during storage of pharmaceutical preparations. Pharm. Res. 1992, 9, 715–726. [Google Scholar] [CrossRef]
- Manning, M.C.; Patel, K.; Borchardt, R.T. Stability of protein pharmaceuticals. Pharm. Res. 1989, 6, 903–918. [Google Scholar] [CrossRef]
- Kroon, D.J.; Baldwin-Ferro, A.; Lalan, P. Identification of sites of degradation in a therapeutic monoclonal antibody by peptide mapping. Pharm. Res. 1992, 9, 1386–1393. [Google Scholar] [CrossRef]
- Li, S.; Nguyen, T.H.; Schoneich, C.; Borchardt, R.T. Aggregation and precipitation of human relaxin induced by metal-catalyzed oxidation. Biochemistry 1995, 34, 5762–5772. [Google Scholar] [CrossRef]
- Lam, X.M.; Yang, J.Y.; Cleland, J.L. Antioxidants for prevention of methionine oxidation in recombinant monoclonal antibody HER2. J. Pharm. Sci. 1997, 86, 1250–1255. [Google Scholar] [CrossRef]
- Ha, E.; Wang, W.; Wang, Y.J. Peroxide formation in polysorbate 80 and protein stability. J. Pharm. Sci. 2002, 91, 2252–2264. [Google Scholar] [CrossRef]
- Knepp, V.M.; Whatley, J.L.; Muchnik, A.; Calderwood, T.S. Identification of antioxidants for prevention of peroxide-mediated oxidation of recombinant human ciliary neurotrophic factor and recombinant human nerve growth factor. PDA J. Pharm. Sci. Technol. 1996, 50, 163–171. [Google Scholar]
- Liu, J.L.; Lu, K.V.; Eris, T.; Katta, V.; Westcott, K.R.; Narhi, L.O.; Lu, H.S. In vitro methionine oxidation of recombinant human leptin. Pharm. Res. 1998, 15, 632–640. [Google Scholar] [CrossRef]
- Fransson, J.; FIorin-Robertsson, E.; Axelsson, K.; Nyhlén, C. Oxidation of human insulin-like growth factor I in formulation studies: Kinetics of methionine oxidation in aqueous solution and in solid state. Pharm. Res. 1996, 13, 1252–1257. [Google Scholar] [CrossRef]
- Shahrokh, Z.; Eberlein, G.; Buckley, D.; Paranandi, M.V.; Aswad, D.W.; Stratton, P.; Mischak, R.; Wang, Y.J. Major degradation products of basic fibroblast growth factor: Detection of succinimide and iso-aspartate in place of aspartate. Pharm. Res. 1994, 11, 936–944. [Google Scholar] [CrossRef]
- Kamat, M.S.; Tolman, G.L.; Brown, J.M. Formulation development of an antifibrin monoclonal antibody radiopharmaceutical. Pharm. Biotechnol. 1996, 9, 343–364. [Google Scholar] [CrossRef]
- Tous, G.I.; Wei, Z.; Feng, J.; Bilbulian, S.; Bowen, S.; Smith, J.; Strouse, R.; McGeehan, P.; Casas-Finet, J.; Schenerman, M.A. Characterization of a novel modification to monoclonal antibodies: Thioether cross-link of heavy and light chains. Anal. Chem. 2005, 77, 2675–2682. [Google Scholar] [CrossRef]
- Hermeling, S.; Crommelin, D.J.A.; Schellekens, H.; Jiskoot, W. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 2004, 21, 897–903. [Google Scholar] [CrossRef]
- Schellekens, H. Immunogenicity of therapeutic proteins: Clinical implications and future prospects. Clin. Ther. 2002, 24, 1720–1740; discussion 1719. [Google Scholar] [CrossRef]
- Casadevall, N.; Nataf, J.; Viron, B.; Kolta, A.; Kiladjian, J.-J.; Martin-Dupont, P.; Michaud, P.; Papo, T.; Ugo, V.; Teyssandier, I.; et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N. Engl. J. Med. 2002, 346, 469–475. [Google Scholar] [CrossRef]
- Joubert, M.K.; Hokom, M.; Eakin, C.; Zhou, L.; Deshpande, M.; Baker, M.P.; Goletz, T.J.; Kerwin, B.A.; Chirmule, N.; Narhi, L.O.; et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J. Biol. Chem. 2012, 287, 25266–25279. [Google Scholar] [CrossRef]
- Demeule, B.; Gurny, R.; Arvinte, T. Where disease pathogenesis meets protein formulation: Renal deposition of immunoglobulin aggregates. Eur. J. Pharm. Biopharm. 2006, 62, 121–130. [Google Scholar] [CrossRef]
- USP, General Chapters: <788> Particulate Matter in Injections; Pharmacopeial Forum: Maryland, MD, USA, 2009; pp. SP32–NF27.
- Harris, R.J.; Kabakoff, B.; Macchi, F.D.; Shen, F.J.; Kwong, M.; Andya, J.D.; Shire, S.J.; Bjork, N.; Totpal, K.; Chen, A.B. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 233–245. [Google Scholar] [CrossRef]
- Hochuli, E. Interferon immunogenicity: Technical evaluation of interferon-alpha 2a. J. Interferon. Cytokine Res. 1997, 17, S15–S21. [Google Scholar]
- Laue, T.M.; Shah, B.; Ridgeway, T.M.; Pelletier, S.L. Computer-aided Interpretation of Sedimentation Data for Proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science; Harding, S.E., Rowe, A.J., Horton, J.C., Eds.; Royal Society of Chemistry: Cambridge, UK, 1992; pp. 90–125. [Google Scholar]
- Hartmann, W.K.; Saptharishi, N.; Yang, X.Y.; Mitra, G.; Soman, G. Characterization and analysis of thermal denaturation of antibodies by size exclusion high-performance liquid chromatography with quadruple detection. Anal. Biochem. 2004, 325, 227–239. [Google Scholar] [CrossRef]
- Chiti, F.; van Nuland, N.A.J.; Taddei, N.; Magherini, F.; Stefani, M.; Ramponi, G.; Dobson, C.M. Conformational stability of muscle acylphosphatase: The role of temperature, denaturant concentration, and pH. Biochemistry 1998, 37, 1447–1455. [Google Scholar] [CrossRef]
- PharmaCircle. Available online: www.pharmacircle.com (accessed on 1 June 2013).
- Lam, X.M.; Oeswein, O.J.; Ongpipattanakul, B.; Shahrokh, Z.; Weissburg, W.S.; Wong, R.P. Stabilized Antibody Formulation. US Patent CA 2292730, 1998. [Google Scholar]
- Johnson, R.E.; Hong, Q.I.; Borgmeyer, J.R.; Kessler, R.K.; Zeng, D.L. Stable pH optimized formulation of a modified antibody. WO Patent WO/2004/019861, 2004. [Google Scholar]
- Warne, N.W. Development of high concentration protein biopharmaceuticals: The use of platform approaches in formulation development. Eur. J. Pharm. Biopharm. 2011, 78, 208–212. [Google Scholar] [CrossRef]
- Lee, L.S. Stabilized Monomeric Protein Compositions. US Patent 5,656,730, 1997. [Google Scholar]
- Worn, A.; Pluckthun, A. Mutual stabilization of VL and VH in single-chain antibody fragments, investigated with mutants engineered for stability. Biochemistry 1998, 37, 13120–13127. [Google Scholar] [CrossRef]
- Horbett, T.A. Protein adsorption on biomaterials. In Biomaterials: Interfacial Phenomena and Applications; Cooper, S.L., Peppas, N.A., Hoffman, A.S., Ratner, B.D., Eds.; American Chemical Society: Washington DC, USA, 1982; pp. 233–244. [Google Scholar]
- Lee, H.J.; McAuley, A.; Schilke, K.F.; McGuire, J. Molecular origins of surfactant-mediated stabilization of protein drugs. Adv. Drug Deliv. Rev. 2011, 63, 1160–1171. [Google Scholar] [CrossRef]
- Kerwin, B.A. Polysorbates 20 and 80 used in the formulation of protein biotherapeutics: Structure and degradation pathways. J. Pharm. Sci. 2008, 97, 2924–2935. [Google Scholar] [CrossRef]
- Chen, B.; Bautista, R.; Yu, K.; Zapata, G.A.; Mulkerrin, M.G.; Chamow, S.M. Influence of histidine on the stability and physical properties of a fully human antibody in aqueous and solid forms. Pharm. Res. 2003, 20, 1952–1960. [Google Scholar] [CrossRef]
- Paborji, M.; Pochopin, N.L.; Coppola, W.P.; Bogardus, J.B. Chemical and physical stability of chimeric L6, a mouse-human monoclonal antibody. Pharm. Res. 1994, 11, 764–771. [Google Scholar] [CrossRef]
- McIntosh, K.A.; Charman, W.N.; Charman, S.A. The application of capillary electrophoresis for monitoring effects of excipients on protein conformation. J. Pharm. Biomed. Anal. 1998, 16, 1097–1105. [Google Scholar] [CrossRef]
- Li, S.; Patapoff, T.W.; Nguyen, T.H.; Borchardt, R.T. Inhibitory effect of sugars and polyols on the metal-catalyzed oxidation of human relaxin. J. Pharm. Sci. 1996, 85, 868–872. [Google Scholar] [CrossRef]
- Li, S.; Patapoff, T.W.; Overcashier, D.; Hsu, C.; Nguyen, T.H.; Borchardt, R.T. Effects of reducing sugars on the chemical stability of human relaxin in the lyophilized state. J. Pharm. Sci. 1996, 85, 873–877. [Google Scholar] [CrossRef]
- Ji, J.A.; Zhang, B.; Cheng, W.; John Wang, Y. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: Mechanisms and stabilization. J. Pharm. Sci. 2009, 98, 4485–4500. [Google Scholar] [CrossRef]
- Gokarn, Y.R.; Matthew Fesinmeyer, R.; Saluja, A.; Razinkov, V.; Chase, S.F.; Laue, T.M.; Brems, D.N. Effective charge measurements reveal selective and preferential accumulation of anions, but not cations, at the protein surface in dilute salt solutions. Protein Sci. 2011, 20, 580–587. [Google Scholar] [CrossRef]
- Fesinmeyer, R.M.; Hogan, S.; Saluja, A.; Brych, S.R.; Kras, E.; Narhi, L.O.; Brems, D.N.; Gokarn, Y.R. Effect of ions on agitation- and temperature-induced aggregation reactions of antibodies. Pharm. Res. 2009, 26, 903–913. [Google Scholar] [CrossRef]
- Laue, T. Proximity energies: A framework for understanding concentrated solutions. J. Mol. Recognit. 2012, 25, 165–173. [Google Scholar] [CrossRef]
- Adams, G.P.; Schier, R.; McCall, A.M.; Crawford, R.S.; Wolf, E.J.; Weiner, L.M.; Marks, J.D. Prolonged in vivo tumour retention of a human diabody targeting the extracellular domain of human HER2/neu. Br. J. Cancer 1998, 77, 1405–1412. [Google Scholar] [CrossRef]
- Di Fede, G.; Bronte, G.; Rizzo, S.; Cervetto, C.R.; Cocorullo, G.; Gulotta, G.; Bazan, V.; Russo, A. Monoclonal antibodies and antibody fragments: State of the art and future perspectives in the treatment of non-haematological tumors. Expert Opin. Biol. Ther. 2011, 11, 1433–1445. [Google Scholar] [CrossRef] [Green Version]
- Stockwin, L.H.; Holmes, S. Antibodies as therapeutic agents: Vive la renaissance! Expert Opin. Biol. Ther. 2003, 3, 1133–1152. [Google Scholar] [CrossRef]
- Bookbinder, L.H.; Hofera, A.; Hallera, M.F.; Zepedab, M.L.; Kellera, G.-A.; Lima, J.E.; Edgingtonc, T.S.; Shepardd, H.M.; Pattone, J.S.; Frosta, G.I. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J. Control Release 2006, 114, 230–241. [Google Scholar] [CrossRef]
- Gatlin, L.A.; Gatlin, C.A.B. Formulation and administration techniques to minimize injection pain and tissue damage associated with parenteral products. In Injectable Drug Development: Techniques to Reduce Pain and Irritation; Gapta, P.K., Brazeau, G.A., Eds.; Interpharm Press: Denver, CO, USA, 1999; pp. 401–425. [Google Scholar]
- Yu, A.W.; Leung, C.B.; Li, P.K.; Lui, S.F.; Lai, K.N. Pain perception following subcutaneous injections of citrate-buffered and phosphate-buffered epoetin alpha. Int. J. Artif. Organs. 1998, 21, 41–43. [Google Scholar]
- Kappelgaard, A.M.; Bojesen, A.; Skydsgaard, K.; Sjögren, I.; Laursen, T. Liquid growth hormone: Preservatives and buffers. Horm. Res. 2004, 62, 98–103. [Google Scholar] [CrossRef]
- Laursen, T.; Hansen, B.; Fisker, S. Pain perception after subcutaneous injections of media containing different buffers. Basic Clin. Pharmacol. Toxicol. 2006, 98, 218–221. [Google Scholar] [CrossRef]
- Hall, D.; Minton, A.P. Macromolecular crowding: Qualitative and semiquantitative successes, quantitative challenges. Biochim. Biophys. Acta 2003, 1649, 127–139. [Google Scholar] [CrossRef]
- Rivas, G.; Minton, A.P. Non-ideal tracer sedimentation equilibrium: A powerful tool for the characterization of macromolecular interactions in crowded solutions. J. Mol. Recognit. 2004, 17, 362–367. [Google Scholar] [CrossRef]
- Yadav, S.; Laue, T.M.; Kalonia, D.S.; Singh, S.N.; Shire, S.J. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol. Pharm. 2012, 9, 791–802. [Google Scholar]
- Schein, C.H. Solubility as a function of protein structure and solvent components. Biotechnology 1990, 8, 308–317. [Google Scholar]
- Shire, S.J.; Shahrokh, Z.; Liu, J. Challenges in the development of high protein concentration formulations. J. Pharm. Sci. 2004, 93, 1390–1402. [Google Scholar] [CrossRef]
- Sukumar, M.; Doyle, B.L.; Combs, J.L.; Pekar, A.H. Opalescent appearance of an IgG1 antibody at high concentrations and its relationship to noncovalent association. Pharm. Res. 2004, 21, 1087–1093. [Google Scholar] [CrossRef]
- Cromwell, M.E.M.; Carpenter, J.F.; Scherer, T.; Randolph, T.J. Opalescence in Antibody Formulations is a Solution Critical Phenomenon. In Proceseedings of the 236th ACS National Meeting, Philadelphia, PA, USA, 2008.
- Saluja, A.; Kalonia, D.S. Nature and consequences of protein-protein interactions in high protein concentration solutions. Int. J. Pharm. 2008, 358, 1–15. [Google Scholar] [CrossRef]
- Zhou, H.X.; Rivas, G.; Minton, A.P. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008, 37, 375–397. [Google Scholar] [CrossRef]
- Shiloach, J.; Martin, N.; Moes, H. Tangential flow filtration. Adv. Biotechnol. Processes. 1988, 8, 97–125. [Google Scholar]
- Shire, S.J.; Liu, J.; Friess, W.; Jörg, S.; Mahler, H.-C. High-concentration antibody formulations. In Formulation and Process Development Strategies for Manufacturing Biopharmaceuticals; Hershenson, F.J.S., Ed.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2010. [Google Scholar]
- Friess, H.; Langrehr, J.M.; Oettle, H.; Raedle, J.; Niedergethmann, M.; Dittrich, C.; Hossfeld, D.K.; Stöger, H.; Neyns, B.; Herzog, P.; et al. A randomized multi-center phase II trial of the angiogenesis inhibitor Cilengitide (EMD 121974) and gemcitabine compared with gemcitabine alone in advanced unresectable pancreatic cancer. BMC Cancer 2006, 6, 285. [Google Scholar] [CrossRef]
- Rathore, N.; Pranay, P.; Bernacki, J.; Eu, B.; Ji, W.; Walls, E. Characterization of protein rheology and delivery forces for combination products. J. Pharm. Sci. 2012, 101, 4472–4480. [Google Scholar] [CrossRef]
- Harris, R.J.; Shire, S.J.; Winter, C. Commercial manufacturing scale formulation and analytical characterization of therapeutic recombinant antibodies. Drug Develop. Res. 2004, 61, 137–154. [Google Scholar] [CrossRef]
- Jimenez, M.; Rivas, G.; Minton, A.P. Quantitative characterization of weak self-association in concentrated solutions of immunoglobulin G via the measurement of sedimentation equilibrium and osmotic pressure. Biochemistry 2007, 46, 8373–8378. [Google Scholar] [CrossRef]
- Minton, A.P. Analytical centrifugation with preparative ultracentrifuges. Anal. Biochem. 1989, 176, 209–216. [Google Scholar] [CrossRef]
- Kroe, R.R.; Laue, T.M. NUTS and BOLTS: Applications of fluorescence-detected sedimentation. Anal. Biochem. 2009, 390, 1–13. [Google Scholar] [CrossRef]
- MacGregor, I.K.; Anderson, A.L.; Laue, T.M. Fluorescence detection for the XLI analytical ultracentrifuge. Biophys. Chem. 2004, 108, 165–185. [Google Scholar] [CrossRef]
- Scherer, T.; Kanai, S.; Liu, J.; Shire, S. Characterization of monoclonal antibodies at high concentrations by light scattering. In Proceedings of AAPS National Biotechnology Conference, San Diego, CA, USA, 2007.
- Ross, P.D.; Minton, A.P. Analysis of non-ideal behavior in concentrated hemoglobin solutions. J. Mol. Biol. 1977, 112, 437–452. [Google Scholar] [CrossRef]
- Yousef, M.A.; Datta, R.; Rodgers, V.G.J. Free-Solvent Model of Osmotic Pressure Revisited: Application to Concentrated IgG Solution under Physiological Conditions. J. Colloid. Interface Sci. 1998, 197, 108–118. [Google Scholar] [CrossRef]
- Paliwal, A.; Asthagiri, D.; Abras, D.; Lenhoff, A.M.; Paulaitis, M.E. Light-scattering studies of protein solutions: Role of hydration in weak protein-protein interactions. Biophys. J. 2005, 89, 1564–1573. [Google Scholar] [CrossRef]
- Neal, B.L.; Asthagiri, D.; Lenhoff, A.M. Molecular origins of osmotic second virial coefficients of proteins. Biophys. J. 1998, 75, 2469–2477. [Google Scholar] [CrossRef]
- Salinas, B.A.; Sathish, H.A.; Bishop, S.M.; Harn, N.; Carpenter, J.F.; Randolph, T.W. Understanding and modulating opalescence and viscosity in a monoclonal antibody formulation. J. Pharm. Sci. 2010, 99, 82–93. [Google Scholar] [CrossRef]
- Saito, S.; Hasegawa, J.; Kobayashi, N.; Kishi, N.; Uchiyama, S.; Fukui, K. Behavior of monoclonal antibodies: Relation between the second virial coefficient (B (2)) at low concentrations and aggregation propensity and viscosity at high concentrations. Pharm. Res. 2012, 29, 397–410. [Google Scholar] [CrossRef]
- Attri, A.K.; Minton, A.P. New methods for measuring macromolecular interactions in solution via static light scattering: Basic methodology and application to nonassociating and self-associating proteins. Anal. Biochem. 2005, 337, 103–110. [Google Scholar] [CrossRef]
- Alford, J.R.; Kendrick, B.S.; Carpenter, J.F.; Randolph, T.W. Measurement of the second osmotic virial coefficient for protein solutions exhibiting monomer-dimer equilibrium. Anal. Biochem. 2008, 377, 128–133. [Google Scholar] [CrossRef]
- Some, D.; Kenrick, S. Characterization of protein-protein interactions via static and dynamic light scattering, in rotein interaction. In Protein Interactions; Cai, J., Ed.; InTech.: Rijeka, Croatia, 2012. [Google Scholar]
- Le Brun, V.; Friess, W.; Bassarab, S.; Garidel, P. Correlation of protein-protein interactions as assessed by affinity chromatography with colloidal protein stability: A case study with lysozyme. Pharm. Dev. Technol. 2010, 15, 421–430. [Google Scholar] [CrossRef]
- Le Brun, V.; Friess, W.; Bassarab, S.; Mühlau, S.; Garidel, P. A critical evaluation of self-interaction chromatography as a predictive tool for the assessment of protein-protein interactions in protein formulation development: A case study of a therapeutic monoclonal antibody. Eur. J. Pharm. Biopharm. 2010, 75, 16–25. [Google Scholar] [CrossRef]
- Deszczynski, M.; Harding, S.E.; Winzor, D.J. Negative second virial coefficients as predictors of protein crystal growth: Evidence from sedimentation equilibrium studies that refutes the designation of those light scattering parameters as osmotic virial coefficients. Biophys. Chem. 2006, 120, 106–113. [Google Scholar] [CrossRef]
- Tessier, P.M.; Lenhoff, A.M.; Sandler, S.I. Rapid measurement of protein osmotic second virial coefficients by self-interaction chromatography. Biophys. J. 2002, 82, 1620–1631. [Google Scholar] [CrossRef]
- Saluja, A.; Matthew Fesinmeyer, R.; Hogan, S.; Brems, D.N. Diffusion and sedimentation interaction parameters for measuring the second virial coefficient and their utility as predictors of protein aggregation. Biophys. J. 2010, 99, 2657–2665. [Google Scholar] [CrossRef]
- Harding, S.E.; Johnson, P. The concentration-dependence of macromolecular parameters. Biochem. J. 1985, 231, 543–547. [Google Scholar]
- Connolly, B.D.; Petry, C.; Yadav, S.; Demeule, B.; Ciaccio, N.; Moore, J.M.R.; Shire, S.J.; Gokarn, Y.R. Weak interactions govern the viscosity of concentrated antibody solutions: High-throughput analysis using the diffusion interaction parameter. Biophys. J. 2012, 103, 69–78. [Google Scholar] [CrossRef]
- Winzor, D.J.; Jones, S.; Harding, S.E. Determination of protein charge by capillary zone electrophoresis. Anal. Biochem. 2004, 333, 225–229. [Google Scholar] [CrossRef]
- Durant, J.A.; Chen, C.; Laue, T.M.; Moody, T.P.; Allison, S.A. Use of T4 lysozyme charge mutants to examine electrophoretic models. Biophys. Chem. 2002, 101–102, 593–609. [Google Scholar] [CrossRef]
- WYATT. Available online: www.wyatt.com (accessed on 1 June 2013).
- Minton, A.P. Molecular crowding: Analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 1998, 295, 127–149. [Google Scholar] [CrossRef]
- Janthur, W.-D.; Cantoni, N.; Mamot, C. Drug Conjugates Such as Antibody Drug Conjugates (ADCs), Immunotoxins and Immunoliposomes Challenge Daily Clinical Practice. Int. J. Mol. Sci. 2012, 13, 16020–16045. [Google Scholar] [CrossRef]
- Adair, J.R.; Howard, P.W.; Hartley, J.A.; Williams, D.G.; Chester, K.A. Antibody-drug conjugates—A perfect synergy. Expert Opin. Biol. Ther. 2012, 12, 1191–1206. [Google Scholar] [CrossRef]
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef]
- Sutherland, M.S.; Sanderson, R.J.; Gordon, K.A.; Andreyka, J.; Cerveny, C.G.; Yu, C.; Lewis, T.S.; Meyer, D.L.; Zabinski, R.F.; Doronina, S.O.; et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J. Biol. Chem. 2006, 281, 10540–10547. [Google Scholar] [CrossRef]
- Okeley, N.M.; Miyamoto, J.B.; Zhang, X.; Sanderson, R.J.; Benjamin, D.R.; Sievers, E.L.; Senter, P.D.; Alley, S.C. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 2010, 16, 888–897. [Google Scholar] [CrossRef]
- Lam, K.C.L.; Rajaraman, G. Assessment of P-glycoprotein substrate and inhibition potential of test compounds in MDR1-transfected MDCK cells. Curr. Protoc. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Krech, T.; Scheuerera, E.; Geffers, R.; Kreipe, H.; Lehmann, U.; Christgen, M. ABCB1/MDR1 contributes to the anticancer drug-resistant phenotype of IPH-926 human lobular breast cancer cells. Cancer Lett. 2012, 315, 153–160. [Google Scholar] [CrossRef]
- Bidwell, G.L., 3rd; Davis, A.N.; Fokt, I.; Priebe, W.; Raucher, D. A thermally targeted elastin-like polypeptide-doxorubicin conjugate overcomes drug resistance. Invest. New Drugs 2007, 25, 313–326. [Google Scholar] [CrossRef]
- Hughes, B. Antibody-drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef]
- Barginear, M.F.; John, V.; Budman, D.R. Trastuzumab-DM1: A clinical update of the novel antibody-drug conjugate for HER2-overexpressing breast cancer. Mol. Med. 2013, 18, 1473–1479. [Google Scholar]
- Rowland, G.F.; O'Neill, G.J.; Davies, D.A. Suppression of tumour growth in mice by a drug-antibody conjugate using a novel approach to linkage. Nature 1975, 255, 487–488. [Google Scholar] [CrossRef]
- Beck, A.; Lambert, J.; Sun, M.; Lin, K. Fourth World Antibody-Drug Conjugate Summit: February 29-March 1, 2012, Frankfurt, Germany. MAbs 2012, 4, 637–647. [Google Scholar] [CrossRef]
- Xie, H.; Audette, C.; Hoffee, M.; Lambert, J.M.; Blättler, W.A. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J. Pharmacol. Exp. Ther. 2004, 308, 1073–1082. [Google Scholar]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins (Basel) 2011, 3, 848–883. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef]
- Le, L.N.; Moore, J.M.R.; Ouyang, J.; Chen, X.; Nguyen, M.D.H.; Galush, W.J. Profiling antibody drug conjugate positional isomers: A system-of-equations approach. Anal. Chem. 2012, 84, 7479–7486. [Google Scholar] [CrossRef]
- Liu, H.; Chumsae, C.; Gaza-Bulseco, G.; Hurkmans, K.; Radziejewski, C.H. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis. Anal. Chem. 2010, 82, 5219–5226. [Google Scholar] [CrossRef]
- Sun, M.M.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.M.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug. Chem. 2005, 16, 1282–1290. [Google Scholar] [CrossRef]
- Dornan, D.; Bennett, F.; Chen, Y.; Dennis, M.; Eaton, D.; Elkins, K.; French, D.; Go, M.A.T.; Jack, A.; Junutula, J.R.; et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood 2009, 114, 2721–2729. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchinsa, B.M.; Kima, C.H.; Kazane, S.A. ; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar]
- Presentini, R.; Terrana, B. Influence of the antibody-peroxidase coupling methods on the conjugate stability and on the methodologies for the preservation of the activity in time. J. Immunoassay 1995, 16, 309–324. [Google Scholar] [CrossRef]
- Fishkin, N.; Maloney, E.K.; Chari, R.V.J.; Singh, R. A novel pathway for maytansinoid release from thioether linked antibody-drug conjugates (ADCs) under oxidative conditions. Chem. Commun. (Camb) 2011, 47, 10752–10754. [Google Scholar]
- Ryan, C.P.; Smith, M.E.B.; Schumacher, F.F.; Grohmann, D.; Papaioannou, D.; Waksman, G.; Werner, F.; Baker, J.R.; Caddick, S. Tunable reagents for multi-functional bioconjugation: Reversible or permanent chemical modification of proteins and peptides by control of maleimide hydrolysis. Chem. Commun. (Camb) 2011, 47, 5452–5454. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Radia, S.; Mastalerz, H.; Walker, M.A.; Firestone, R.A.; Dalton King, H.; Hofstead, S.J.; Willner, D.; Lasch, S.J.; Trail, P.A. Doxorubicin immunoconjugates containing bivalent, lysosomally-cleavable dipeptide linkages. Bioorg. Med. Chem. Lett. 2002, 12, 1529–1532. [Google Scholar] [CrossRef]
- Sapra, P.; Damelin, M.; DiJoseph, J.; Marquette, K.; Geles, K.G.; Golas, J.; Dougher, M.; Narayanan, B.; Giannakou, A.; Khandke, K.; et al. Long-term tumor regression induced by an antibody-drug conjugate that targets 5T4, an oncofetal antigen expressed on tumor-initiating cells. Mol. Cancer Ther. 2013, 12, 38–47. [Google Scholar]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Michael Elliott, J.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin's lymphoma: Target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef]
- Oflazoglu, E.; Stone, I.J.; Gordon, K.; Wood, C.G.; Repasky, E.A.; Grewal, I.S.; Law, C.-L.; Gerber, H.-P. Potent anticarcinoma activity of the humanized anti-CD70 antibody h1F6 conjugated to the tubulin inhibitor auristatin via an uncleavable linker. Clin. Cancer Res. 2008, 14, 6171–6180. [Google Scholar] [CrossRef]
- Hollander, I.; Kunz, A.; Hamann, P.R. Selection of reaction additives used in the preparation of monomeric antibody-calicheamicin conjugates. Bioconjug. Chem. 2008, 19, 358–361. [Google Scholar] [CrossRef]
- Wakankar, A.A.; Feeney, M.B.; Rivera, J.; Chen, Y.; Kim, M.; Sharma, V.K.; John Wang, Y. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: Changes due to modification and conjugation processes. Bioconjug. Chem. 2010, 21, 1588–1595. [Google Scholar] [CrossRef]
- Quiles, S.; Raisch, K.P.; Sanford, L.L.; Bonner, J.A.; Safavy, A. Synthesis and preliminary biological evaluation of high-drug-load paclitaxel-antibody conjugates for tumor-targeted chemotherapy. J. Med. Chem. 2010, 53, 586–594. [Google Scholar] [CrossRef]
- King, H.D.; Yurgaitis, D.; Willner, D.; Firestone, R.A.; Yang, M.B.; Lasch, S.J.; Hellström, K.E.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: Inhibition of aggregation by methoxytriethyleneglycol chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar] [CrossRef]
- Kafi, K.; Betting, D.J.; Yamada, R.E.; Bacica, M.; Steward, K.K.; Timmerman, J.M. Maleimide conjugation markedly enhances the immunogenicity of both human and murine idiotype-KLH vaccines. Mol. Immunol. 2009, 46, 448–456. [Google Scholar] [CrossRef]
- Christie, R.J.; Anderson, D.J.; Grainger, D.W. Comparison of hydrazone heterobifunctional cross-linking agents for reversible conjugation of thiol-containing chemistry. Bioconjug. Chem. 2010, 21, 1779–1787. [Google Scholar] [CrossRef]
- Bagshawe, K.D. Targeting: The ADEPT story so far. Curr. Drug Targets 2009, 10, 152–157. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Porta, C.; Mutti, L. New agents in the management of advanced mesothelioma. Semin. Oncol. 2005, 32, 336–350. [Google Scholar] [CrossRef]
- Pavlakis, N.; Vogelzang, N.J. Ranpirnase—An antitumour ribonuclease: Its potential role in malignant mesothelioma. Expert Opin. Biol. Ther. 2006, 6, 391–399. [Google Scholar] [CrossRef]
- Rosenblum, M.G.; Barth, S. Development of novel, highly cytotoxic fusion constructs containing granzyme B: Unique mechanisms and functions. Curr. Pharm. Des. 2009, 15, 2676–2692. [Google Scholar] [CrossRef]
- Kurschus, F.C.; Kleinschmidt, M.; Fellows, E.; Dornmair, K.; Rudolph, R.; Lilie, H.; Jenne, D.E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004, 562, 87–92. [Google Scholar] [CrossRef]
- Weidle, U.H.; Georges, G.; Brinkmann, U. Fully human targeted cytotoxic fusion proteins: New anticancer agents on the horizon. Cancer Genomics Proteomics 2012, 9, 119–133. [Google Scholar]
- Larrick, J.W.; Cresswell, P. Modulation of cell surface iron transferrin receptors by cellular density and state of activation. J. Supramol. Struct. 1979, 11, 579–586. [Google Scholar] [CrossRef]
- Kordower, J.H.; Charles, V.; Bayer, R.; Bartus, R.T.; Putney, S.; Walus, L.R.; Friden, P.M. Intravenous administration of a transferrin receptor antibody-nerve growth factor conjugate prevents the degeneration of cholinergic striatal neurons in a model of Huntington disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9077–9080. [Google Scholar] [CrossRef]
- Granholm, A.C.; Albeck, D.; Bäckman, C.; Curtis, M.; Ebendal, T.; Friden, P.; Henry, M.; Hoffer, B.; Kordower, J.; Rose, G.M.; et al. A non-invasive system for delivering neural growth factors across the blood-brain barrier: A review. Rev. Neurosci. 1998, 9, 31–55. [Google Scholar]
- McGrath, J.P.; Cao, X.; Schutz, A.; Lynch, P.; Ebendal, T.; Josephina Coloma, M.; Morrison, S.L.; Putney, S.D. Bifunctional fusion between nerve growth factor and a transferrin receptor antibody. J. Neurosci. Res. 1997, 47, 123–133. [Google Scholar] [CrossRef]
- Penichet, M.L.; Kang, Y.-S.; Pardridge, W.M.; Morrison, S.L.; Shin, S.-U. An antibody-avidin fusion protein specific for the transferrin receptor serves as a delivery vehicle for effective brain targeting: Initial applications in anti-HIV antisense drug delivery to the brain. J. Immunol. 1999, 163, 4421–4426. [Google Scholar]
- Ng, P.P.; Dela Cruz, J.S.; Sorour, D.N.; Stinebaugh, J.M.; Shin, S.-U.; Shin, D.S.; Morrison, S.L.; Penichet, M.L. An anti-transferrin receptor-avidin fusion protein exhibits both strong proapoptotic activity and the ability to deliver various molecules into cancer cells. Proc. Natl. Acad. Sci. USA 2002, 99, 10706–10711. [Google Scholar] [CrossRef]
- Xuan, C.; Steward, K.K.; Timmerman, J.M.; Morrison, S.L. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood 2010, 115, 2864–2871. [Google Scholar] [CrossRef]
- Koehn, T.A.; Trimble, L.L.; Alderson, K.L.; Erbe, A.K.; McDowell, K.A.; Grzywacz, B.; Hank, J.A.; Sondel, P.M. Increasing the clinical efficacy of NK and antibody-mediated cancer immunotherapy: Potential predictors of successful clinical outcome based on observations in high-risk neuroblastoma. Front. Pharmacol. 2012, 3, 91. [Google Scholar]
- Lode, H.N.; Xiang, R.; Becker, J.C.; Gillies, S.D.; Reisfel, R.A. Immunocytokines: A promising approach to cancer immunotherapy. Pharmacol. Ther. 1998, 80, 277–292. [Google Scholar] [CrossRef]
- Johnson, E.; Dean, S.M.; Sondel, P.M. Antibody-based immunotherapy in high-risk neuroblastoma. Expert Rev. Mol. Med. 2007, 9, 1–21. [Google Scholar]
- Dela Cruz, J.S.; Ryan Trinh, K.; Morrison, S.L.; Penichet, M.L. Recombinant anti-human HER2/neu IgG3-(GM-CSF) fusion protein retains antigen specificity and cytokine function and demonstrates antitumor activity. J. Immunol. 2000, 165, 5112–5121. [Google Scholar]
- Cho, H.M.; Rosenblatt, J.D.; Kang, Y.-S.; Luisa Iruela-Arispe, M.; Morrison, S.L.; Penichet, M.L.; Kwon, Y.-G.; Kim, T.-W.; Webster, K.A.; Nechustan, H. Enhanced inhibition of murine tumor and human breast tumor xenografts using targeted delivery of an antibody-endostatin fusion protein. Mol. Cancer Ther. 2005, 4, 956–967. [Google Scholar]
- Zhang, H.; Lu, S.; Morrison, S.L.; Tomlinson, S. Targeting of functional antibody-decay-accelerating factor fusion proteins to a cell surface. J. Biol. Chem. 2001, 276, 27290–27295. [Google Scholar] [CrossRef]
- Fung, V.P. Method for producing recombinant proteins. US Patent 7,294,481, 2007. [Google Scholar]
- Sassenfeld, H.M.; Remmele, R.L., Jr.; McCoy, R.E. Increased recovery of active proteins. US Patent 7,157,55, 2007. [Google Scholar]
- Bouchon, A.; Facchetti, F.; Weigand, M.A.; Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001, 410, 1103–1107. [Google Scholar] [CrossRef]
- Iwai, H.; Kohsaka, H. Blockade of Triggering receptor expressed on myeloid cells-1 as a new therapy of arthritis. Nihon Rinsho Meneki Gakkai Kaishi 2012, 35, 81–86. [Google Scholar] [CrossRef]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef]
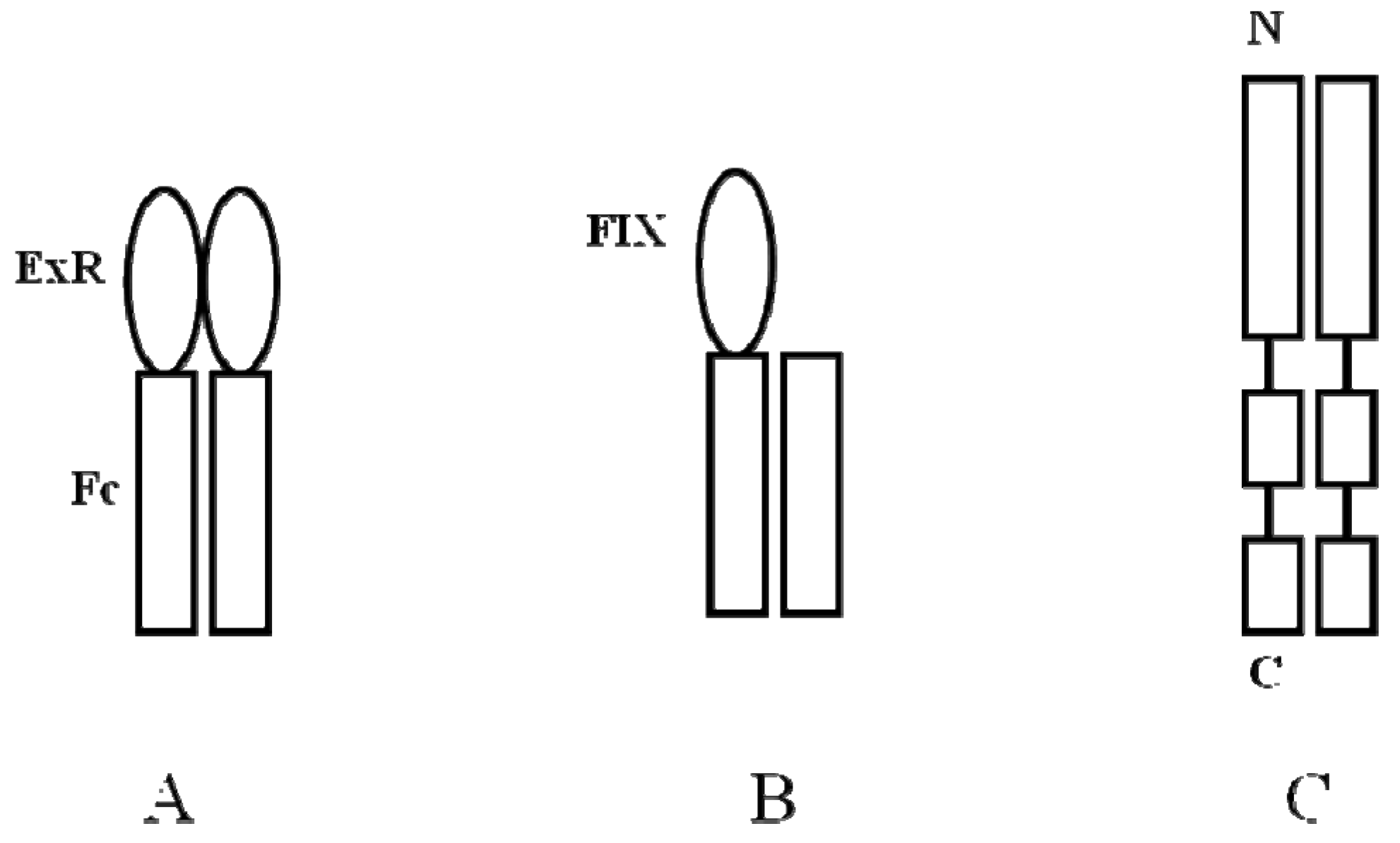
- Peters, R.T.; Low, S.C.; Kamphaus, G.D.; Dumont, J.A.; Amari, J.V.; Lu, Q.; Zarbis-Papastoitsis, G.; Reidy, T.J.; Merricks, E.P.; Nichols, T.C.; et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood 2010, 115, 2057–2064. [Google Scholar] [CrossRef]
- Hall, M.P.; Gegg, C.; Walker, K.; Spahr, C.; Ortiz, R.; Patel, V.; Yu, S.; Zhang, L.; Lu, H.; DeSilva, B.; et al. Ligand-binding mass spectrometry to study biotransformation of fusion protein drugs and guide immunoassay development: Strategic approach and application to peptibodies targeting the thrombopoietin receptor. AAPS J. 2010, 12, 576–585. [Google Scholar] [CrossRef]
- Gokarn, Y.R.; Matthew Fesinmeyer, R.; Saluja, A.; Cao, S.; Dankberg, J.; Goetze, A.; Remmele, R.L., Jr.; Narhi, L.O.; Brems, D.N. Ion-specific modulation of protein interactions: Anion-induced, reversible oligomerization of a fusion protein. Protein Sci. 2009, 18, 169–179. [Google Scholar]
- Nisonoff, A.; Rivers, M.M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef]
- May, C.; Sapra, P.; Gerber, H.P. Advances in bispecific biotherapeutics for the treatment of cancer. Biochem. Pharmacol. 2012, 84, 1105–1112. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Vallera, D.A.; Todhunter, D.A.; Kuroki, D.W.; Shu, Y.; Sicheneder, A.; Chen, H. A bispecific recombinant immunotoxin, DT2219, targeting human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma. Clin. Cancer Res. 2005, 11, 3879–3888. [Google Scholar] [CrossRef]
- Dorvillius, M.; Garambois, V.; Pourquier, D.; Gutowski, M.; Rouanet, P.; Mani, J.-C.; Pugnière, M.; Hynes, N.E.; Pèlegrin, A. Targeting of human breast cancer by a bispecific antibody directed against two tumour-associated antigens: ErbB-2 and carcinoembryonic antigen. Tumour Biol. 2002, 23, 337–347. [Google Scholar] [CrossRef]
- Jimenez, X.; Lu, D.; Brennan, L.; Persaud, K.; Liu, M.; Miao, H.; Witte, L.; Zhu, Z. A recombinant, fully human, bispecific antibody neutralizes the biological activities mediated by both vascular endothelial growth factor receptors 2 and 3. Mol. Cancer Ther. 2005, 4, 427–434. [Google Scholar]
- Lu, D.; Zhang, H.; Koo, H.; Tonra, J.; Balderes, P.; Prewett, M.; Corcoran, E.; Mangalampalli, V.; Bassi, R.; Anselma, D.; et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J. Biol. Chem. 2005, 280, 19665–19672. [Google Scholar] [CrossRef]
- Tanaka, K.; Isselbacher, K.J.; Khoury, G.; Jay, G. Reversal of oncogenesis by the expression of a major histocompatibility complex class I gene. Science 1985, 228, 26–30. [Google Scholar]
- Hartmann, F.; Renner, C.; Jung, W.; Deisting, C.; Juwana, M.; Eichentopf, B.; Kloft, M.; Pfreundschuh, M. Treatment of refractory Hodgkin's disease with an anti-CD16/CD30 bispecific antibody. Blood 1997, 89, 2042–2047. [Google Scholar]
- Taylor, R.P.; Sutherland, W.M.; Martin, E.N.; Ferguson, P.J.; Reinagel, M.L.; Gilbert, E.; Lopez, K.; Incardona, N.L.; Ochs, H.D. Bispecific monoclonal antibody complexes bound to primate erythrocyte complement receptor 1 facilitate virus clearance in a monkey model. J. Immunol. 1997, 158, 842–850. [Google Scholar]
- Reinagel, M.L.; Taylor, R.P. Transfer of immune complexes from erythrocyte CR1 to mouse macrophages. J. Immunol. 2000, 164, 1977–1985. [Google Scholar]
- French, R.R.; Penney, C.A.; Browning, A.C.; Stirpe, F.; George, A.J.; Glennie, M.J. Delivery of the ribosome-inactivating protein, gelonin, to lymphoma cells via CD22 and CD38 using bispecific antibodies. Br. J. Cancer 1995, 71, 986–994. [Google Scholar] [CrossRef]
- Ford, C.H.; Osborne, P.A.; Rego, B.G.; Mathew, A. Bispecific antibody targeting of doxorubicin to carcinoembryonic antigen-expressing colon cancer cell lines in vitro and in vivo. Int. J. Cancer 2001, 92, 851–855. [Google Scholar] [CrossRef]
- Zhu, H.; Jain, R.K.; Baxter, L.T. Tumor pretargeting for radioimmunodetection and radioimmunotherapy. J. Nucl. Med. 1998, 39, 65–76. [Google Scholar]
- Kipriyanov, S.M.; Le Gall, F. Recent advances in the generation of bispecific antibodies for tumor immunotherapy. Curr. Opin. Drug Discov. Develop. 2004, 7, 233–242. [Google Scholar]
- Mack, M.; Riethmuller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef]
- Mack, M.; Gruber, R.; Schmidt, S.; Riethmüller, G.; Kufer, P. Biologic properties of a bispecific single-chain antibody directed against 17-1A (EpCAM) and CD3: Tumor cell-dependent T cell stimulation and cytotoxic activity. J. Immunol. 1997, 158, 3965–39670. [Google Scholar]
- Milstein, C.; Cuello, A.C. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef]
- Staerz, U.D.; Bevan, M.J. Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc. Natl. Acad. Sci. USA 1986, 83, 1453–1457. [Google Scholar] [CrossRef]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar]
- Brennan, M.; Davison, P.F.; Paulus, H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 1985, 229, 81–83. [Google Scholar]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Ridgway, J.B.; Presta, L.G.; Carter, P. 'Knobs-into-holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.-M.; Huston, J.S. SEEDbodies: Fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 2010, 23, 195–202. [Google Scholar] [CrossRef]
- Holliger, P.; Prospero, T.; Winter, G. "Diabodies": Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; Von der Lieth, C.-W.; Ronald Matys, E.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef]
- De Jonge, J.; Heirman, C.; de Veerman, M.; Van Meirvenne, S.; Moser, M.; Leo, O.; Thielemans, K. In vivo retargeting of T cell effector function by recombinant bispecific single chain Fv (anti-CD3 x anti-idiotype) induces long-term survival in the murine BCL1 lymphoma model. J. Immunol. 1998, 161, 1454–1461. [Google Scholar]
- Kufer, P.; Lutterbuse, R.; Baeuerle, P.A. A revival of bispecific antibodies. Trends Biotechnol. 2004, 22, 238–244. [Google Scholar] [CrossRef]
- Topp, M.S.; Kufer, P.; Gökbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.-A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Jonah Rainey, G.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.-R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef]
- F-star. Available online: www.f-star.com (accessed on 1 June 2013).
- IMMUNOCORE. Available online: www.immunocore.com (accessed on 1 June 2013).
- Hartmann, F.; Renner, C.; Jung, W.; da Costa, L.; Tembrink, S.; Held, G.; Sek, A.; König, J.; Bauer, S.; Kloft, M.; Pfreundschuh, M. Anti-CD16/CD30 bispecific antibody treatment for Hodgkin's disease: Role of infusion schedule and costimulation with cytokines. Clin. Cancer Res. 2001, 7, 1873–1881. [Google Scholar]
- Schmitt, M.; Schmitt, A.; Reinhardt, P.; Thess, B.; Manfras, B.; Lindhofer, H.; Riechelmann, H.; Wiesneth, M.; Gronau, S. Opsonization with a trifunctional bispecific (alphaCD3 x alphaEpCAM) antibody results in efficient lysis in vitro and in vivo of EpCAM positive tumor cells by cytotoxic T lymphocytes. Int. J. Oncol. 2004, 25, 841–848. [Google Scholar]
- Jager, M.; Schoberth, A.; Ruf, P.; Hess, J.; Hennig, M.; Schmalfeldt, B.; Wimberger, P.; Ströhlein, M.; Theissen, B.; Heiss, M.M.; et al. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res. 2012, 72, 24–32. [Google Scholar] [CrossRef]
- Pluen, A.; Boucher, Y.; Ramanujan, S.; McKee, T.D.; Gohongi, T.; di Tomaso, E.; Brown, E.B.; Izumi, Y.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar] [CrossRef]
- Khawli, L.A.; Biela, B.; Hu, P.; Epstein, A.L. Comparison of recombinant derivatives of chimeric TNT-3 antibody for the radioimaging of solid tumors. Hybrid. Hybridomics 2003, 22, 1–9. [Google Scholar] [CrossRef]
- Tahtis, K.; Lee, F.-T.; Smyth, F.E.; Power, B.E.; Renner, C.; Brechbiel, M.W.; Old, L.J.; Hudson, P.J.; Scott, A.M. Biodistribution properties of (111)indium-labeled C-functionalized trans-cyclohexyl diethylenetriaminepentaacetic acid humanized 3S193 diabody and F(ab')(2) constructs in a breast carcinoma xenograft model. Clin. Cancer Res. 2001, 7, 1061–1072. [Google Scholar]
- Thurber, G.M.; Wittrup, K.D. Quantitative spatiotemporal analysis of antibody fragment diffusion and endocytic consumption in tumor spheroids. Cancer Res. 2008, 68, 3334–3341. [Google Scholar] [CrossRef]
- Ottiger, M.; Thiel, M.A.; Feige, U.; Lichtlen, P.; Urech, D.M. Efficient intraocular penetration of topical anti-TNF-alpha single-chain antibody (ESBA105) to anterior and posterior segment without penetration enhancer. Invest. Ophthalmol. Vis. Sci. 2009, 50, 779–786. [Google Scholar]
- Cardoso, R.M.; Zwick, M.B.; Stanfield, R.L.; Kunert, R.; Binley, J.M.; Katinger, H.; Burton, D.R. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 2005, 22, 163–173. [Google Scholar] [CrossRef]
- Stijlemans, B.; Wyns, L.; Senter, P.; Revets, H.; De Baetselier, P.; Muyldermans, S.; Magez, S. Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies. African trypanosomes as paradigm. J. Biol. Chem. 2004, 279, 1256–1261. [Google Scholar]
- Nuttall, S.D.; Humberstone, K.S.; Krishnan, U.V.; Carmichael, J.A.; Doughty, L.; Hattarki, M.; Coley, A.M.; Casey, J.L.; Anders, R.F.; Foley, M.; et al. Selection and affinity maturation of IgNAR variable domains targeting Plasmodium falciparum AMA1. Proteins 2004, 55, 187–197. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Varghese, J.N.; Carmichael, J.A.; Irving, R.A.; Hudson, P.J.; Nuttall, S.D. Structural evidence for evolution of shark Ig new antigen receptor variable domain antibodies from a cell-surface receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 12444–12449. [Google Scholar]
- Choudhry, V.; Zhang, M.-Y.; Dimitrova, D.; Prabakaran, P.; Dimitrov, A.S.; Fouts, T.R.; Dimitrov, D.S. Antibody-based inhibitors of HIV infection. Expert Opin. Biol. Ther. 2006, 6, 523–531. [Google Scholar] [CrossRef]
- Chen, W.; Dimitrov, D.S. Human monoclonal antibodies and engineered antibody domains as HIV-1 entry inhibitors. Curr. Opin. HIV AIDS 2009, 4, 112–117. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Albrecht, H.; Burke, P.A.; Natarajan, A.; Xiong, C.-Y.; Kalicinsky, M.; DeNardo, G.L.; DeNardo, S.J. Production of soluble ScFvs with C-terminal-free thiol for site-specific conjugation or stable dimeric ScFvs on demand. Bioconjug. Chem. 2004, 15, 16–26. [Google Scholar] [CrossRef]
- Lu, D.; Jimenez, X.; Zhanga, H.; Atkins, A.; Brennanb, L.; Balderes, P.; Bohlend, P.; Witte, L.; Zhu, Z. Di-diabody: A novel tetravalent bispecific antibody molecule by design. J. Immunol. Methods 2003, 279, 219–232. [Google Scholar] [CrossRef]
- Robinson, M.K.; Doss, M.; Shaller, C.; Narayanan, D.; Marks, J.D.; Adler, L.P.; González Trotter, D.E.; Adams, G.P. Quantitative immuno-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res. 2005, 65, 1471–1478. [Google Scholar] [CrossRef]
- Olafsen, T.; Cheung, C.-W.; Yazaki, P.J.; Li, L.; Sundaresan, G.; Gambhir, S.S.; Sherman, M.A.; Williams, L.E.; Shively, J.E.; Raubitschek, A.A.; et al. Covalent disulfide-linked anti-CEA diabody allows site-specific conjugation and radiolabeling for tumor targeting applications. Protein Eng. Des. Sel. 2004, 17, 21–27. [Google Scholar] [CrossRef]
- Hu, S.; Shively, L.; Raubitschek, A.; Sherman, M.; Williams, L.E.; Wong, J.Y.C.; Shively, J.E.; Wu, A.M. Minibody: A novel engineered anti-carcinoembryonic antigen antibody fragment (single-chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996, 56, 3055–3061. [Google Scholar]
- Nakamura, T.; Peng, K.-W.; Vongpunsawad, S.; Harvey, M.; Mizuguchi, H.; Hayakawa, T.; Cattaneo, R.; Russell, S.J. Antibody-targeted cell fusion. Nat. Biotechnol. 2004, 22, 331–336. [Google Scholar] [CrossRef]
- Brignole, C.; Pastorino, F.; Marimpietri, D.; Pagnan, G.; Pistorio, A.; Allen, T.M.; Pistoia, V.; Ponzoni, M. Immune cell-mediated antitumor activities of GD2-targeted liposomal c-myb antisense oligonucleotides containing CpG motifs. J. Natl. Cancer Inst. 2004, 96, 1171–1180. [Google Scholar] [CrossRef]
- van Broekhoven, C.L.; Parish, C.R.; Demange, C.; Britton, W.J.; Altin, J.G. Targeting dendritic cells with antigen-containing liposomes: A highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 2004, 64, 4357–4365. [Google Scholar] [CrossRef]
- Sharma, S.K.; Barbara Pedley, R.; Bhatia, J.; Boxer, G.M.; El-Emir, E.; Qureshi, U.; Tolner, B.; Lowe, H.; Paul Michael, N.; Minton, N.; et al. Sustained tumor regression of human colorectal cancer xenografts using a multifunctional mannosylated fusion protein in antibody-directed enzyme prodrug therapy. Clin. Cancer Res. 2005, 11, 814–825. [Google Scholar]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef]
- Holt, L.J.; Herring, C.; Jespers, L.S.; Woolven, B.P.; Tomlinson, I.M. Domain antibodies: Proteins for therapy. Trends Biotechnol. 2003, 21, 484–490. [Google Scholar] [CrossRef]
- Ryckaert, S.; Pardon, E.; Steyaert, J.; Callewaert, N. Isolation of antigen-binding camelid heavy chain antibody fragments (nanobodies) from an immune library displayed on the surface of Pichia pastoris. J. Biotechnol. 2010, 145, 93–98. [Google Scholar] [CrossRef]
- Muyldermans, S.; Baral, T.N.; Cortez Retamozzo, V.; De Baetselier, P.; De Genst, E.; Kinnec, J.; Leonhardt, H.; Magez, S.; Nguyen, V.K.; Revets, H.; et al. Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 2009, 128, 178–183. [Google Scholar] [CrossRef]
- Vandenbroucke, K.; de Haard, H.; Beirnaert, E.; Dreier, T.; Lauwereys, M.; Huyck, L.; Van Huysse, J.; Demetter, P.; Steidler, L.; Remaut, E.; et al. Orally administered L. lactis secreting an anti-TNF Nanobody demonstrate efficacy in chronic colitis. Mucosal. Immunol. 2010, 3, 49–56. [Google Scholar] [CrossRef]
- Sanz, L.; Blanco, B.; Alvarez-Vallina, L. Antibodies and gene therapy: Teaching old 'magic bullets' new tricks. Trends Immunol. 2004, 25, 85–91. [Google Scholar] [CrossRef]
- Afanasieva, T.A.; Wittmer, M.; Vitaliti, A.; Ajmo, M.; Neri, D.; Klemenz, R. Single-chain antibody and its derivatives directed against vascular endothelial growth factor: Application for antiangiogenic gene therapy. Gene Ther. 2003, 10, 1850–1859. [Google Scholar] [CrossRef]
- Blanco, B.; Holliger, P.; Vile, R.G.; Álvarez-Vallina, L. Induction of human T lymphocyte cytotoxicity and inhibition of tumor growth by tumor-specific diabody-based molecules secreted from gene-modified bystander cells. J. Immunol. 2003, 171, 1070–1077. [Google Scholar]
- Jendreyko, N.; Popkov, M.; Beerli, R.R.; Chung, J.; McGavern, D.B.; Rader, C.; Barbas, C.F., III. Intradiabodies, bispecific, tetravalent antibodies for the simultaneous functional knockout of two cell surface receptors. J. Biol. Chem. 2003, 278, 47812–47819. [Google Scholar]
- Grosse-Hovest, L.; Wick, W.; Minoia, R.; Weller, M.; Rammensee, H.-G.; Brem, G.; Jung, G. Supraagonistic, bispecific single-chain antibody purified from the serum of cloned, transgenic cows induces T-cell-mediated killing of glioblastoma cells in vitro and in vivo. Int. J. Cancer 2005, 117, 1060–1064. [Google Scholar] [CrossRef]
- Chambers, R.S. High-throughput antibody production. Curr. Opin. Chem. Biol. 2005, 9, 46–50. [Google Scholar] [CrossRef]
- McKeating, J.A.; Gow, J.; Goudsmit, J.; Pearl, L.H.; Mulder, C.; Weiss, R.A. Characterization of HIV-1 neutralization escape mutants. AIDS 1989, 3, 777–784. [Google Scholar] [CrossRef]
- Keck, Z.Y.; Li, S.H.; Xia, J.; von Hahn, T.; Balfe, P.; McKeating, J.A.; Witteveldt, J.; Patel, A.H.; Alter, H.; Rice, C.M.; et al. Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J. Virol. 2009, 83, 6149–6160. [Google Scholar] [CrossRef]
- Zharikova, D.; Mozdzanowska, K.; Feng, J.; Zhang, M.; Gerhard, W. Influenza type A virus escape mutants emerge in vivo in the presence of antibodies to the ectodomain of matrix protein 2. J. Virol. 2005, 79, 6644–6654. [Google Scholar] [CrossRef]
- Prabakaran, M.; Prabhu, N.; He, F.; Qian, H.; Ho, H.-T.; Qiang, J.; Meng, T.; Goutama, M.; Kwang, J. Combination therapy using chimeric monoclonal antibodies protects mice from lethal H5N1 infection and prevents formation of escape mutants. PLoS One 2009, 4, e5672. [Google Scholar]
- Haurum, J.S. Recombinant human polyclonal antibodies: A new class of therapeutic antibodies against viral infections. Curr. Pharm. Des. 2006, 12, 2007–2015. [Google Scholar] [CrossRef]
- de Kruif, J.; Bakker, A.B.H.; Marissen, W.E.; Arjen Kramer, R.; Throsby, M.; Rupprecht, C.E.; Goudsmit, J. A human monoclonal antibody cocktail as a novel component of rabies postexposure prophylaxis. Annu. Rev. Med. 2007, 58, 359–368. [Google Scholar] [CrossRef]
- Elliott, E.V.; Pindar, A.; Stevenson, F.K.; Stevenson, G.T. Synergistic cytotoxic effects of antibodies directed against different cell surface determinants. Immunology 1978, 34, 405–409. [Google Scholar]
- Hellstrom, I.; Brown, J.P.; Hellstrom, K.E. Monoclonal antibodies to two determinants of melanoma-antigen p97 act synergistically in complement-dependent cytotoxicity. J. Immunol. 1981, 127, 157–160. [Google Scholar]
- Ziegler-Heitbrock, H.W.; Reiter, C.; Trenkmann, J.; Fütterer, A.; Riethmüller, G. Protection of mice against tetanus toxin by combination of two human monoclonal antibodies recognizing distinct epitopes on the toxin molecule. Hybridoma (Larchmt) 1986, 5, 21–31. [Google Scholar] [CrossRef]
- Bakker, A.B.; Python, C.; Kissling, C.J.; Pandyad, P.; Marissen, W.E.; Brinka, M.F.; Lagerwerf, F.; Worsta, S.; van Corven, E.; Kostense, S.; et al. First administration to humans of a monoclonal antibody cocktail against rabies virus: Safety, tolerability, and neutralizing activity. Vaccine 2008, 26, 5922–5927. [Google Scholar] [CrossRef]
- Mascola, J.R.; Louder, M.K.; VanCott, T.C.; Sapan, C.V.; Lambert, J.S.; Muenz, L.R.; Bunow, B.; Birx, D.L.; Robb, M.L. Potent and synergistic neutralization of human immunodeficiency virus (HIV) type 1 primary isolates by hyperimmune anti-HIV immunoglobulin combined with monoclonal antibodies 2F5 and 2G12. J. Virol. 1997, 71, 7198–7206. [Google Scholar]
- Armbruster, C.; Stiegler, G.M.; Vcelar, B.A.; Jäger, W.; Köller, U.; Jilch, R.; Ammann, C.G.; Pruenster, M.; Stoiber, H.; Katinger, H.W.D. Passive immunization with the anti-HIV-1 human monoclonal antibody (hMAb) 4E10 and the hMAb combination 4E10/2F5/2G12. J. Antimicrob. Chemother. 2004, 54, 915–920. [Google Scholar] [CrossRef]
- Trkola, A.; Kuster, H.; Rusert, P.; Joos, B.; Fischer, M.; Leemann, C.; Manrique, A.; Huber, M.; Rehr, M.; Oxenius, A.; et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat. Med. 2005, 11, 615–622. [Google Scholar] [CrossRef]
- Spiridon, C.I.; Ghetie, M.-A.; Uhr, J.; Marches, R.; Li, J.-L.; Shen, G.-L.; Vitetta, E.S. Targeting multiple Her-2 epitopes with monoclonal antibodies results in improved antigrowth activity of a human breast cancer cell line in vitro and in vivo. Clin. Cancer Res. 2002, 8, 1720–1730. [Google Scholar]
- Strauss, S.J.; Morschhauser, F.; Rech, J.; Repp, R.; Solal-Celigny, P.; Zinzani, P.L.; Engert, A.; Coiffier, B.; Hoelzer, D.F.; Wegener, W.A.; et al. Multicenter phase II trial of immunotherapy with the humanized anti-CD22 antibody, epratuzumab, in combination with rituximab, in refractory or recurrent non-Hodgkin's lymphoma. J. Clin. Oncol. 2006, 24, 3880–3886. [Google Scholar] [CrossRef]
- Nowakowski, A.; Wang, C.; Powers, D.B.; Amersdorfer, P.; Smith, T.J.; Montgomery, V.A.; Sheridan, R.; Blake, R.; Smith, L.A.; Marks, J.D. Potent neutralization of botulinum neurotoxin by recombinant oligoclonal antibody. Proc. Natl. Acad. Sci. USA 2002, 99, 11346–11350. [Google Scholar] [CrossRef]
- Jefferis, R. Glycosylation of recombinant antibody therapeutics. Biotechnol. Prog. 2005, 21, 11–16. [Google Scholar] [CrossRef]
- Burton, D.R.; Desrosiers, R.C.; Doms, R.W.; Koff, W.C.; Kwong, P.D.; Moore, J.P.; Nabel, G.J.; Sodronski, J.; Wilson, I.A.; Wyatt, R. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 2004, 5, 233–236. [Google Scholar] [CrossRef]
- Marasco, W.A.; Sui, J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat. Biotechnol. 2007, 25, 1421–1434. [Google Scholar] [CrossRef]
- Brock, T.D. (Ed.) Milestones in Microbiology; 1556–1940; ASM Press: Washington DC, USA, 1998.
- Casadevall, A.; Scharff, M.D. Serum therapy revisited: Animal models of infection and development of passive antibody therapy. Antimicrob. Agents Chemother. 1994, 38, 1695–1702. [Google Scholar] [CrossRef]
- Logtenberg, T. Antibody cocktails: Next-generation biopharmaceuticals with improved potency. Trends Biotechnol. 2007, 25, 390–394. [Google Scholar] [CrossRef]
- Glassy, M.C.; McKnight, M. The rate-limiting step in obtaining human monoclonal antibody drug pharmaceuticals. Expert Opin. Investig. Drugs 1995, 4, 225–228. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goswami, S.; Wang, W.; Arakawa, T.; Ohtake, S. Developments and Challenges for mAb-Based Therapeutics. Antibodies 2013, 2, 452-500. https://doi.org/10.3390/antib2030452
Goswami S, Wang W, Arakawa T, Ohtake S. Developments and Challenges for mAb-Based Therapeutics. Antibodies. 2013; 2(3):452-500. https://doi.org/10.3390/antib2030452
Chicago/Turabian StyleGoswami, Sumit, Wei Wang, Tsutomu Arakawa, and Satoshi Ohtake. 2013. "Developments and Challenges for mAb-Based Therapeutics" Antibodies 2, no. 3: 452-500. https://doi.org/10.3390/antib2030452