Nickel and Epigenetic Gene Silencing


{kind=link}
Abstract
:1. Introduction
2. Epigenetics and Gene Silencing
2.1. Histone Modification
2.2. DNA Methylation
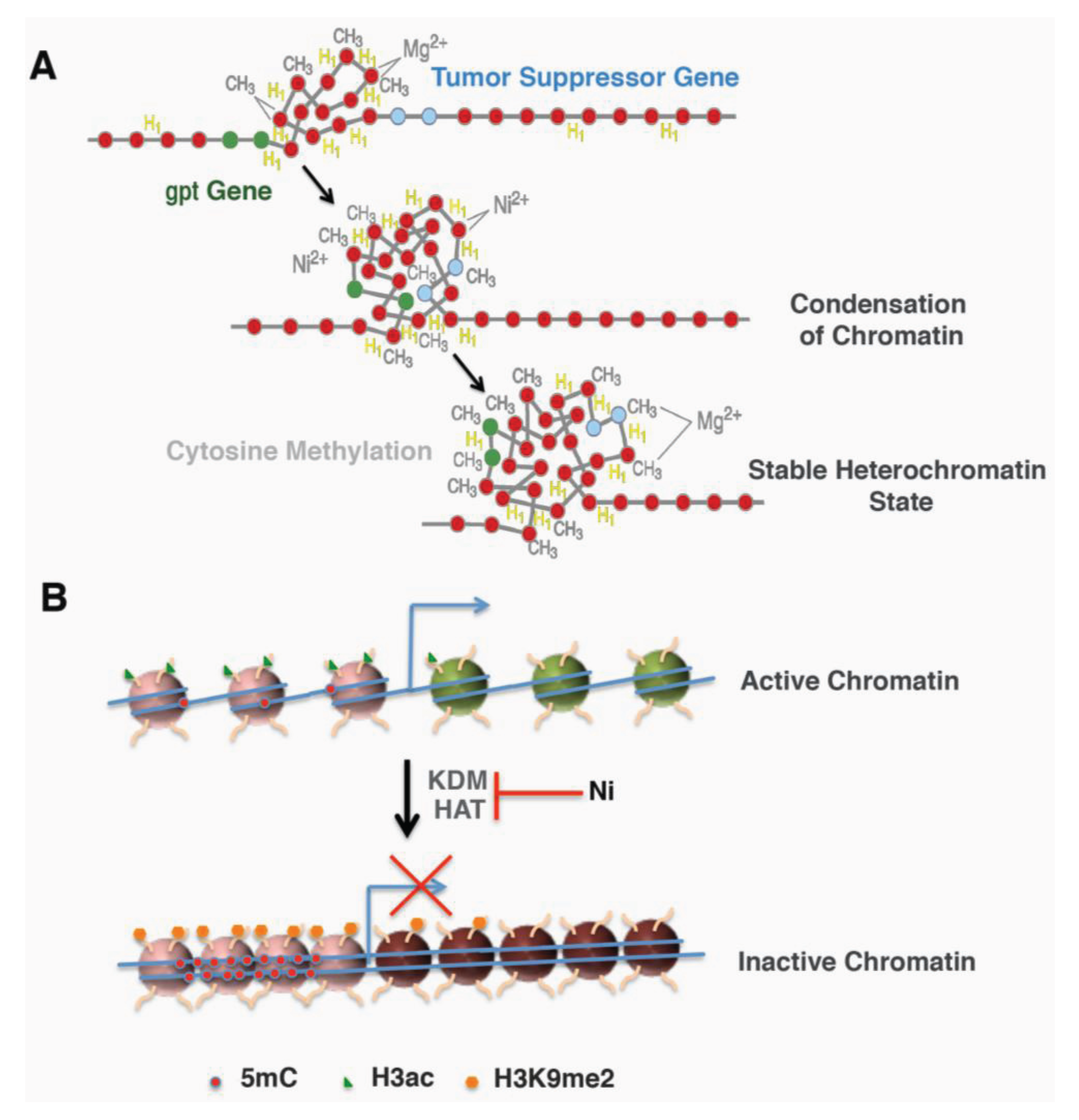
3. Nickel and Gene Silencing
3.1. Nickel and Heterochromatinization

3.2. Nickel and DNA Methylation
3.3. Nickel and Histone Modification
4. Beyond Chromatin: microRNAs and Ni Induced Gene Silencing
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Polednak, A.P. Mortality among welders, including a group exposed to nickel oxides. Arch. Environ. Health (US) 1981, 36, 235–242. [Google Scholar] [CrossRef]
- Grimsrud, T.K.; Berge, S.R.; Resmann, F.; Norseth, T.; Andersen, A. Assessment of historical exposures in a nickel refinery in Norway. Scand. J. Work Environ. Health 2000, 26, 338–345. [Google Scholar] [CrossRef]
- Doll, R.; Morgan, L.G.; Speizer, F.E. Cancers of the lung and nasal sinuses in nickel workers. Br. J. Cancer 1970, 24, 623–632. [Google Scholar] [CrossRef]
- Roberts, R.S.; Julian, J.A.; Muir, D.C.; Shannon, H.S. Cancer mortality associated with the high-temperature oxidation of nickel subsulfide. IARC Sci. Publ. 1984, 53, 23–35. [Google Scholar]
- Roberts, R.S.; Julian, J.A.; Muir, D.C.; Shannon, H.S. A study of mortality in workers engaged in the mining, smelting, and refining of nickel. II. Mortality from cancer of the respiratory tract and kidney. Toxicol. Ind. Health (USA) 1989, 5, 975–993. [Google Scholar]
- Grimsrud, T.K.; Berge, S.R.; Haldorsen, T.; Andersen, A. Exposure to different forms of nickel and risk of lung cancer. Am. J. Epidemiol. 2002, 156, 1123–1132. [Google Scholar] [CrossRef]
- Sunderman, F.W. Carcinogenicity of nickel compounds in animals. IARC Sci. Publ. 1984, 53, 127–142. [Google Scholar]
- Lumb, G.; Sunderman, F.W. The mechanism of malignant tumor induction by nickel subsulfide. Ann. Clin. Lab. Sci. 1988, 18, 353–366. [Google Scholar]
- Kasprzak, K.S.; Sunderman, F.W.; Salnikow, K. Nickel carcinogenesis. Mutat. Res. 2003, 533, 67–97. [Google Scholar] [CrossRef]
- Patierno, S.R.; Dirscherl, L.A.; Xu, J. Transformation of rat tracheal epithelial cells to immortal growth variants by particulate and soluble nickel compounds. Mutat. Res. 1993, 300, 179–193. [Google Scholar] [CrossRef]
- Landolph, J.R. Molecular mechanisms of transformation of C3H/10T1/2 C1 8 mouse embryo cells and diploid human fibroblasts by carcinogenic metal compounds. Environ. Health Perspect. 1994, 102, 119–125. [Google Scholar]
- Lu, H.; Shi, X.; Costa, M.; Huang, C. Carcinogenic effect of nickel compounds. Mol. Cell. Biochem. 2005, 279, 45–67. [Google Scholar] [CrossRef]
- Miura, T.; Patierno, S.R.; Sakuramoto, T.; Landolph, J.R. Morphological and neoplastic transformation of C3H/10T1/2 Cl 8 mouse embryo cells by insoluble carcinogenic nickel compounds. Environ. Mol. Mutagen. 1989, 14, 65–78. [Google Scholar] [CrossRef]
- IARC, Chromium, Nickel and Welding. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Lyon, France, 1990; Volume 49.
- DOLL, R.; Andersen, A.; Cooper, W.; Cosmatos, I.; Cragle, D.; Easton, D.; Enterline, P.; Goldberg, M.; Metcalfe, L.; Norseth, T. Report of the international committee on nickel carcinogenesis in man. Scand. J. Work Environ. Health 1990, 16, 1–82. [Google Scholar] [CrossRef]
- Costa, M.; Davidson, T.L.; Chen, H.; Ke, Q.; Zhang, P.; Yan, Y.; Huang, C.; Kluz, T. Nickel carcinogenesis: Epigenetics and hypoxia signaling. Mutat. Res. 2005, 592, 79–88. [Google Scholar] [CrossRef]
- Salnikow, K.; Zhitkovich, A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, arsenic, and chromium. Chem. Res. Toxicol. 2008, 21, 28–44. [Google Scholar] [CrossRef]
- Chervona, Y.; Arita, A.; Costa, M. Carcinogenic metals and the epigenome: Understanding the effect of nickel, arsenic, and chromium. Metallomics 2012, 4, 619–627. [Google Scholar] [CrossRef]
- Conway, K.; Costa, M. Nonrandom chromosomal alterations in nickel-transformed Chinese hamster embryo cells. Cancer Res. 1989, 49, 6032–6038. [Google Scholar]
- Conway, K.; Costa, M. The involvement of heterochromatic damage in nickel-induced transformation. Biol. Trace Elem. Res. 1989, 21, 437–444. [Google Scholar] [CrossRef]
- Clemens, F.; Verma, R.; Ramnath, J.; Landolph, J.R. Amplification of the Ect2 proto-oncogene and over-expression of Ect2 mRNA and protein in nickel compound and methylcholanthrene-transformed 10T1/2 mouse fibroblast cell lines. Toxicol. Appl. Pharmacol. 2005, 206, 138–149. [Google Scholar] [CrossRef]
- Salnikow, K.; Blagosklonny, M.V.; Ryan, H.; Johnson, R.; Costa, M. Carcinogenic nickel induces genes involved with hypoxic stress. Cancer Res. 2000, 60, 38–41. [Google Scholar]
- Davidson, T.; Salnikow, K.; Costa, M. Hypoxia inducible factor-1 alpha-independent suppression of aryl hydrocarbon receptor-regulated genes by nickel. Mol. Pharmacol. 2003, 64, 1485–1493. [Google Scholar] [CrossRef]
- Salnikow, K.; Davidson, T.; Zhang, Q.; Chen, L.C.; Su, W.; Costa, M. The involvement of hypoxia-inducible transcription factor-1-dependent pathway in nickel carcinogenesis. Cancer Res. 2003, 63, 3524–3530. [Google Scholar]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenomebiological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Arita, A.; Costa, M. Epigenetics in metal carcinogenesis: Nickel, arsenic, chromium and cadmium. Metallomics 2009, 1, 222–228. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef]
- Latham, J.A.; Dent, S.Y.R. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef]
- Cohen, I.; Poręba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.-H.; LeProust, E.M.; Park, I.-H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Li, X.-Q.; Guo, Y.-Y.; De, W. DNA methylation and microRNAs in cancer. World J. Gastroenterol. 2012, 18, 882–888. [Google Scholar] [CrossRef]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef]
- Klein, C.B.; Conway, K.; Wang, X.W.; Bhamra, R.K.; Lin, X.H.; Cohen, M.D.; Annab, L.; Barrett, J.C.; Costa, M. Senescence of nickel-transformed cells by an X chromosome: Possible epigenetic control. Science 1991, 251, 796–799. [Google Scholar]
- Klein, C.B.; Rossman, T.G. Transgenic Chinese hamster V79 cell lines which exhibit variable levels of gpt mutagenesis. Environ. Mol. Mutagen. 1990, 16, 1–12. [Google Scholar] [CrossRef]
- Kargacin, B.; Klein, C.B.; Costa, M. Mutagenic responses of nickel oxides and nickel sulfides in Chinese hamster V79 cell lines at the xanthine-guanine phosphoribosyl transferase locus. Mutat. Res. 1993, 300, 63–72. [Google Scholar] [CrossRef]
- Lee, Y.W.; Pons, C.; Tummolo, D.M.; Klein, C.B.; Rossman, T.G.; Christie, N.T. Mutagenicity of soluble and insoluble nickel compounds at the gpt locus in G12 Chinese hamster cells. Environ. Mol. Mutagen. 1993, 21, 365–371. [Google Scholar] [CrossRef]
- Verma, R.; Ramnath, J.; Clemens, F.; Kaspin, L.C.; Landolph, J.R. Molecular biology of nickel carcinogenesis: Identification of differentially expressed genes in morphologically transformed C3H10T1/2 Cl 8 mouse embryo fibroblast cell lines induced by specific insoluble nickel compounds. Mol. Cell. Biochem. 2004, 255, 203–216. [Google Scholar] [CrossRef]
- Landolph, J.R.; Verma, A.; Ramnath, J.; Clemens, F. Molecular biology of deregulated gene expression in transformed C3H/10T1/2 mouse embryo cell lines induced by specific insoluble carcinogenic nickel compounds. Environ. Health Perspect. 2002, 110, 845–850. [Google Scholar] [CrossRef]
- DeSilva, A.; Verma, R.; Landolph, J.R. Silencing of Expression the Beta Centaurin-2 and the FAD Synthetae Genes in Nickel Transformed C3H/10T1/2 Cell Lines. In Meal Ions in Biology and Medicine; Collery, P., Maymaud, I., Theophanides, T., Khassanova, I., Collery, T., Eds.; John Libbey Eurotext: Paris, France, 2008; Volume 10, pp. 65–67. [Google Scholar]
- Lee, Y.W.; Klein, C.B.; Kargacin, B.; Salnikow, K.; Kitahara, J.; Dowjat, K.; Zhitkovich, A.; Christie, N.T.; Costa, M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: A new model for epigenetic carcinogens. Mol. Cell. Biol. 1995, 15, 2547–2557. [Google Scholar]
- Ellen, T.P.; Kluz, T.; Harder, M.E.; Xiong, J.; Costa, M. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry 2009, 48, 4626–4632. [Google Scholar] [CrossRef]
- Yan, Y.; Kluz, T.; Zhang, P.; Chen, H.-B.; Costa, M. Analysis of specific lysine histone H3 and H4 acetylation and methylation status in clones of cells with a gene silenced by nickel exposure. Toxicol. Appl. Pharmacol. 2003, 190, 272–277. [Google Scholar] [CrossRef]
- Wu, C.-H.; Tang, S.-C.; Wang, P.-H.; Lee, H.; Ko, J.-L. Nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation. J. Biol. Chem. 2012, 287, 25292–25302. [Google Scholar] [CrossRef]
- Ji, W.; Yang, L.; Yu, L.; Yuan, J.; Hu, D.; Zhang, W.; Yang, J.; Pang, Y.; Li, W.; Lu, J.; et al. Epigenetic silencing of O6-methylguanine DNA methyltransferase gene in NiS-transformed cells. Carcinogenesis 2008, 29, 1267–1275. [Google Scholar] [CrossRef]
- Yasaei, H.; Gilham, E.; Pickles, J.C.; Roberts, T.P.; O’Donovan, M.; Newbold, R.F. Carcinogen-specific mutational and epigenetic alterations in INK4A, INK4B and p53 tumor-suppressor genes drive induced senescence bypass in normal diploid mammalian cells. Oncogene 2013, 32, 171–179. [Google Scholar] [CrossRef]
- Govindarajan, B.; Klafter, R.; Miller, M.S.; Mansur, C.; Mizesko, M.; Bai, X.; LaMontagne, K.; Arbiser, J.L. Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol. Med. 2002, 8, 1–8. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Li, M.; Wu, Y.; Fan, Y.; Zhou, Y.; Tan, L.; Shao, Z.; Shi, H. Methylation of RAR-β2, RASSF1A, and CDKN2A genes induced by nickel subsulfide and nickel-carcinogenesis in rats. Biomed. Environ. Sci. 2011, 24, 163–171. [Google Scholar]
- Broday, L.; Peng, W.; Kuo, M.H.; Salnikow, K.; Zoroddu, M.; Costa, M. Nickel compounds are novel inhibitors of histone H4 acetylation. Cancer Res. 2000, 60, 238–241. [Google Scholar]
- Golebiowski, F.; Kasprzak, K.S. Inhibition of core histones acetylation by carcinogenic nickel(II). Mol. Cell. Biochem. 2005, 279, 133–139. [Google Scholar] [CrossRef]
- Ke, Q.; Davidson, T.; Chen, H.; Kluz, T.; Costa, M. Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis 2006, 27, 1481–1488. [Google Scholar] [CrossRef]
- Kang, J.; Zhang, Y.; Chen, J.; Chen, H.; Lin, C.; Wang, Q.; Ou, Y. Nickel-induced histone hypoacetylation: The role of reactive oxygen species. Toxicol. Sci. 2003, 74, 279–286. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Wang, Y.-F.; Huang, W.-R.; Huang, Y.-T. Nickel induces oxidative stress and genotoxicity in human lymphocytes. Toxicol. Appl. Pharmacol. 2003, 189, 153–159. [Google Scholar] [CrossRef]
- Salnikow, K.; Gao, M.; Voitkun, V.; Huang, X.; Costa, M. Altered oxidative stress responses in nickel-resistant mammalian cells. Cancer Res. 1994, 54, 6407–6412. [Google Scholar]
- Sutherland, J.E.; Peng, W.; Zhang, Q.; Costa, M. The histone deacetylase inhibitor trichostatin A reduces nickel-induced gene silencing in yeast and mammalian cells. Mutat. Res. 2001, 479, 225–233. [Google Scholar] [CrossRef]
- Zhang, Q.; Salnikow, K.; Kluz, T.; Chen, L.C.; Su, W.C.; Costa, M. Inhibition and reversal of nickel-induced transformation by the histone deacetylase inhibitor trichostatin A. Toxicol. Appl. Pharmacol. 2003, 192, 201–211. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Peana, M.; Medici, S.; Casella, L.; Monzani, E.; Costa, M. Nickel binding to histone H4. Dalton Trans. 2010, 39, 787–793. [Google Scholar] [CrossRef]
- Chen, H.; Ke, Q.; Kluz, T.; Yan, Y.; Costa, M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol. Cell. Biol. 2006, 26, 3728–3737. [Google Scholar] [CrossRef]
- Chen, H.; Kluz, T.; Zhang, R.; Costa, M. Hypoxia and nickel inhibit histone demethylase JMJD1A and repress Spry2 expression in human bronchial epithelial BEAS-2B cells. Carcinogenesis 2010, 31, 2136–2144. [Google Scholar] [CrossRef]
- Chen, H.; Giri, N.C.; Zhang, R.; Yamane, K.; Zhang, Y.; Maroney, M.; Costa, M. Nickel ions inhibit histone demethylase JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in the catalytic centers. J. Biol. Chem. 2010, 285, 7374–7383. [Google Scholar]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Ma, L.; Huang, S.; Wang, R.; Gao, R.; Wu, Y.; Shi, H.; Zhang, J. The alteration of MiR-222 and its target genes in nickel-induced tumor. Biol. Trace Elem. Res. 2013, 152, 267–274. [Google Scholar] [CrossRef]
- Ji, W.; Yang, L.; Yuan, J.; Yang, L.; Zhang, M.; Qi, D.; Duan, X.; Xuan, A.; Zhang, W.; Lu, J.; et al. MicroRNA-152 targets DNA methyltransferase 1 in NiS-transformed cells via a feedback mechanism. Carcinogenesis 2013, 34, 446–453. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, H.; Shamy, M.; Costa, M. Nickel and Epigenetic Gene Silencing. Genes 2013, 4, 583-595. https://doi.org/10.3390/genes4040583
Sun H, Shamy M, Costa M. Nickel and Epigenetic Gene Silencing. Genes. 2013; 4(4):583-595. https://doi.org/10.3390/genes4040583
Chicago/Turabian StyleSun, Hong, Magdy Shamy, and Max Costa. 2013. "Nickel and Epigenetic Gene Silencing" Genes 4, no. 4: 583-595. https://doi.org/10.3390/genes4040583
APA StyleSun, H., Shamy, M., & Costa, M. (2013). Nickel and Epigenetic Gene Silencing. Genes, 4(4), 583-595. https://doi.org/10.3390/genes4040583