Antisense Gene Silencing: Therapy for Neurodegenerative Disorders?

Abstract
:1. Introduction
2. Origin of Antisense Molecules
2.1. RNA Interference
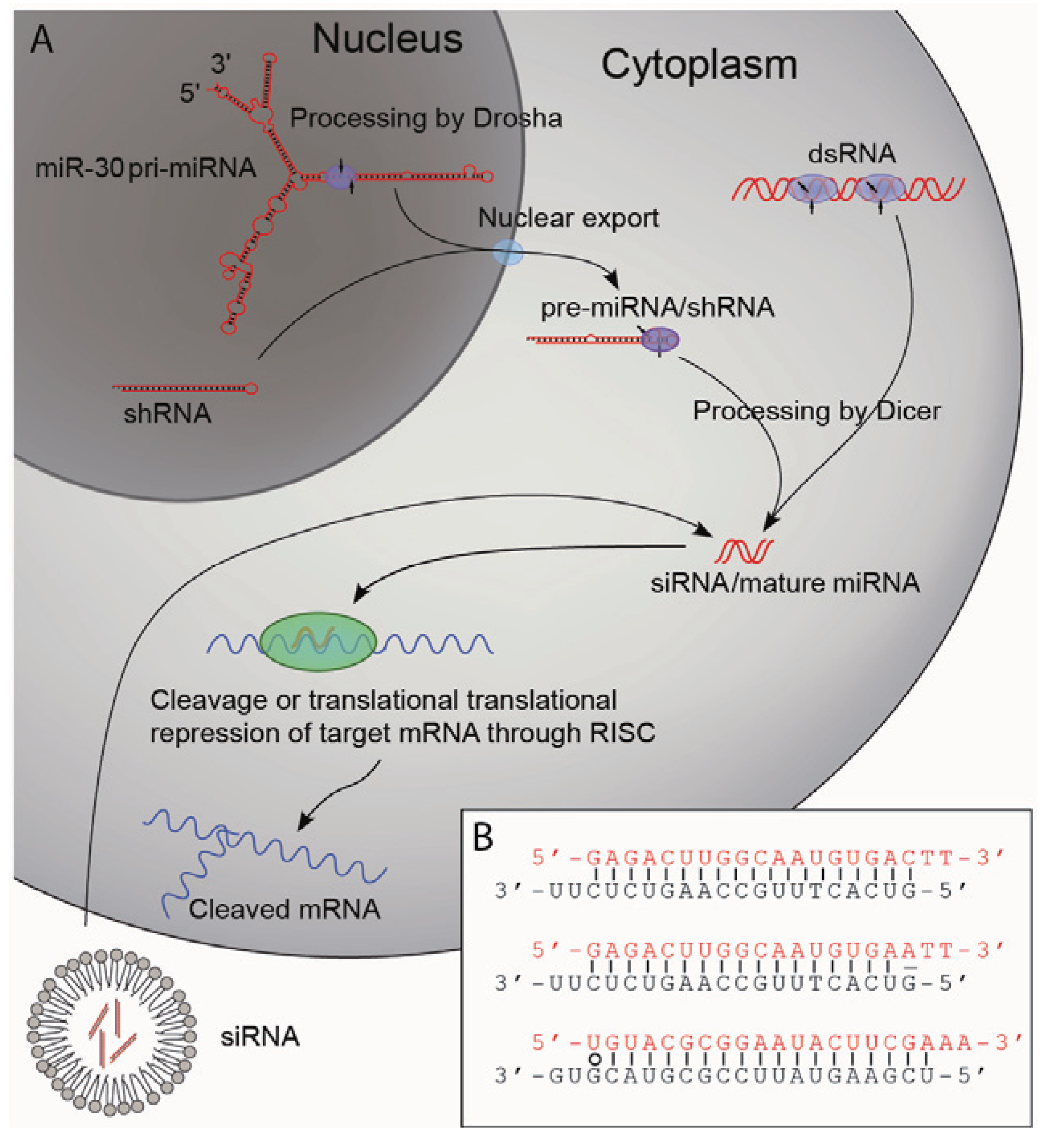
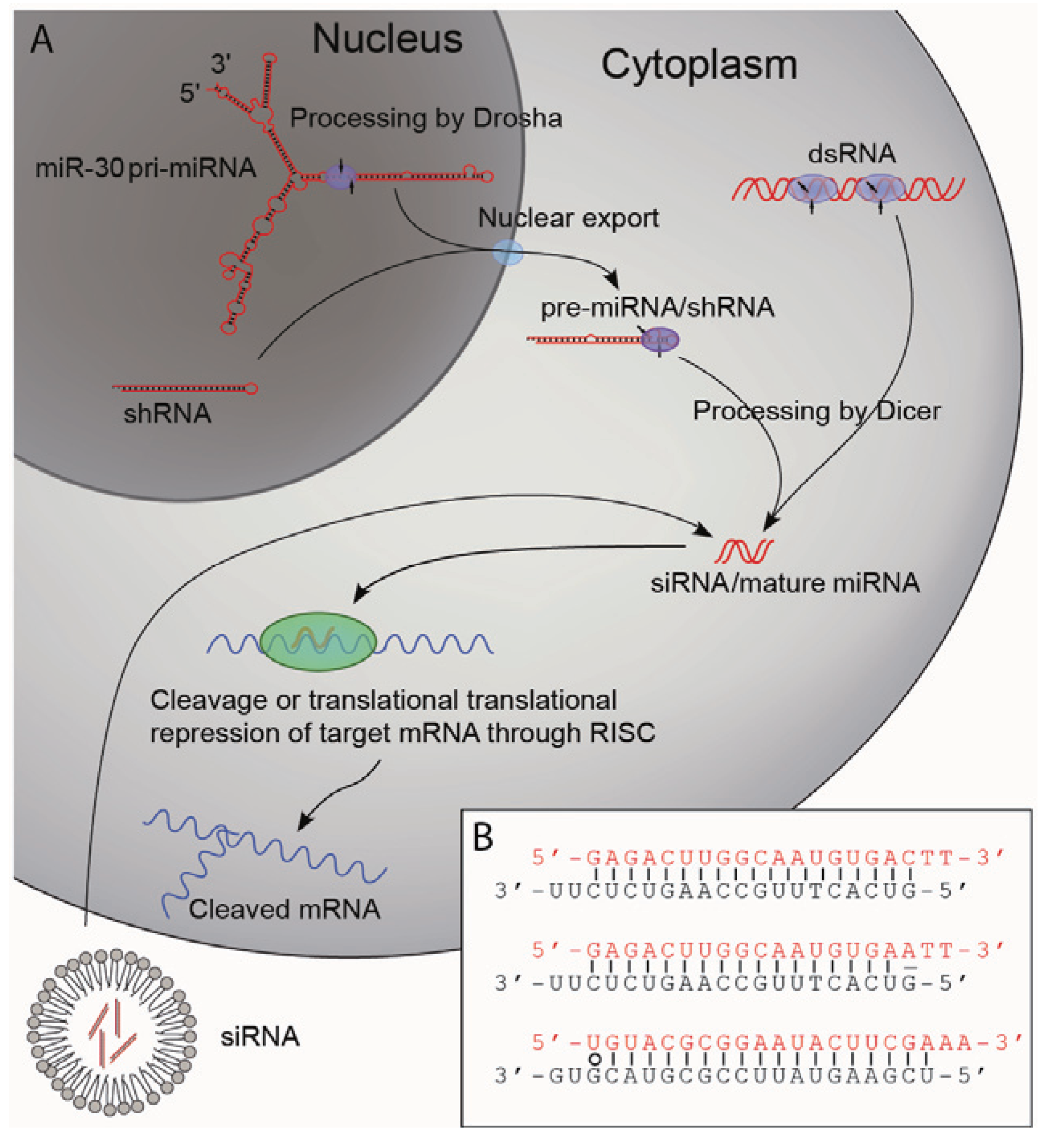
2.2. Biogenesis of Small RNAs
3. Delivery of Small RNAs
3.1. Non-Viral Delivery
3.2. Viral Delivery

{kind=link}
{kind=link}
| Vector | Retrovirus | Lentivirus | HSV | ssAAV, scAAV | Adenovirus |
|---|---|---|---|---|---|
| Genome | RNA | RNA | DNA | DNA | dsRNA |
| Cloning capacity | 8–10 kb | 8–10 kb | 150 kb | <5 kb, 2.2 kb | Up to 35 kb |
| Pseudotype/serotype | VSV-G LCMV-G Ebola etc. | VSV-G LCMV-G RV-G RB-G MV-G Ebola etc. | Mainly HSV-1 | 1–12, Chimeric and engineered | >50 naturally occurring. Type 2 and 5 used for vectors |
| Immuno-genecity | Low | Low | Highly | Mild | Highly |
| Pre-existing immunity | Limited | Limited | Yes | Limited | Yes |
| Transduces non-dividing cells | No | Yes | Yes | Yes | Yes |
| Insertion into chromatin | Yes | Yes | No (Episomal) | Yes/No (Episomal/integrated) | No (Episomal) |
| References | [49,50,51] | [49,52,53,54,55] | [56] | [57,58,59,60,61,62,63,64,65] | [66] |
4. Therapeutic Applications of Antisense Technology
| Disorder | RNAi method | Target | Mechanism | Disease model | References |
|---|---|---|---|---|---|
| Huntington’s disease | siRNA AAV-shRNA/miRNA | htt | Removal of toxic protein | Cell culture Transgenic mouse models Monkey | [7,34,94,95,96,97,98,99,100,101] |
| SCA1 | AAV-shRNA | ATXN1 | Removal of toxic proteijn | Transgenic mouse model | [102] |
| SCA3 | LV-shRNA AAV-shRNA AAV-miRNA | ATXN3 | Removal of toxic protein | Rat model Transgenic mouse models | [6,96,103,104] |
| SCA6 | siRNA miRNA | CACNA1 | Removal of toxic protein | Cell culture | [105] |
| Parkinson’s disease | siRNA LV-shRNA AAV-shRNA | α-synuclein LRRK2 GAD67 | Removal of toxic protein Modulation of neuronal transmission | Cell culture Mouse model Rat model | [106,107,108,109,110,111] |
| ALS | siRNA shRNA LV-shRNA Mouse transgenesis, shRNA | SOD1 | Removal of toxic protein | Cell culture Mouse model Transgenic mouse models | [112,113,114,115,116] |
| Alzheimer’s disease | siRNA shRNA LV-shRNA HSV-shRNA | APP PS1 DMT1 BACE1 CDK5 | Removal of toxic protein Indirect modulation of APP expression. Modulation of APP processing. Modulation of Tau phosphorylation. | Cell culture Mouse model Transgenic mouse models | [56,117,118,119,120,121,122,123,124] |
| Multiple sclerosis | LV-miRNA | Act1 | Modulation of interleukin-17 signalling | MS mouse disease model (EAE mouse) | [125] |
| Prion disease | Mouse transgenesis, shRNA | PrP(C) | Removal of wt protein to avoid conversion to toxic species. | Mouse model | [126] |
4.1. Monogenic Disorders
4.1.1. PolyQ Disorders
4.1.2. Parkinson’s Disease
4.1.3. Amyotrophic Lateral Sclerosis
4.1.4. Alzheimer’s Disease and Frontotemporal Lobar Degeneration
4.2. Non-Monogenic Disorders
4.2.1. Parkinson’s Disease

4.2.2. Multiple Sclerosis
4.2.3. Prion Disease
5. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Fire, X.S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 19, 806–811. [Google Scholar]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- U.S. National Institues of Health. Available online: http://www.ClinicalTrials.gov/ (accessed on 2 July 2013).
- Xia, H.; Mao, Q.; Paulson, H.L.; Davidson, B.L. siRNA-mediated gene silencing in vitro and in vivo. Nat. Biotechnol. 2002, 20, 1006–1010. [Google Scholar]
- Alves, S.; Nascimento-Ferreira, I.; Dufour, N.; Hassig, R.; Auregan, G.; Nobrega, C.; Brouillet, E.; Hantraye, P.; de Lima, M.C.P.; Déglon, N.; et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: No role for wild-type ataxin-3? Hum. Mol. Genet. 2010, 19, 2380–2394. [Google Scholar] [CrossRef]
- McBride, J.L.; Pitzer, M.R.; Boudreau, R.L.; Dufour, B.; Hobbs, T.; Ojeda, S.R.; Davidson, B.L. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 2011, 19, 2152–2162. [Google Scholar] [CrossRef]
- Carroll, J.B.; Warby, S.C.; Southwell, A.L.; Doty, C.N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C.F.; Freier, S.M.; Hayden, M.R. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther. 2011, 19, 2178–2185. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Chen, K.; Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007, 8, 93–103. [Google Scholar] [CrossRef]
- Manchester University. The miRBase. Available online: http://www.mirbase.org/cgi-bin/mirna_summary.pl?org=has/ (accessed on 2 July 2013).
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N.; Micro, R.N. A maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef]
- Cullen, B.R. Transcription and processing of human microRNA precursors. Mol. Cell. 2004, 16, 861–865. [Google Scholar] [CrossRef]
- Knight, S.W.; Bass, B.L. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 2001, 293, 2269–2271. [Google Scholar] [CrossRef]
- Ketting, R.F.; Fischer, S.E.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef]
- Tang, G. siRNA and miRNA: an insight into RISCs. Trends Biochem. Sci. 2005, 30, 106–114. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- Piao, X.; Zhang, X.; Wu, L.; Belasco, J.G. CCR4-NOT deadenylates mRNA associated with RNA-induced silencing complexes in human cells. Mol. Cell. Biol. 2010, 30, 1486–1494. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.; Nam, J.W.; Farh, K.K.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: Functional sites with centered pairing. Mol. Cell. 2010, 38, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Boudreau, R.L.; Spengler, R.M.; Davidson, B.L. Rational design of therapeutic siRNAs: Minimizing off-targeting potential to improve the safety of RNAi therapy for Huntington’s disease. Mol. Ther. 2011, 19, 2169–2177. [Google Scholar] [CrossRef]
- Hohjoh, H. Enhancement of RNAi activity by improved siRNA duplexes. FEBS Lett. 2004, 557, 193–198. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 19, 550–553. [Google Scholar] [CrossRef]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002, 2, 243–247. [Google Scholar] [CrossRef]
- An, D.S.; Xie, Y.; Mao, S.H.; Morizono, K.; Kung, S.K.; Chen, I.S. Efficient lentiviral vectors for short hairpin RNA delivery into human cells. Hum. Gene Ther. 2003, 14, 1207–1212. [Google Scholar] [CrossRef]
- Yu, J.Y.; DeRuiter, S.L.; Turner, D.L. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6047–6052. [Google Scholar] [CrossRef]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Grimm, D.; Wang, L.; Lee, J.S.; Schurmann, N.; Gu, S.; Borner, K.; Storm, T.A.; Kay, M.A. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J. Clin. Invest. 2010, 120, 3106–3119. [Google Scholar] [CrossRef]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef]
- Gou, D.; Narasaraju, T.; Chintagari, N.R.; Jin, N.; Wang, P.; Liu, L. Gene silencing in alveolar type II cells using cell-specific promoter in vitro and in vivo. Nucleic Acids Res. 2004, 32, e134. [Google Scholar] [CrossRef]
- Giering, J.C.; Grimm, D.; Storm, T.A.; Kay, M.A. Expression of shRNA From a Tissue-specific pol II Promoter Is an Effective and Safe RNAi Therapeutic. Mol. Ther. 2008, 16, 1630–1636. [Google Scholar] [CrossRef]
- Zhou, H.; Xia, X.G.; Xu, Z. An RNA polymerase II construct synthesizes short-hairpin RNA with a quantitative indicator and mediates highly efficient RNAi. Nucleic Acids Res. 2005, 33, e62. [Google Scholar] [CrossRef]
- Stegmeier, F.; Hu, G.; Rickles, R.J.; Hannon, G.J.; Elledge, S.J. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13212–13217. [Google Scholar]
- Silva, J.M.; Li, M.Z.; Chang, K.; Ge, W.; Golding, M.C.; Rickles, R.J.; Siolas, D.; Hu, G.; Paddison, P.J.; Schlabach, M.R.; et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 2005, 37, 1281–1288. [Google Scholar]
- Nielsen, T.T.; Marion, I.; Hasholt, L.; Lundberg, C. Neuron-specific RNA interference using lentiviral vectors. J. Gene Med. 2009, 11, 559–569. [Google Scholar]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Heidel, J.D.; Bartlett, D.W.; Davis, M.E.; Triche, T.J. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005, 65, 8984–8992. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G.; et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Buchschacher, G.L., Jr. Introduction to retroviruses and retroviral vectors. Somat. Cell Mol. Genet. 2001, 26, 1–11. [Google Scholar] [CrossRef]
- Mann, R.; Mulligan, R.C.; Baltimore, D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 1983, 33, 153–159. [Google Scholar] [CrossRef]
- Beyer, W.R.; Westphal, M.; Ostertag, W.; von Laer, D. Oncoretrovirus and lentivirus vectors pseudotyped with lymphocytic choriomeningitis virus glycoprotein: Generation, concentration, and broad host range. J. Virol. 2002, 76, 1488–1495. [Google Scholar]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Cronin, J.; Zhang, X.Y.; Reiser, J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- Bartz, S.R.; Vodicka, M.A. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods 1997, 12, 337–342. [Google Scholar] [CrossRef]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar]
- Kahl, C.A.; Marsh, J.; Fyffe, J.; Sanders, D.A.; Cornetta, K. Human immunodeficiency virus type 1-derived lentivirus vectors pseudotyped with envelope glycoproteins derived from Ross River virus and Semliki Forest virus. J. Virol. 2004, 78, 1421–1430. [Google Scholar] [CrossRef]
- Hong, C.S.; Goins, W.F.; Goss, J.R.; Burton, E.A.; Glorioso, J.C. Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer’s disease-related amyloid-beta peptide in vivo. Gene Ther. 2006, 13, 1068–1079. [Google Scholar] [CrossRef]
- Dong, J.Y.; Fan, P.D.; Frizzell, R.A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 1996, 7, 2101–2112. [Google Scholar] [CrossRef]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef]
- Kotin, R.M.; Linden, R.M.; Berns, K.I. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 1992, 11, 5071–5078. [Google Scholar]
- Kaplitt, M.G.; Leone, P.; Samulski, R.J.; Xiao, X.; Pfaff, D.W.; O'Malley, K.L.; During, M.J. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet. 1994, 8, 148–154. [Google Scholar] [CrossRef]
- McCown, T.J.; Xiao, X.; Li, J.; Breese, G.R.; Samulski, R.J. Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res. 1996, 713, 99–107. [Google Scholar]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar] [CrossRef]
- Bartel, M.A.; Weinstein, J.R.; Schaffer, D.V. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Ther. 2012, 19, 694–700. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef]
- Weinberg, M.S.; Samulski, R.J.; McCown, T.J. Adeno-associated virus (AAV) gene therapy for neurological disease. Neuropharmacology 2013, 69, 82–88. [Google Scholar] [CrossRef]
- Teichler, Z.D. US gene therapy in crisis. Trends Genet. 2000, 16, 272–275. [Google Scholar] [CrossRef]
- Cann, A. Principles of Molecular Virology, 3rd ed.; Academic Press: Waltham, MA, USA, 2001. [Google Scholar]
- Bukrinsky, M.I.; Haggerty, S.; Dempsey, M.P.; Sharova, N.; Adzhubel, A.; Spitz, L.; Lewis, P.; Goldfarb, D.; Emerman, M.; Stevenson, M. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature 1993, 365, 666–669. [Google Scholar] [CrossRef]
- Emi, N.; Friedmann, T.; Yee, J.K. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J. Virol. 1991, 65, 1202–1207. [Google Scholar]
- Kang, Y.; Stein, C.S.; Heth, J.A.; Sinn, P.L.; Penisten, A.K.; Staber, P.D.; Ratliff, K.L.; Shen, H.; Barker, C.K.; Martins, I. In vivo gene transfer using a nonprimate lentiviral vector pseudotyped with Ross River Virus glycoproteins. J. Virol. 2002, 76, 9378–9388. [Google Scholar]
- Georgievska, B.; Kirik, D.; Bjorklund, A. Overexpression of glial cell line-derived neurotrophic factor using a lentiviral vector induces time- and dose-dependent downregulation of tyrosine hydroxylase in the intact nigrostriatal dopamine system. J. Neuro. Sci. 2004, 24, 6437–6445. [Google Scholar]
- Deglon, N.; Tseng, J.L.; Bensadoun, J.C.; Zurn, A.D.; Arsenijevic, Y.; de Pereira, A.L.; Zufferey, R.; Trono, D.; Aebischer, P. Self-inactivating lentiviral vectors with enhanced transgene expression as potential gene transfer system in Parkinson’s disease. Hum. Gene Ther. 2000, 11, 179–190. [Google Scholar] [CrossRef]
- Jakobsson, J.; Nielsen, T.T.; Staflin, K.; Georgievska, B.; Lundberg, C. Efficient transduction of neurons using Ross River glycoprotein-pseudotyped lentiviral vectors. Gene Ther. 2006, 13, 966–973. [Google Scholar] [CrossRef]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar]
- Azzouz, M.; Le, T.; Ralph, G.S.; Walmsley, L.; Monani, U.R.; Lee, D.C.; Wilkes, F.; Mitrophanous, K.A.; Kingsman, S.M.; Burghes, A.H.; et al. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2004, 114, 1726–1731. [Google Scholar]
- Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.; Mitrophanous, K.A.; Kingsman, S.M.; Carmeliet, P.; Mazarakis, N.D. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004, 429, 413–417. [Google Scholar] [CrossRef]
- Powell, S.K.; Kaloss, M.A.; Pinkstaff, A.; McKee, R.; Burimski, I.; Pensiero, M.; Otto, E.; Stemmer, W.P.; Soong, N.W. Breeding of retroviruses by DNA shuffling for improved stability and processing yields. Nat. Biotechnol. 2000, 18, 1279–1282. [Google Scholar] [CrossRef]
- Merten, C.A.; Stitz, J.; Braun, G.; Poeschla, E.M.; Cichutek, K.; Buchholz, C.J. Directed evolution of retrovirus envelope protein cytoplasmic tails guided by functional incorporation into lentivirus particles. J. Virol. 2005, 79, 834–840. [Google Scholar] [CrossRef]
- Hwang, B.Y.; Schaffer, D.V. Engineering a serum-resistant and thermostable vesicular stomatitis virus G glycoprotein for pseudotyping retroviral and lentiviral vectors. Gene Ther. 2013. [Google Scholar] [CrossRef]
- Bell, P.; Wang, L.; Lebherz, C.; Flieder, D.B.; Bove, M.S.; Wu, D.; Gao, G.P.; Wilson, J.M.; Wivel, N.A. No evidence for tumorigenesis of AAV vectors in a large-scale study in mice. Mol. Ther. 2005, 12, 299–306. [Google Scholar] [CrossRef]
- Deyle, D.R.; Russell, D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther. 2009, 11, 442–447. [Google Scholar]
- Bell, P.; Moscioni, A.D.; McCarter, R.J.; Wu, D.; Gao, G.; Hoang, A.; Sanmiguel, J.C.; Sun, X.; Wivel, N.A.; Raper, S.E.; et al. Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Mol. Ther. 2006, 14, 34–44. [Google Scholar] [CrossRef]
- UCL Institute of Child Health and Great Ormond Street Hospital for Children NHS Trust. GOSH Announces Leukaemia Case Following Gene Therapy for X-SCID. Available online: http://www.ich.ucl.ac.uk/pressoffice/pressrelease_00591/ (accessed on 2 July 2013).
- Weinberg, M.S.; Blake, B.L.; Samulski, R.J.; McCown, T.J. The influence of epileptic neuropathology and prior peripheral immunity on CNS transduction by rAAV2 and rAAV5. Gene Ther. 2011, 18, 961–968. [Google Scholar] [CrossRef]
- Dodiya, H.B.; Bjorklund, T.; Stansell, J., III; Mandel, R.J.; Kirik, D.; Kordower, J.H. Differential transduction following basal ganglia administration of distinct pseudotyped AAV capsid serotypes in nonhuman primates. Mol. Ther. 2010, 18, 579–587. [Google Scholar] [CrossRef]
- Hadaczek, P.; Kohutnicka, M.; Krauze, M.T.; Bringas, J.; Pivirotto, P.; Cunningham, J.; Bankiewicz, K. Convection-enhanced delivery of adeno-associated virus type 2 (AAV2) into the striatum and transport of AAV2 within monkey brain. Hum. Gene Ther. 2006, 17, 291–302. [Google Scholar] [CrossRef]
- Markakis, E.A.; Vives, K.P.; Bober, J.; Leichtle, S.; Leranth, C.; Beecham, J.; Elsworth, J.D.; Roth, R.H.; Samulski, R.J.; Redmond, D.E., Jr. Comparative transduction efficiency of AAV vector serotypes 1–6 in the substantia nigra and striatum of the primate brain. Mol. Ther. 2010, 18, 588–593. [Google Scholar] [CrossRef]
- Masamizu, Y.; Okada, T.; Ishibashi, H.; Takeda, S.; Yuasa, S.; Nakahara, K. Efficient gene transfer into neurons in monkey brain by adeno-associated virus 8. Neuroreport 2010, 21, 447–451. [Google Scholar] [CrossRef]
- Hadaczek, P.; Forsayeth, J.; Mirek, H.; Munson, K.; Bringas, J.; Pivirotto, P.; McBride, J.L.; Davidson, B.L.; Bankiewicz, K.S. Transduction of nonhuman primate brain with adeno-associated virus serotype 1, vector trafficking and immune response. Hum. Gene Ther. 2009, 20, 225–237. [Google Scholar] [CrossRef]
- Klein, R.L.; Dayton, R.D.; Tatom, J.B.; Henderson, K.M.; Henning, P.P. AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol. Ther. 2008, 16, 89–96. [Google Scholar] [CrossRef]
- Harding, T.C.; Dickinson, P.J.; Roberts, B.N.; Yendluri, S.; Gonzalez-Edick, M.; Lecouteur, R.A.; Jooss, K.U. Enhanced gene transfer efficiency in the murine striatum and an orthotopic glioblastoma tumor model, using AAV-7- and AAV-8-pseudotyped vectors. Hum. Gene Ther. 2006, 17, 807–820. [Google Scholar] [CrossRef]
- Rahim, A.A.; Wong, A.M.; Hoefer, K.; Buckley, S.M.; Mattar, C.N.; Cheng, S.H.; Chan, J.K.; Cooper, J.D.; Waddington, S.N. Intravenous administration of AAV2/9 to the fetal and neonatal mouse leads to differential targeting of CNS cell types and extensive transduction of the nervous system. FASEB J. 2011, 25, 3505–3518. [Google Scholar] [CrossRef]
- Schmidt, C. RNAi momentum fizzles as pharma shifts priorities. Nat. Biotechnol. 2011, 29, 93–94. [Google Scholar] [CrossRef]
- Wang, Y.L.; Liu, W.; Wada, E.; Murata, M.; Wada, K.; Kanazawa, I. Clinico-pathological rescue of a model mouse of Huntington’s disease by siRNA. Neurosci. Res. 2005, 53, 241–249. [Google Scholar] [CrossRef]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 19, 5820–5825. [Google Scholar]
- Hu, J.; Matsui, M.; Gagnon, K.T.; Schwartz, J.C.; Gabillet, S.; Arar, K.; Wu, J.; Bezprozvanny, I.; Corey, D.R. Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol. 2009, 27, 478–484. [Google Scholar] [CrossRef]
- Fiszer, A.; Mykowska, A.; Krzyzosiak, W.J. Inhibition of mutant huntingtin expression by RNA duplex targeting expanded CAG repeats. Nucleic. Acid. Res. 2011, 39, 5578–5585. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Corey, D.R. Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol. 2010, 17, 1183–1188. [Google Scholar] [CrossRef]
- Van Bilsen, P.H.; Jaspers, L.; Lombardi, M.S.; Odekerken, J.C.; Burright, E.N.; Kaemmerer, W.F. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum. Gene Ther. 2008, 19, 710–719. [Google Scholar] [CrossRef]
- Warby, S.C.; Montpetit, A.; Hayden, A.R.; Carroll, J.B.; Butland, S.L.; Visscher, H.; Collins, J.A.; Semaka, A.; Hudson, T.J.; Hayden, M.R. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Genet. 2009, 84, 351–366. [Google Scholar] [CrossRef]
- Pfister, E.L.; Kennington, L.; Straubhaar, J.; Wagh, S.; Liu, W.; DiFiglia, M.; Landwehrmeyer, B.; Vonsattel, J.P.; Zamore, P.D.; Aronin, N. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 2009, 19, 774–778. [Google Scholar] [CrossRef]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef]
- Nobrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Hirai, H.; Deglon, N.; de Almeida, L.P. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS One 2013, 8, e52396. [Google Scholar] [CrossRef]
- Rodriguez-Lebron, E.; do Carmo, C.M.; Molina-Luna, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. 2013. [Google Scholar] [CrossRef]
- Tsou, W.L.; Soong, B.W.; Paulson, H.L.; Rodriguez-Lebron, E. Splice isoform-specific suppression of the Cav2.1 variant underlying spinocerebellar ataxia type 6. Neurobiol. Dis. 2011, 43, 533–542. [Google Scholar] [CrossRef]
- Fountaine, T.M.; Wade-Martins, R. RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 2007, 85, 351–363. [Google Scholar] [CrossRef]
- Sapru, M.K.; Yates, J.W.; Hogan, S.; Jiang, L.; Halter, J.; Bohn, M.C. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 2006, 198, 382–390. [Google Scholar] [CrossRef]
- De Ynigo-Mojado, L.; Martin-Ruiz, I.; Sutherland, J.D. Efficient allele-specific targeting of LRRK2 R1441 mutations mediated by RNAi. PLoS One 2011, 6, e21352. [Google Scholar] [CrossRef]
- Sibley, C.R.; Seow, Y.; Curtis, H.; Weinberg, M.S.; Wood, M.J. Silencing of Parkinson’s disease-associated genes with artificial mirtron mimics of miR-1224. Nucleic. Acid. Res. 2012, 40, 9863–9875. [Google Scholar] [CrossRef]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Horvath, L.; van Marion, I.; Tai, K.; Nielsen, T.T.; Lundberg, C. Knockdown of GAD67 protein levels normalizes neuronal activity in a rat model of Parkinson’s disease. J. Gene Med. 2011, 13, 188–197. [Google Scholar]
- Ding, H.; Schwarz, D.S.; Keene, A.; Affar, E.; Fenton, L.; Xia, X.; Shi, Y.; Zamore, P.D.; Xu, Z.; et al. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2003, 2, 209–217. [Google Scholar] [CrossRef]
- Ralph, G.S.; Radcliffe, P.A.; Day, D.M.; Carthy, J.M.; Leroux, M.A.; Lee, D.C.; Wong, L.F.; Bilsland, L.G.; Greensmith, L.; Kingsman, S.M.; et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005, 11, 429–433. [Google Scholar] [CrossRef]
- Xia, X.; Zhou, H.; Huang, Y.; Xu, Z. Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol. Dis. 2006, 23, 578–586. [Google Scholar] [CrossRef]
- Towne, C.; Raoul, C.; Schneider, B.L.; Aebischer, P. Systemic AAV6 delivery mediating RNA interference against SOD1: Neuromuscular transduction does not alter disease progression in fALS mice. Mol. Ther. 2008, 16, 1018–1025. [Google Scholar] [CrossRef]
- Towne, C.; Setola, V.; Schneider, B.L.; Aebischer, P. Neuroprotection by gene therapy targeting mutant SOD1 in individual pools of motor neurons does not translate into therapeutic benefit in fALS mice. Mol. Ther. 2011, 19, 274–283. [Google Scholar] [CrossRef]
- Xie, Z.; Romano, D.M.; Kovacs, D.M.; Tanzi, R.E. Effects of RNA interference-mediated silencing of gamma-secretase complex components on cell sensitivity to caspase-3 activation. J. Biol. Chem. 2004, 279, 34130–34137. [Google Scholar]
- Kandimalla, R.J.; Wani, W.Y.; Binukumar, B.K.; Gill, K.D. siRNA against presenilin 1 (PS1) down regulates amyloid beta42 production in IMR-32 cells. J. Biomed. Sci. 2012, 19, 2. [Google Scholar] [CrossRef]
- Zhao, X.; Wen, L.; Li, G.; Ba, Q.; Cui, Y.; Han, Z.; Jia, Y.; Xu, Y. Lentivirus-mediated APP695-RNAi reduces apoptosis in APP transgenic mouse neurons. Neuroreport 2011, 22, 804–808. [Google Scholar]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef]
- Senechal, Y.; Prut, L.; Kelly, P.H.; Staufenbiel, M.; Natt, F.; Hoyer, D.; Wiessner, C.; Dev, K.K. Increased exploratory activity of APP23 mice in a novel environment is reversed by siRNA. Brain Res. 2008, 1243, 124–133. [Google Scholar] [CrossRef]
- Singer, O.; Marr, R.A.; Rockenstein, E.; Crews, L.; Coufal, N.G.; Gage, F.H.; Verma, I.M.; Masliah, E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat. Neurosci. 2005, 8, 1343–1349. [Google Scholar] [CrossRef]
- Zheng, W.; Xin, N.; Chi, Z.H.; Zhao, B.L.; Zhang, J.; Li, J.Y.; Wang, Z.Y. Divalent metal transporter 1 is involved in amyloid precursor protein processing and Abeta generation. FASEB J. 2009, 23, 4207–4217. [Google Scholar] [CrossRef]
- Piedrahita, D.; Hernandez, I.; Lopez-Tobon, A.; Fedorov, D.; Obara, B.; Manjunath, B.S.; Boudreau, R.L.; Davidson, B.; Laferla, F.; Gallego-Gomez, J.C.; et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J. Neurosci. 2010, 30, 13966–13976. [Google Scholar] [CrossRef]
- Nakahara, J.; Maeda, M.; Aiso, S.; Suzuki, N. Current concepts in multiple sclerosis: Autoimmunity versus oligodendrogliopathy. Clin. Rev. Allergy Immunol. 2012, 42, 26–34. [Google Scholar] [CrossRef]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Ambrose, C.M.; Duyao, M.P.; Barnes, G.; Bates, G.P.; Lin, C.S.; Srinidhi, J.; Baxendale, S.; Hummerich, H.; Lehrach, H.; Altherr, M.; et al. Structure and expression of the Huntington’s disease gene: Evidence against simple inactivation due to an expanded CAG repeat. Somat. Cell. Mol. Genet. 1994, 20, 27–38. [Google Scholar] [CrossRef]
- Kowall, N.W.; Ferrante, R.J.; Martin, J.B. Patterns of cell loss in Huntington’s disease. Trends Neurosci. 1987, 10, 24–29. [Google Scholar] [CrossRef]
- Rosas, H.D.; Liu, A.K.; Hersch, S.; Glessner, M.; Ferrante, R.J.; Salat, D.H.; van der Kouwe, A.; Jenkins, B.G.; Dale, A.M.; Fischl, B. Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology 2002, 58, 695–701. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar]
- Palfi, S.; Brouillet, E.; Jarraya, B.; Bloch, J.; Jan, C.; Shin, M.; Conde, F.; Li, X.J.; Aebischer, P.; Hantraye, P.; et al. Expression of mutated huntingtin fragment in the putamen is sufficient to produce abnormal movement in non-human primates. Mol. Ther. 2007, 15, 1444–1451. [Google Scholar] [CrossRef]
- Wellington, C.L.; Ellerby, L.M.; Hackam, A.S.; Margolis, R.L.; Trifiro, M.A.; Singaraja, R.; McCutcheon, K.; Salvesen, G.S.; Propp, S.S.; Bromm, M.; et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem. 1998, 273, 9158–9167. [Google Scholar] [CrossRef]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef]
- Bird, T.D. Hereditary Ataxia Overview; Bird, T.D., Pagon, R.A., Adam, M.P., Dolan, C.R., Fong, C.T., Stephens, K., Eds.; University of Washington: Seattle, WA, USA, 2012. [Google Scholar]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef]
- Cookson, M.R.; van der Brug, M. Cell systems and the toxic mechanism(s) of alpha-synuclein. Exp. Neurol. 2008, 209, 5–11. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Chen, S.; Ge, X.; Chen, Y.; Lv, N.; Liu, Z.; Yuan, W. Advances with RNA interference in Alzheimer’s disease research. Drug Des. Dev. Ther. 2013, 7, 117–125. [Google Scholar]
- Josephs, K.A.; Hodges, J.R.; Snowden, J.S.; Mackenzie, I.R.; Neumann, M.; Mann, D.M.; Dickson, D.W. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta. Neuropathol. 2011, 122, 137–153. [Google Scholar] [CrossRef] [Green Version]
- Carta, A.R.; Fenu, S.; Pala, P.; Tronci, E.; Morelli, M. Selective modifications in GAD67 mRNA levels in striatonigral and striatopallidal pathways correlate to dopamine agonist priming in 6-hydroxydopamine-lesioned rats. Eur. J. Neurosci. 2003, 18, 2563–2572. [Google Scholar] [CrossRef]
- Bergman, H.; Deuschl, G. Pathophysiology of Parkinson’s disease: From clinical neurology to basic neuroscience and back. Mov. Disord. 2002, 17, S28–S40. [Google Scholar] [CrossRef]
- Winkler, C.; Bentlage, C.; Cenci, M.A.; Nikkhah, G.; Bjorklund, A. Regulation of neuropeptide mRNA expression in the basal ganglia by intrastriatal and intranigral transplants in the rat Parkinson model. Neuroscience 2003, 118, 1063–1077. [Google Scholar] [CrossRef]
- Soghomonian, J.J.; Gonzales, C.; Chesselet, M.F. Messenger RNAs encoding glutamate-decarboxylases are differentially affected by nigrostriatal lesions in subpopulations of striatal neurons. Brain Res. 1992, 576, 68–79. [Google Scholar] [CrossRef]
- Wichmann, T.; DeLong, M.R. Pathophysiology of Parkinson’s disease: The MPTP primate model of the human disorder. Ann. N. Y. Acad. Sci. 2003, 991, 199–213. [Google Scholar] [CrossRef]
- Vila, M.; Levy, R.; Herrero, M.T.; Ruberg, M.; Faucheux, B.; Obeso, J.A.; Agid, Y.; Hirsch, E.C. Consequences of nigrostriatal denervation on the functioning of the basal ganglia in human and nonhuman primates: An in situ hybridization study of cytochrome oxidase subunit I mRNA. J. Neurosci. 1997, 17, 765–773. [Google Scholar]
- Jakobsson, J.; Rosenqvist, N.; Marild, K.; Agoston, V.; Lundberg, C. Evidence for disease-regulated transgene expression in the brain with use of lentiviral vectors. J. Neurosci. Res. 2006, 84, 58–67. [Google Scholar] [CrossRef]
- Voorn, P.; Roest, G.; Groenewegen, H.J. Increase of enkephalin and decrease of substance P immunoreactivity in the dorsal and ventral striatum of the rat after midbrain 6-hydroxydopamine lesions. Brain Res. 1987, 412, 391–396. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef]
- Yan, Y.; Ding, X.; Li, K.; Ciric, B.; Wu, S.; Xu, H.; Gran, B.; Rostami, A.; Zhang, G.X. CNS-specific therapy for ongoing EAE by silencing IL-17 pathway in astrocytes. Mol. Ther. 2012, 20, 1338–1348. [Google Scholar] [CrossRef]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; de Armond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Pfeifer, A.; Eigenbrod, S.; Al-Khadra, S.; Hofmann, A.; Mitteregger, G.; Moser, M.; Bertsch, U.; Kretzschmar, H. Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J. Clin. Invest. 2006, 116, 3204–3210. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nielsen, T.T.; Nielsen, J.E. Antisense Gene Silencing: Therapy for Neurodegenerative Disorders? Genes 2013, 4, 457-484. https://doi.org/10.3390/genes4030457
Nielsen TT, Nielsen JE. Antisense Gene Silencing: Therapy for Neurodegenerative Disorders? Genes. 2013; 4(3):457-484. https://doi.org/10.3390/genes4030457
Chicago/Turabian StyleNielsen, Troels T., and Jørgen E. Nielsen. 2013. "Antisense Gene Silencing: Therapy for Neurodegenerative Disorders?" Genes 4, no. 3: 457-484. https://doi.org/10.3390/genes4030457