Signaling Pathways from the Endoplasmic Reticulum and Their Roles in Disease

Abstract
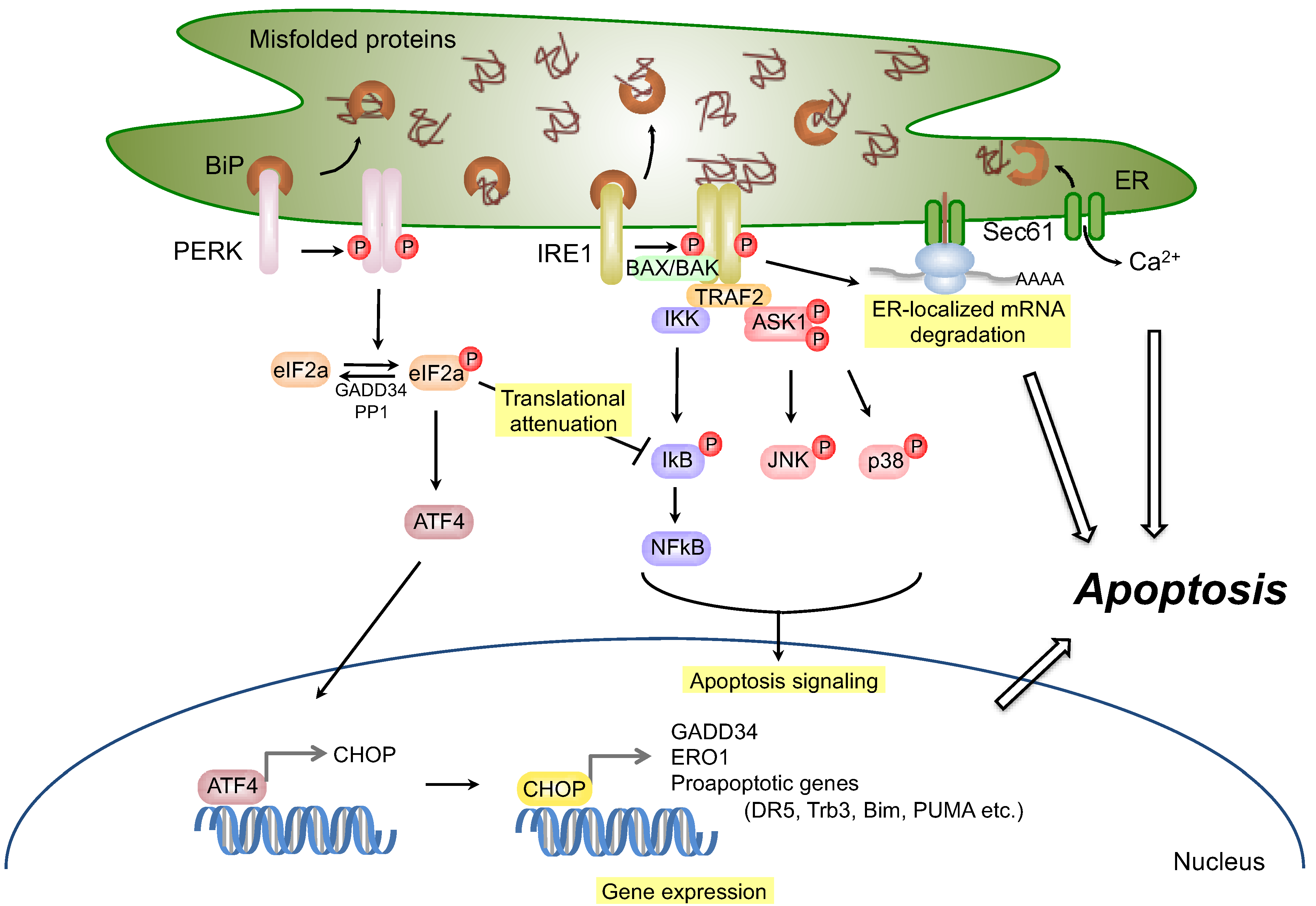
:1. Introduction
2. The Signaling Pathways from Three ER Stress Sensors during the UPR
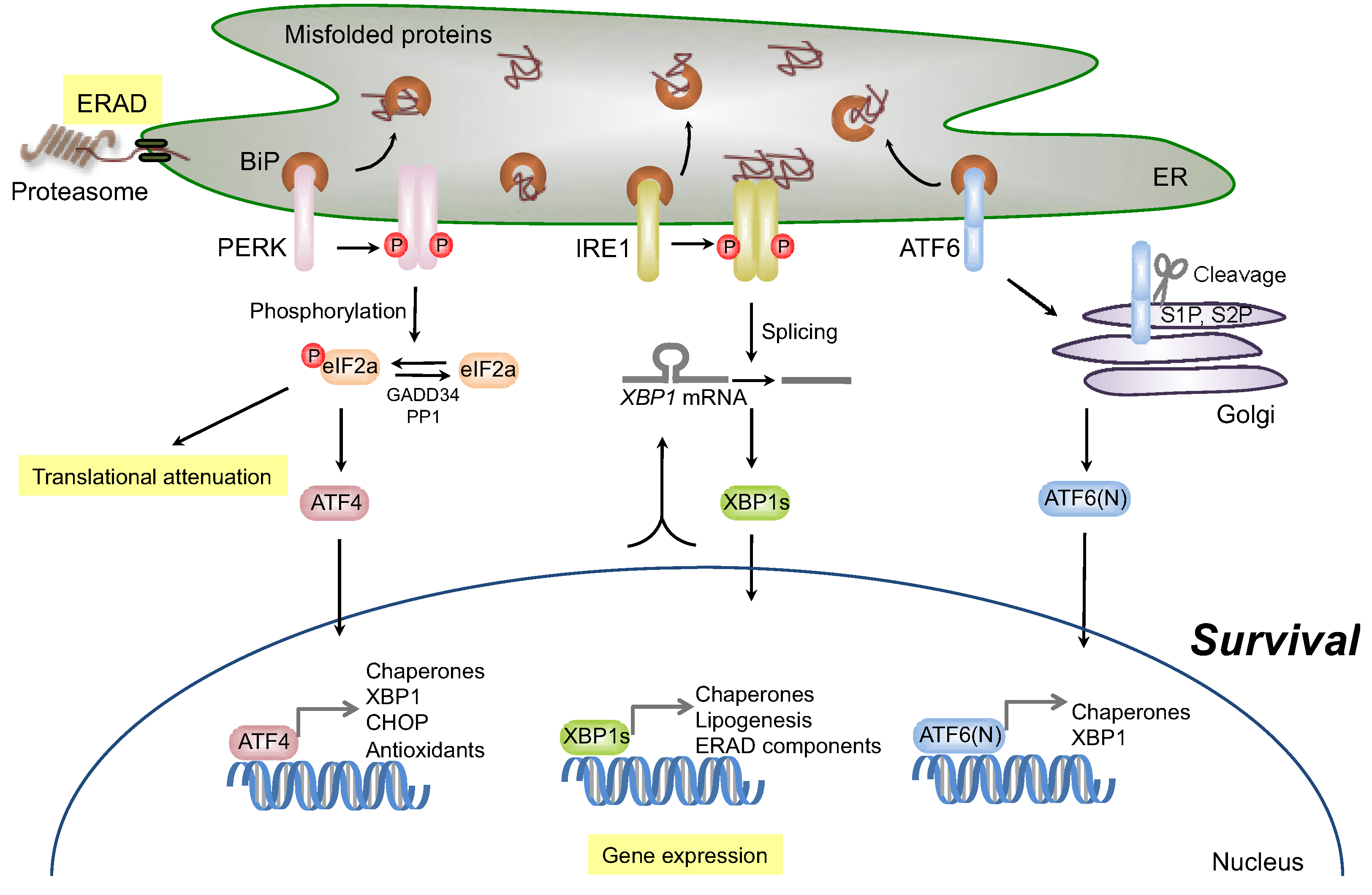
2.1. Signaling through Activating Transcription Factor-6 (ATF6)
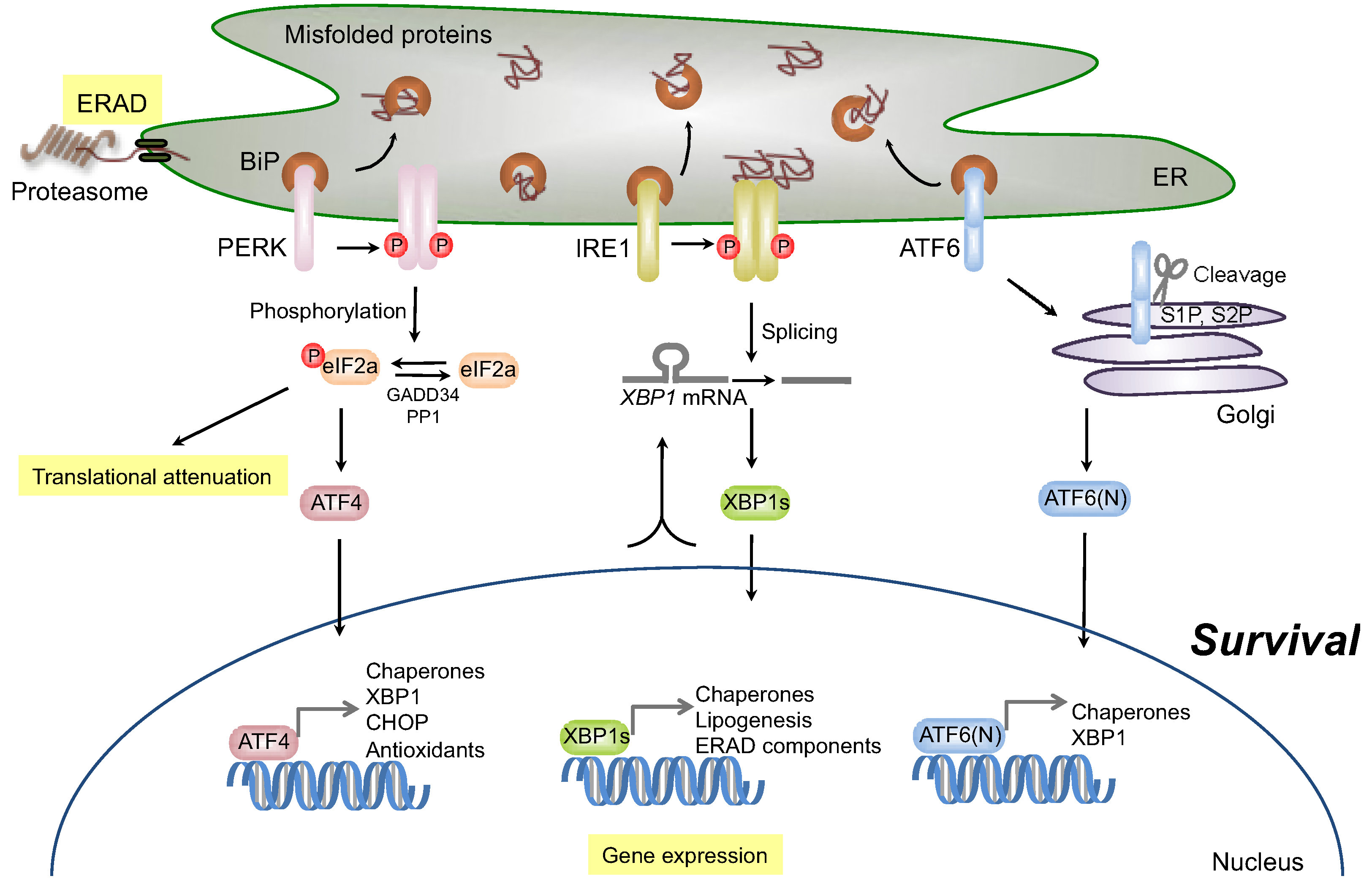
2.2. Signaling through Inositol-Requiring Transmembrane Kinase/Endoribonuclease 1 (IRE1)
2.2.1. Survival Signaling via IRE1
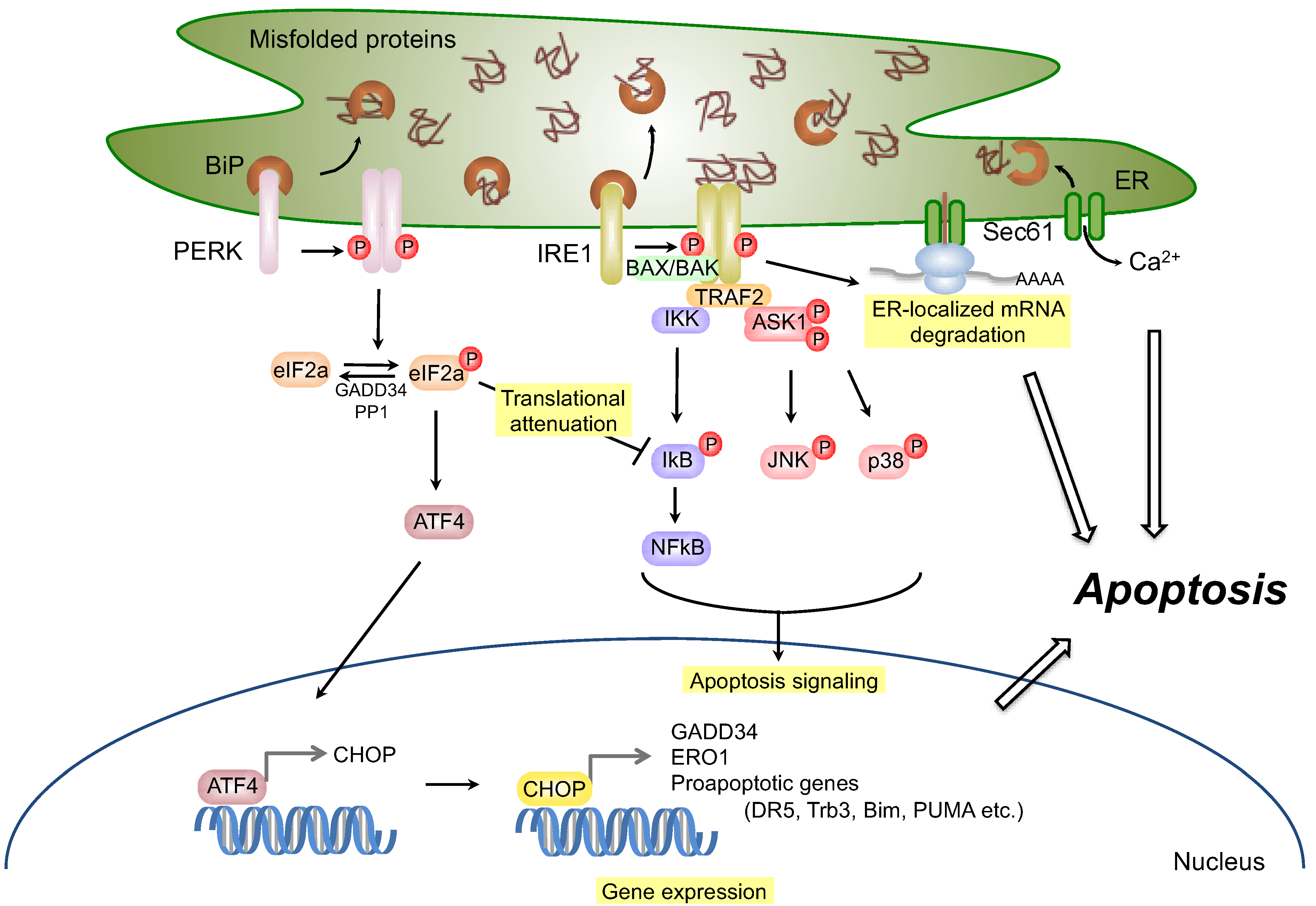
2.2.2. Apoptosis Signaling via IRE1
2.3. Signaling through PERK
2.3.1. Survival Signaling via PERK
2.3.2. Apoptosis Signaling via PERK
3. ER-Associated Degradation
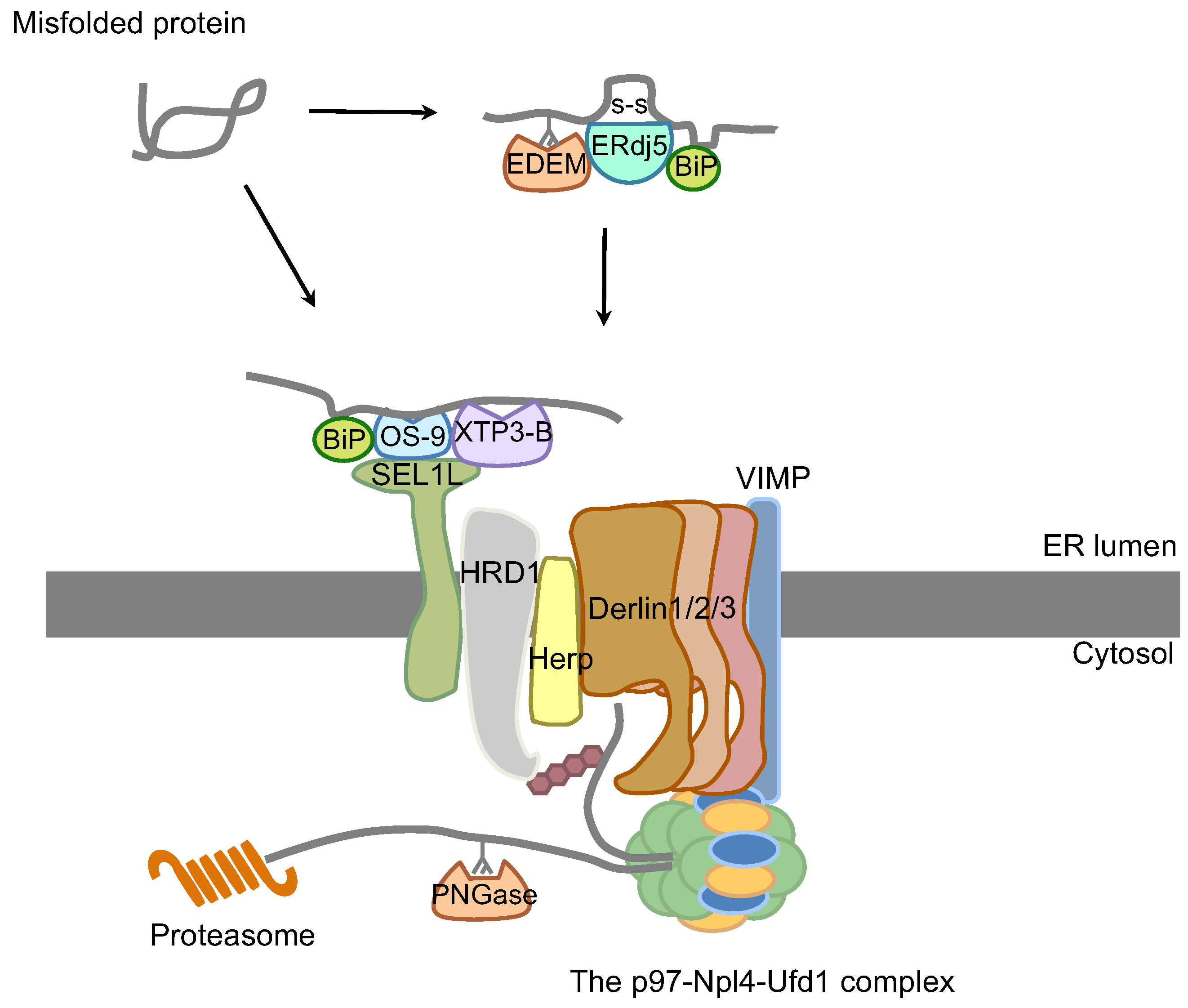
4. ER Stress and Related Diseases
4.1. Neurodegenerative Disease

{kind=link}
{kind=link}
{kind=link}
| Diseases | Key factors | The UPR signaling related physiological function & pathogenesis | References |
|---|---|---|---|
| Neurodegenerative disease | |||
| Alzheimer’s disease | Amyloid β (Aβ), IRE1, XBP1, PERK, eIF2α, CHOP |
| [90,91,92,93,94,95] |
| PolyQ diseases | Polyglutamine (polyQ), p97, IRE1, TRAF2, ASK1, JNK |
| [96,97] |
| [7] | ||
| Amyotrophic lateral sclerosis | SOD1, Derlin-1, ASK1 |
| [98] |
| XBP1 |
| [99] | |
| Metabolic disease | |||
| Hypertriglyceridemia | CREBH |
| [23,100] |
| Inflammatory disease | |||
| Inflammatory bowel disease | IRE1β, XBP1 |
| [43,101] |
| Diabetes mellitus | |||
| Type 1 diabetes | CHOP, NO |
| [102] |
| Type 2 diabetes | PERK, eIF2α, ATF6 |
| [50,54,103] |
| CHOP |
| [61] | |
| XBP1 |
| [42] | |
| IRE1α |
| [44,104] | |
| Wolcott-Rallison syndrome | PERK |
| [105] |
| Wolfram syndrome | WFS1 ATF6 |
| [106] |
| [107,108] | ||
| Cancer | |||
| Cancer | BiP, PERK, eIF2α, IRE1, XBP1 |
| [109,110,111,112,113,114,115,116,117,118] |
| Cardiovascular disease | |||
| Atherosclerosis | CHOP |
| [119,120,121] |
4.2. Metabolic Disease
4.3. Inflammatory Disease
4.4. Diabetes Mellitus
4.5. Cancer
4.6. Cardiovascular Disease
4.7. Therapeutic Approach
5. Concluding Remarks
Acknowledgements
Conflict of Interest
References
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the er transmembrane kinase/ribonuclease ire1 under er stress. Nat. Cell Biol. 2001, 3, 158–164. [Google Scholar]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the er to activation of jnk protein kinases by transmembrane protein kinase ire1. Science 2000, 287, 664–666. [Google Scholar]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. Ask1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic bax and bak modulate the unfolded protein response by a direct interaction with ire1alpha. Science 2006, 312, 572–576. [Google Scholar] [CrossRef]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. Bax and bak regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic bax and bak: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to er stress is mediated by differential stabilities of pro-survival and pro-apoptotic mrnas and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 2002, 109, 525–532. [Google Scholar]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. Chop is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Haze, K.; Okada, T.; Yoshida, H.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Identification of the g13 (camp-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 2001, 355, 19–28. [Google Scholar] [CrossRef]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of atf6 to bip in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of atf6 senses endoplasmic reticulum (er) stress and causes translocation of atf6 from the er to the golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Atf6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef]
- Dorner, A.J.; Wasley, L.C.; Raney, P.; Haugejorden, S.; Green, M.; Kaufman, R.J. The stress response in chinese hamster ovary cells. Regulation of erp72 and protein disulfide isomerase expression and secretion. J. Biol. Chem. 1990, 265, 22029–22034. [Google Scholar]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor atf6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. Atf6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with nf-y and yy1. Mol. Cell. Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Atf6 activated by proteolysis binds in the presence of nf-y (cbf) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian er quality control proteins is mediated by single or combined action of atf6alpha and xbp1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of crebh to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Kondo, S.; Murakami, T.; Tatsumi, K.; Ogata, M.; Kanemoto, S.; Otori, K.; Iseki, K.; Wanaka, A.; Imaizumi, K. Oasis, a creb/atf-family member, modulates upr signalling in astrocytes. Nat. Cell Biol. 2005, 7, 186–194. [Google Scholar] [CrossRef]
- Nagamori, I.; Yabuta, N.; Fujii, T.; Tanaka, H.; Yomogida, K.; Nishimune, Y.; Nojima, H. Tisp40, a spermatid specific bzip transcription factor, functions by binding to the unfolded protein response element via the rip pathway. Genes Cells Devoted Mol. Cell. Mech. 2005, 10, 575–594. [Google Scholar] [CrossRef]
- Stirling, J.; O’Hare, P. Creb4, a transmembrane bzip transcription factor and potential new substrate for regulation and cleavage by s1p. Mol. Biol. Cell 2006, 17, 413–426. [Google Scholar] [CrossRef]
- Kondo, S.; Saito, A.; Hino, S.; Murakami, T.; Ogata, M.; Kanemoto, S.; Nara, S.; Yamashita, A.; Yoshinaga, K.; Hara, H.; et al. Bbf2h7, a novel transmembrane bzip transcription factor, is a new type of endoplasmic reticulum stress transducer. Mol. Cell. Biol. 2007, 27, 1716–1729. [Google Scholar] [CrossRef]
- DenBoer, L.M.; Hardy-Smith, P.W.; Hogan, M.R.; Cockram, G.P.; Audas, T.E.; Lu, R. Luman is capable of binding and activating transcription from the unfolded protein response element. Biochem. Biophys. Res. Commun. 2005, 331, 113–119. [Google Scholar]
- Liang, G.; Audas, T.E.; Li, Y.; Cockram, G.P.; Dean, J.D.; Martyn, A.C.; Kokame, K.; Lu, R. Luman/creb3 induces transcription of the endoplasmic reticulum (er) stress response protein herp through an er stress response element. Mol. Cell. Biol. 2006, 26, 7999–8010. [Google Scholar]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian ire1 reveals diversity in the er stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef]
- Cox, J.S.; Walter, P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef]
- Mori, K.; Kawahara, T.; Yoshida, H.; Yanagi, H.; Yura, T. Signalling from endoplasmic reticulum to nucleus: Transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells Devoted Mol. Cell. Mech. 1996, 1, 803–817. [Google Scholar]
- Urano, F.; Bertolotti, A.; Ron, D. Ire1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 2000, 113, 3697–3702. [Google Scholar]
- Gardner, B.M.; Walter, P. Unfolded proteins are ire1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar]
- Promlek, T.; Ishiwata-Kimata, Y.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor ire1 in different ways. Mol. Biol. Cell 2011, 22, 3520–3532. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. Xbp1 mrna is induced by atf6 and spliced by ire1 in response to er stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. Xbp1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Masaki, T.; Yoshida, M.; Noguchi, S. Targeted disruption of cre-binding factor treb5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem. Biophys. Res. Commun. 1999, 261, 350–356. [Google Scholar]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of ire1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor xbp-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Lee, A.H.; Heidtman, K.; Hotamisligil, G.S.; Glimcher, L.H. Dual and opposing roles of the unfolded protein response regulated by ire1alpha and xbp1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 8885–8890. [Google Scholar]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in ire1beta-deficient mice. J. Clin. Invest. 2001, 107, 585–593. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. Ire1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated ire1-dependent decay of messenger rnas in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mrnas during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. Ire1alpha cleaves select micrornas during er stress to derepress translation of proapoptotic caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef]
- Kang, M.J.; Chung, J.; Ryoo, H.D. Cdk5 and mekk1 mediate pro-apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat. Cell Biol. 2012, 14, 409–415. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of bip and er stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar]
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. Atf4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar]
- Yamaguchi, H.; Wang, H.G. Chop is involved in endoplasmic reticulum stress-induced apoptosis by enhancing dr5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. Trb3, a novel er stress-inducible gene, is induced via atf4-chop pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. Er stress triggers apoptosis by activating bh3-only protein bim. Cell 2007, 129, 1337–1349. [Google Scholar]
- Cazanave, S.C.; Elmi, N.A.; Akazawa, Y.; Bronk, S.F.; Mott, J.L.; Gores, G.J. Chop and ap-1 cooperatively mediate puma expression during lipoapoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G236–G243. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. Chop induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 2008, 118, 3378–3389. [Google Scholar] [CrossRef]
- Hegde, R.S.; Ploegh, H.L. Quality and quantity control at the endoplasmic reticulum. Curr. Opin. Cell Biol. 2010, 22, 437–446. [Google Scholar]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of er proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef]
- Denic, V.; Quan, E.M.; Weissman, J.S. A luminal surveillance complex that selects misfolded glycoproteins for er-associated degradation. Cell 2006, 126, 349–359. [Google Scholar] [CrossRef]
- Gauss, R.; Jarosch, E.; Sommer, T.; Hirsch, C. A complex of yos9p and the hrd ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat. Cell Biol. 2006, 8, 849–854. [Google Scholar]
- Gauss, R.; Sommer, T.; Jarosch, E. The hrd1p ligase complex forms a linchpin between er-lumenal substrate selection and cdc48p recruitment. EMBO J. 2006, 25, 1827–1835. [Google Scholar] [CrossRef]
- Clerc, S.; Hirsch, C.; Oggier, D.M.; Deprez, P.; Jakob, C.; Sommer, T.; Aebi, M. Htm1 protein generates the n-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 2009, 184, 159–172. [Google Scholar] [CrossRef]
- Quan, E.M.; Kamiya, Y.; Kamiya, D.; Denic, V.; Weibezahn, J.; Kato, K.; Weissman, J.S. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell 2008, 32, 870–877. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human hrd1 is an e3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar]
- Nadav, E.; Shmueli, A.; Barr, H.; Gonen, H.; Ciechanover, A.; Reiss, Y. A novel mammalian endoplasmic reticulum ubiquitin ligase homologous to the yeast hrd1. Biochem. Biophys. Res. Commun. 2003, 303, 91–97. [Google Scholar] [CrossRef]
- Omura, T.; Kaneko, M.; Okuma, Y.; Orba, Y.; Nagashima, K.; Takahashi, R.; Fujitani, N.; Matsumura, S.; Hata, A.; Kubota, K.; et al. A ubiquitin ligase hrd1 promotes the degradation of pael receptor, a substrate of parkin. J. Neurochem. 2006, 99, 1456–1469. [Google Scholar] [CrossRef]
- Hosokawa, N.; Tremblay, L.O.; Sleno, B.; Kamiya, Y.; Wada, I.; Nagata, K.; Kato, K.; Herscovics, A. Edem1 accelerates the trimming of alpha1,2-linked mannose on the c branch of n-glycans. Glycobiology 2010, 20, 567–575. [Google Scholar] [CrossRef]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A.; et al. Edem3, a soluble edem homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar]
- Olivari, S.; Galli, C.; Alanen, H.; Ruddock, L.; Molinari, M. A novel stress-induced edem variant regulating endoplasmic reticulum-associated glycoprotein degradation. J. Biol. Chem. 2005, 280, 2424–2428. [Google Scholar]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Araki, K.; Nagata, K. Protein folding and quality control in the er. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. Erdj5 is required as a disulfide reductase for degradation of misfolded proteins in the er. Science 2008, 321, 569–572. [Google Scholar] [CrossRef]
- Lilley, B.N.; Ploegh, H.L. A membrane protein required for dislocation of misfolded proteins from the er. Nature 2004, 429, 834–840. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus us11 gene product dislocates mhc class i heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef]
- Wiertz, E.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar] [CrossRef]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the er lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef]
- Alder, N.N.; Shen, Y.; Brodsky, J.L.; Hendershot, L.M.; Johnson, A.E. The molecular mechanisms underlying bip-mediated gating of the sec61 translocon of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 389–399. [Google Scholar] [CrossRef]
- Hammadi, M.; Oulidi, A.; Gackiere, F.; Katsogiannou, M.; Slomianny, C.; Roudbaraki, M.; Dewailly, E.; Delcourt, P.; Lepage, G.; Lotteau, S.; et al. Modulation of er stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: Involvement of grp78. FASEB J. 2013, 27, 1600–1609. [Google Scholar] [CrossRef]
- Schauble, N.; Lang, S.; Jung, M.; Cappel, S.; Schorr, S.; Ulucan, O.; Linxweiler, J.; Dudek, J.; Blum, R.; Helms, V.; et al. Bip-mediated closing of the sec61 channel limits Ca2+ leakage from the er. EMBO J. 2012, 31, 3282–3296. [Google Scholar] [CrossRef]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal er protein by the ubiquitin-ligase hrd1p. Cell 2010, 143, 579–591. [Google Scholar]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef]
- Ye, Y.; Shibata, Y.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Rapoport, T.A. Recruitment of the p97 atpase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14132–14138. [Google Scholar]
- Greenblatt, E.J.; Olzmann, J.A.; Kopito, R.R. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2011, 18, 1147–1152. [Google Scholar]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; Veerhuis, R.; van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in alzheimer's disease, but not in aged tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef]
- Lee do, Y.; Lee, K.S.; Lee, H.J.; Kim do, H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of perk signaling attenuates abeta-mediated er stress. PLoS One 2010, 5, e10489. [Google Scholar] [CrossRef]
- Ghribi, O.; Herman, M.M.; DeWitt, D.A.; Forbes, M.S.; Savory, J. Abeta(1–42) and aluminum induce stress in the endoplasmic reticulum in rabbit hippocampus, involving nuclear translocation of gadd 153 and nf-kappab. Brain Res. Mol. Brain Res. 2001, 96, 30–38. [Google Scholar]
- Song, S.; Lee, H.; Kam, T.I.; Tai, M.L.; Lee, J.Y.; Noh, J.Y.; Shim, S.M.; Seo, S.J.; Kong, Y.Y.; Nakagawa, T.; et al. E2–25k/hip-2 regulates caspase-12 in er stress-mediated abeta neurotoxicity. J. Cell Biol. 2008, 182, 675–684. [Google Scholar]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R. Global changes to the ubiquitin system in huntington’s disease. Nature 2007, 448, 704–708. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Lindquist, S. Impaired erad and er stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008, 22, 3308–3319. [Google Scholar]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. Als-linked mutant sod1 induces er stress- and ask1-dependent motor neuron death by targeting derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. Xbp-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor camp responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. Xbp1 links er stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef]
- Oyadomari, S.; Takeda, K.; Takiguchi, M.; Gotoh, T.; Matsumoto, M.; Wada, I.; Akira, S.; Araki, E.; Mori, M. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 10845–10850. [Google Scholar] [CrossRef]
- Scheuner, D.; Vander Mierde, D.; Song, B.; Flamez, D.; Creemers, J.W.; Tsukamoto, K.; Ribick, M.; Schuit, F.C.; Kaufman, R.J. Control of mrna translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat. Med. 2005, 11, 757–764. [Google Scholar] [CrossRef]
- Lipson, K.L.; Ghosh, R.; Urano, F. The role of ire1alpha in the degradation of insulin mrna in pancreatic beta-cells. PLoS One 2008, 3, e1648. [Google Scholar] [CrossRef]
- Delepine, M.; Nicolino, M.; Barrett, T.; Golamaully, M.; Lathrop, G.M.; Julier, C. Eif2ak3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with wolcott-rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. Wfs1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates er stress signaling in rodent and human cells. J. Clin. Invest. 2010, 120, 744–755. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Urano, F.; Weir, G.C.; Gromada, J.; Burcin, M. Wolfram syndrome 1 and adenylyl cyclase 8 interact at the plasma membrane to regulate insulin production and secretion. Nat. Cell Biol. 2012, 14, 1105–1112. [Google Scholar] [CrossRef]
- Lee, A.S. Grp78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar]
- Jamora, C.; Dennert, G.; Lee, A.S. Inhibition of tumor progression by suppression of stress protein grp78/bip induction in fibrosarcoma b/c10me. Proc. Natl. Acad. Sci. USA 1996, 93, 7690–7694. [Google Scholar] [CrossRef]
- Fu, Y.; Lee, A.S. Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol. Ther. 2006, 5, 741–744. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, Y.; Jia, Z.; Li, Q.; Gong, W.; Wang, L.; Wei, D.; Yao, J.; Fang, S.; Xie, K. Association of elevated grp78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin. Exp. Metastasis 2006, 23, 401–410. [Google Scholar]
- Tsutsumi, S.; Namba, T.; Tanaka, K.I.; Arai, Y.; Ishihara, T.; Aburaya, M.; Mima, S.; Hoshino, T.; Mizushima, T. Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells. Oncogene 2006, 25, 1018–1029. [Google Scholar] [CrossRef]
- Fels, D.R.; Koumenis, C. The perk/eif2alpha/atf4 module of the upr in hypoxia resistance and tumor growth. Cancer Biol. Ther. 2006, 5, 723–728. [Google Scholar]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. Perk promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef]
- Auf, G.; Jabouille, A.; Guerit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef]
- Fujimoto, T.; Yoshimatsu, K.; Watanabe, K.; Yokomizo, H.; Otani, T.; Matsumoto, A.; Osawa, G.; Onda, M.; Ogawa, K. Overexpression of human x-box binding protein 1 (xbp-1) in colorectal adenomas and adenocarcinomas. Anticancer Res. 2007, 27, 127–131. [Google Scholar]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. Xbp1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef]
- Gao, J.; Ishigaki, Y.; Yamada, T.; Kondo, K.; Yamaguchi, S.; Imai, J.; Uno, K.; Hasegawa, Y.; Sawada, S.; Ishihara, H.; et al. Involvement of endoplasmic stress protein c/ebp homologous protein in arteriosclerosis acceleration with augmented biological stress responses. Circulation 2011, 124, 830–839. [Google Scholar] [CrossRef]
- Thorp, E.; Li, G.; Seimon, T.A.; Kuriakose, G.; Ron, D.; Tabas, I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of apoe−/− and ldlr−/− mice lacking chop. Cell Metab. 2009, 9, 474–481. [Google Scholar] [CrossRef]
- Tsukano, H.; Gotoh, T.; Endo, M.; Miyata, K.; Tazume, H.; Kadomatsu, T.; Yano, M.; Iwawaki, T.; Kohno, K.; Araki, K.; et al. The endoplasmic reticulum stress-c/ebp homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1925–1932. [Google Scholar] [CrossRef]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The er stress factor xbp1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet 2011, 20, 2144–2160. [Google Scholar]
- Schapansky, J.; Olson, K.; van der Ploeg, R.; Glazner, G. Nf-kappab activated by er calcium release inhibits abeta-mediated expression of chop protein: Enhancement by ad-linked mutant presenilin 1. Exp. Neurol. 2007, 208, 169–176. [Google Scholar] [CrossRef]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of hrd1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef]
- Yang, H.; Zhong, X.; Ballar, P.; Luo, S.; Shen, Y.; Rubinsztein, D.C.; Monteiro, M.J.; Fang, S. Ubiquitin ligase hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 2007, 313, 538–550. [Google Scholar] [CrossRef]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective er stress in disease manifestations of fals mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar]
- Wang, L.; Popko, B.; Roos, R.P. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2011, 20, 1008–1015. [Google Scholar] [CrossRef]
- Fujisawa, T.; Homma, K.; Yamaguchi, N.; Kadowaki, H.; Tsuburaya, N.; Naguro, I.; Matsuzawa, A.; Takeda, K.; Takahashi, Y.; Goto, J.; et al. A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked sod1 mutants. Ann. Neurol. 2012, 72, 739–749. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferre, P.; Foufelle, F. Grp78 expression inhibits insulin and er stress-induced srebp-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 2009, 119, 1201–1215. [Google Scholar] [CrossRef]
- Wang, Y.; Vera, L.; Fischer, W.H.; Montminy, M. The creb coactivator crtc2 links hepatic er stress and fasting gluconeogenesis. Nature 2009, 460, 534–537. [Google Scholar]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y. Atf6 modulates srebp2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Hatzivassiliou, G.; Grigoriadou, C.; Romero, M.; Cavener, D.R.; Thompson, C.B.; Diehl, J.A. Perk-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 16314–16319. [Google Scholar] [CrossRef]
- Sha, H.; He, Y.; Chen, H.; Wang, C.; Zenno, A.; Shi, H.; Yang, X.; Zhang, X.; Qi, L. The ire1alpha-xbp1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009, 9, 556–564. [Google Scholar]
- Batchvarova, N.; Wang, X.Z.; Ron, D. Inhibition of adipogenesis by the stress-induced protein chop (gadd153). EMBO J. 1995, 14, 4654–4661. [Google Scholar]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of pi3k, p85alpha and p85beta, interact with xbp-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef]
- Winnay, J.N.; Boucher, J.; Mori, M.A.; Ueki, K.; Kahn, C.R. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of x-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 2010, 16, 438–445. [Google Scholar] [CrossRef]
- Jurczak, M.J.; Lee, A.H.; Jornayvaz, F.R.; Lee, H.Y.; Birkenfeld, A.L.; Guigni, B.A.; Kahn, M.; Samuel, V.T.; Glimcher, L.H.; Shulman, G.I. Dissociation of inositol-requiring enzyme (ire1alpha)-mediated c-jun n-terminal kinase activation from hepatic insulin resistance in conditional x-box-binding protein-1 (xbp1) knock-out mice. J. Biol. Chem. 2012, 287, 2558–2567. [Google Scholar]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor xbp1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through ire1alpha-mediated mrna decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar]
- Wang, S.; Chen, Z.; Lam, V.; Han, J.; Hassler, J.; Finck, B.N.; Davidson, N.O.; Kaufman, R.J. Ire1alpha-xbp1s induces pdi expression to increase mtp activity for hepatic vldl assembly and lipid homeostasis. Cell Metab. 2012, 16, 473–486. [Google Scholar] [CrossRef]
- Garg, A.D.; Kaczmarek, A.; Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Er stress-induced inflammation: Does it aid or impede disease progression? Trends Mol. Med. 2012, 18, 589–598. [Google Scholar] [CrossRef]
- Kim, D.H.; Feinbaum, R.; Alloing, G.; Emerson, F.E.; Garsin, D.A.; Inoue, H.; Tanaka-Hino, M.; Hisamoto, N.; Matsumoto, K.; Tan, M.W.; et al. A conserved p38 map kinase pathway in caenorhabditis elegans innate immunity. Science 2002, 297, 623–626. [Google Scholar] [CrossRef]
- Richardson, C.E.; Kooistra, T.; Kim, D.H. An essential role for xbp-1 in host protection against immune activation in c. Elegans. Nature 2010, 463, 1092–1095. [Google Scholar] [CrossRef]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Chu, W.S.; Das, S.K.; Wang, H.; Chan, J.C.; Deloukas, P.; Froguel, P.; Baier, L.J.; Jia, W.; McCarthy, M.I.; Ng, M.C.; et al. Activating transcription factor 6 (atf6) sequence polymorphisms in type 2 diabetes and pre-diabetic traits. Diabetes 2007, 56, 856–862. [Google Scholar] [CrossRef]
- Meex, S.J.; van Greevenbroek, M.M.; Ayoubi, T.A.; Vlietinck, R.; van Vliet-Ostaptchouk, J.V.; Hofker, M.H.; Vermeulen, V.M.; Schalkwijk, C.G.; Feskens, E.J.; Boer, J.M.; et al. Activating transcription factor 6 polymorphisms and haplotypes are associated with impaired glucose homeostasis and type 2 diabetes in dutch caucasians. J. Clin. Endocrinol. Metab. 2007, 92, 2720–2725. [Google Scholar] [CrossRef]
- Thameem, F.; Farook, V.S.; Bogardus, C.; Prochazka, M. Association of amino acid variants in the activating transcription factor 6 gene (atf6) on 1q21-q23 with type 2 diabetes in pima indians. Diabetes 2006, 55, 839–842. [Google Scholar] [CrossRef]
- Usui, M.; Yamaguchi, S.; Tanji, Y.; Tominaga, R.; Ishigaki, Y.; Fukumoto, M.; Katagiri, H.; Mori, K.; Oka, Y.; Ishihara, H. Atf6alpha-null mice are glucose intolerant due to pancreatic beta-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism 2012, 61, 1118–1128. [Google Scholar]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Marhfour, I.; Lopez, X.M.; Lefkaditis, D.; Salmon, I.; Allagnat, F.; Richardson, S.J.; Morgan, N.G.; Eizirik, D.L. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012, 55, 2417–2420. [Google Scholar] [CrossRef]
- Elouil, H.; Bensellam, M.; Guiot, Y.; Vander Mierde, D.; Pascal, S.M.; Schuit, F.C.; Jonas, J.C. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia 2007, 50, 1442–1452. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Takeda, K.; Kadowaki, H.; Ueda, I.; Namba, Y.; Ouchi, Y.; Nishitoh, H.; Ichijo, H. Involvement of ask1-p38 pathway in the pathogenesis of diabetes triggered by pancreatic ss cell exhaustion. Biochim. Biophys. Acta 2013, 1830, 3656–3663. [Google Scholar] [CrossRef]
- Lipson, K.L.; Fonseca, S.G.; Urano, F. Endoplasmic reticulum stress-induced apoptosis and auto-immunity in diabetes. Curr. Mol. Med. 2006, 6, 71–77. [Google Scholar] [CrossRef]
- Hardy, C.; Khanim, F.; Torres, R.; Scott-Brown, M.; Seller, A.; Poulton, J.; Collier, D.; Kirk, J.; Polymeropoulos, M.; Latif, F.; et al. Clinical and molecular genetic analysis of 19 wolfram syndrome kindreds demonstrating a wide spectrum of mutations in wfs1. Am. J. Hum. Genet. 1999, 65, 1279–1290. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Schonthal, A.H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 2013, 85, 653–666. [Google Scholar]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef]
- Sawada, T.; Minamino, T.; Fu, H.Y.; Asai, M.; Okuda, K.; Isomura, T.; Yamazaki, S.; Asano, Y.; Okada, K.; Tsukamoto, O.; et al. X-box binding protein 1 regulates brain natriuretic peptide through a novel ap1/cre-like element in cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 48, 1280–1289. [Google Scholar] [CrossRef]
- Zhao, H.; Liao, Y.; Minamino, T.; Asano, Y.; Asakura, M.; Kim, J.; Asanuma, H.; Takashima, S.; Hori, M.; Kitakaze, M. Inhibition of cardiac remodeling by pravastatin is associated with amelioration of endoplasmic reticulum stress. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2008, 31, 1977–1987. [Google Scholar] [CrossRef]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of c/ebp homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef]
- Tsukamoto, O.; Minamino, T.; Okada, K.; Shintani, Y.; Takashima, S.; Kato, H.; Liao, Y.; Okazaki, H.; Asai, M.; Hirata, A.; et al. Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem. Biophys. Res. Commun. 2006, 340, 1125–1133. [Google Scholar] [CrossRef]
- Weekes, J.; Morrison, K.; Mullen, A.; Wait, R.; Barton, P.; Dunn, M.J. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 2003, 3, 208–216. [Google Scholar] [CrossRef]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.M.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013, 144, 989–1000. [Google Scholar] [CrossRef]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce er stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eif2alpha dephosphorylation protects cells from er stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Lin, W.; Kunkler, P.E.; Harding, H.P.; Ron, D.; Kraig, R.P.; Popko, B. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am. J. Pathol. 2008, 173, 1508–1517. [Google Scholar] [CrossRef]
- Reijonen, S.; Putkonen, N.; Norremolle, A.; Lindholm, D.; Korhonen, L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by n-terminal mutant huntingtin proteins. Exp. Cell Res. 2008, 314, 950–960. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef]
- Cnop, M.; Ladriere, L.; Hekerman, P.; Ortis, F.; Cardozo, A.K.; Dogusan, Z.; Flamez, D.; Boyce, M.; Yuan, J.; Eizirik, D.L. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J. Biol. Chem. 2007, 282, 3989–3997. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kadowaki, H.; Nishitoh, H. Signaling Pathways from the Endoplasmic Reticulum and Their Roles in Disease. Genes 2013, 4, 306-333. https://doi.org/10.3390/genes4030306
Kadowaki H, Nishitoh H. Signaling Pathways from the Endoplasmic Reticulum and Their Roles in Disease. Genes. 2013; 4(3):306-333. https://doi.org/10.3390/genes4030306
Chicago/Turabian StyleKadowaki, Hisae, and Hideki Nishitoh. 2013. "Signaling Pathways from the Endoplasmic Reticulum and Their Roles in Disease" Genes 4, no. 3: 306-333. https://doi.org/10.3390/genes4030306
APA StyleKadowaki, H., & Nishitoh, H. (2013). Signaling Pathways from the Endoplasmic Reticulum and Their Roles in Disease. Genes, 4(3), 306-333. https://doi.org/10.3390/genes4030306