Use of RNA Interference by In Utero Electroporation to Study Cortical Development: The Example of the Doublecortin Superfamily

Abstract
:1. Introduction
2. Introduction of shRNA in the Developing Brain: Pros and Cons
2.1. Silencing of Gene Expression
2.2. Gene Redundancy

{kind=link}
{kind=link}
| Protein | Expression | Function |
|---|---|---|
| DCX | The developing neocortex [21,29], olfactory cells, developing retina [20], adult neurogenic regions [30,31] | Neuronal migration during development [4,32,33]; migration of adult SVZ cells [34]; branching of neurites, dendrites [32,35,36]; epilepsy [17] |
| DCLK1 | Developing neocortex [20,37,38], olfactory cells [20], adult brain [31,39], developing retina [20], neuronal progenitors [31] | Neuronal migration during development [32,39]; neurogenesis [38]; apoptosis [40,41]; hippocampal activity, anxious behavior, contextual fear memories [42,43,44] |
| DCLK2 | Developing neocortex [45,46], adult brain [45], developing retina [20] | Hippocampal lamination [47]; branching of dendrites (hippocampus) [47] |
| DCLK3 | Adult brain [48] | |
| RP1 | Retina, part of the photoreceptor axoneme [49] | Microtubule organization [49], organization of the photoreceptor outer segment [50] |
| RP1L1 | Developing retina [20,28] | |
| DCDC2 | Cortex [26], choroid plexus, cerebellum [20] | Neuronal migration [26], structure and function of primary cilia [51] |
| DCDC2B | Developing neocortex [20] | |
| DCDC2C | Ubiquitous expression [20] | |
| DCDC5 | Thalamus, posterior hypothalamus, septum, developing retina, olfactory cells, choroid plexus [20] | Mitosis [52] |
2.3. Controls, Off-Targets
| PROS | CONS |
|---|---|
|
|
2.4. Rescue
2.5. Targeting Different Areas or Different Cell Populations
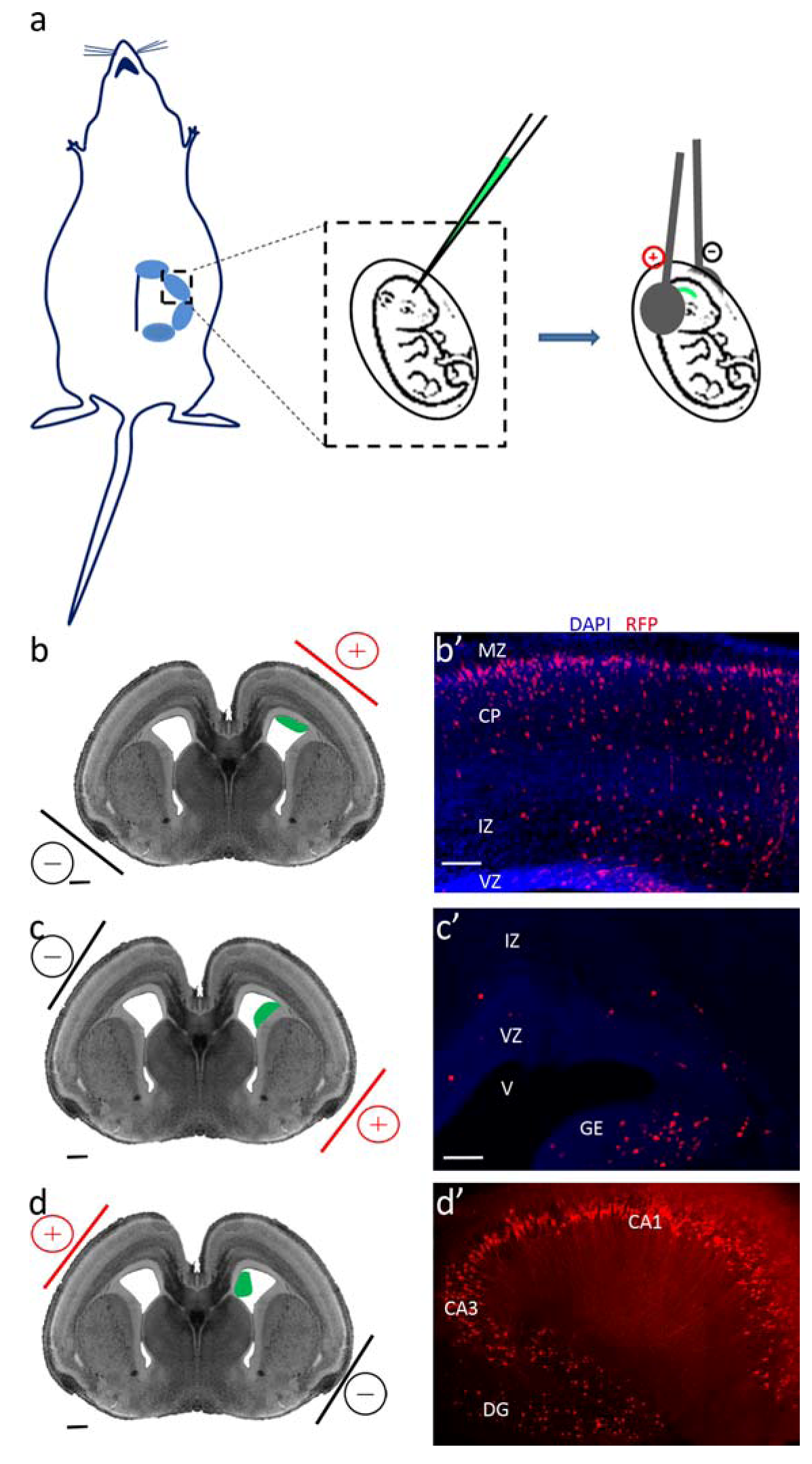
| In utero electroporation | |
| Electroporated area | Labeled population |
| Ventricular zone of the dorsal telencephalon | Cortical progenitors and projection neurons |
| Lateral telencephalon/corticostriatal junction | Neurons of the amygdala and piriform cortex |
| Ganglionic eminences of the ventral telencephalon | Cortical interneurons |
| Hippocampal neuroepithelium | Hippocampal neurons |
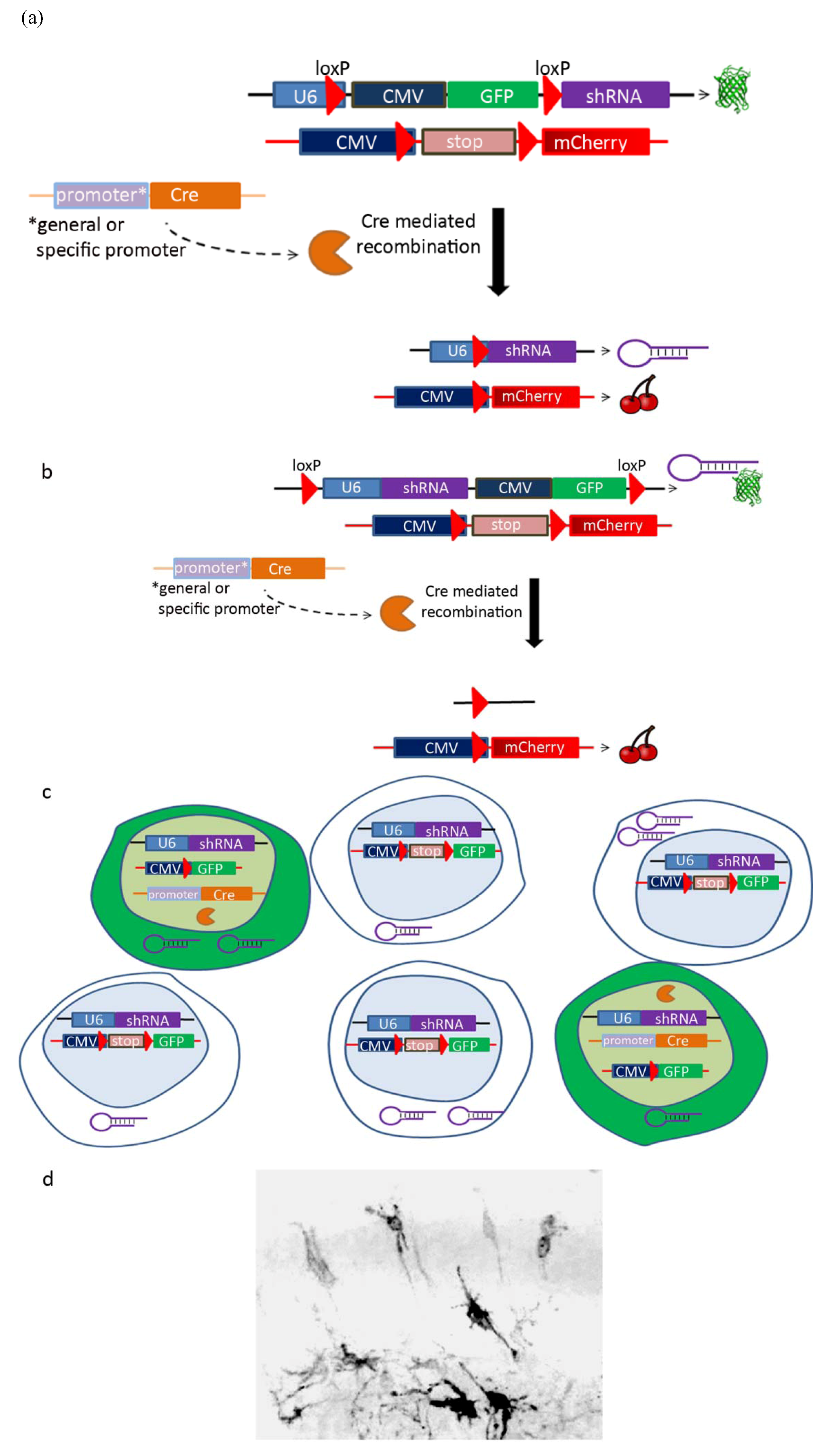
| Cre-Lox system | |
| Promoter | Labeled population |
| BLBP, GFAP or GLAST | Radial glia [78] |
| Tubulin α | Post-mitotic neurons and neuronal progenitors [78] |
| Nestin | All ventricular zone progenitors [79] |
2.6. Cell-Autonomous versus Non Cell-Autonomous Features
3. Conclusions and Perspectives
Acknowledgements
References and Notes
- Dorsett, Y.; Tuschl, T. SiRNAs: Applications in functional genomics and potential as therapeutics. Nature reviews. Drug Discov. 2004, 3, 318–329. [Google Scholar]
- Montgomery, M.K.; Xu, S.; Fire, A. RNA as a target of double-stranded RNA-mediated genetic interference in caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1998, 95, 15502–15507. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Bai, J.; Ramos, R.L.; Ackman, J.B.; Thomas, A.M.; Lee, R.V.; LoTurco, J.J. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 2003, 6, 1277–1283. [Google Scholar]
- Mittal, V. Improving the efficiency of RNA interference in mammals. Nat. Rev. Genet. 2004, 5, 355–365. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Zeng, L.; Gu, S.; Li, Y.; Zhao, E.; Xu, J.; Ye, X.; Wu, Q.; Wang, L.; Xie, Y.; Mao, Y. Identification of a novel human doublecortin-domain-containing gene (DCDC1) expressed mainly in testis. J. Hum. Genet. 2003, 48, 393–396. [Google Scholar] [CrossRef]
- Carmell, M.A.; Hannon, G.J. RNase III enzymes and the initiation of gene silencing. Nat. Struct. Mol. Biol. 2004, 11, 214–218. [Google Scholar] [CrossRef]
- Nykanen, A.; Haley, B.; Zamore, P.D. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell 2001, 107, 309–321. [Google Scholar]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Luhrmann, R.; Tuschl, T. Single-stranded antisense siRNAs guide target RNA cleavage in RANi. Cell 2002, 110, 563–574. [Google Scholar]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar]
- Taxman, D.J.; Livingstone, L.R.; Zhang, J.; Conti, B.J.; Iocca, H.A.; Williams, K.L.; Lich, J.D.; Ting, J.P.; Reed, W. Criteria for effective design, construction, and gene knockdown by shRAN vector. BMC Biotechnol. 2006, 6, 7. [Google Scholar] [CrossRef]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational siRAN design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar]
- Yu, J.Y.; DeRuiter, S.L.; Turner, D.L. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6047–6052. [Google Scholar]
- Krichevsky, A.M.; Kosik, K.S. RNAi functions in cultured mammalian neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 11926–11929. [Google Scholar] [CrossRef]
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar]
- Coquelle, F.M.; Levy, T.; Bergmann, S.; Wolf, S.G.; Bar-El, D.; Sapir, T.; Brody, Y.; Orr, I.; Barkai, N.; Eichele, G.; Reiner, O. Common and divergent roles for members of the mouse dcx superfamily. Cell Cycle 2006, 5, 976–983. [Google Scholar] [CrossRef]
- Reiner, O.; Coquelle, F.M.; Peter, B.; Levy, T.; Kaplan, A.; Sapir, T.; Orr, I.; Barkai, N.; Eichele, G.; Bergmann, S. The evolving doublecortin (dcx) superfamily. BMC Genomics 2006, 7, 188. [Google Scholar]
- des Portes, V.; Pinard, J.M.; Billuart, P.; Vinet, M.C.; Koulakoff, A.; Carrie, A.; Gelot, A.; Dupuis, E.; Motte, J.; Berwald-Netter, Y.; et al. A novel cns gene required for neuronal migration and involved in x-linked subcortical laminar hetrotropia and lissencephaly syndrome. Cell 1998, 92, 51–61. [Google Scholar] [CrossRef]
- Gleeson, J.G.; Allen, K.M.; Fox, J.W.; Lamperti, E.D.; Berkovic, S.; Scheffer, I.; Cooper, E.C.; Dobyns, W.B.; Minnerath, S.R.; Ross, M.E.; et al. Doublecortin, a brain-specific gene mutated in human x-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 1998, 92, 63–72. [Google Scholar]
- Le Hellard, S.; Havik, B.; Espeseth, T.; Breilid, H.; Lovlie, R.; Luciano, M.; Gow, A.J.; Harris, S.E.; Starr, J.M.; Wibrand, K.; et al. Variants in doublecortin- and calmodulin kinase like 1, a gene up-regulated by BDNF, are associated with memory and general cognitive abilities. PLoS One 2009, 4, e7534. [Google Scholar]
- Lionel, A.C.; Crosbie, J.; Barbosa, N.; Goodale, T.; Thiruvahindrapuram, B.; Rickaby, J.; Gazzellone, M.; Carson, A.R.; Howe, J.L.; Wang, Z.; et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 2011, 3, 95ra75. [Google Scholar] [CrossRef]
- Schumacher, J.; Anthoni, H.; Dahdouh, F.; Konig, I.R.; Hillmer, A.M.; Kluck, N.; Manthey, M.; Plume, E.; Warnke, A.; Remschmidt, H.; et al. Strong genetic evidence of DCDC2 as a susceptibility gene for dyslexia. Am. J. Hum. Genet. 2006, 78, 52–62. [Google Scholar] [CrossRef]
- Meng, H.; Smith, S.D.; Hager, K.; Held, M.; Liu, J.; Olson, R.K.; Pennington, B.F.; Defries, J.C.; Gelernter, J.; O’Reilly-Pol, T.; et al. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc. Natl. Acad. Sci. USA 2005, 102, 17053–17058. [Google Scholar]
- Sullivan, L.S.; Heckenlively, J.R.; Bowne, S.J.; Zuo, J.; Hide, W.A.; Gal, A.; Denton, M.; Inglehearn, C.F.; Blanton, S.H.; Daiger, S.P. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat. Genet. 1999, 22, 255–259. [Google Scholar]
- Conte, I.; Lestingi, M.; den Hollander, A.; Alfano, G.; Ziviello, C.; Pugliese, M.; Circolo, D.; Caccioppoli, C.; Ciccodicola, A.; Banfi, S. Identification and characterisation of the retinitis pigmentosa 1-like1 gene (rp1l1): A novel candidate for retinal degenerations. Eur. J. Hum. Genet. 2003, 11, 155–162. [Google Scholar]
- Francis, F.; Koulakoff, A.; Boucher, D.; Chafey, P.; Schaar, B.; Vinet, M.C.; Friocourt, G.; McDonnell, N.; Reiner, O.; Kahn, A.; et al. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 1999, 23, 247–256. [Google Scholar]
- Nacher, J.; Crespo, C.; McEwen, B.S. Doublecortin expression in the adult rat telencephalon. Eur. J. Neurosci. 2001, 14, 629–644. [Google Scholar]
- Saaltink, D.J.; Havik, B.; Verissimo, C.S.; Lucassen, P.J.; Vreugdenhil, E. Doublecortin and doublecortin-like are expressed in overlapping and non-overlapping neuronal cell population: Implications for neurogenesis. J. Comp. Neurol. 2012, 520, 2805–2823. [Google Scholar]
- Friocourt, G.; Liu, J.S.; Antypa, M.; Rakic, S.; Walsh, C.A.; Parnavelas, J.G. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J. Neurosci. 2007, 27, 3875–3883. [Google Scholar] [CrossRef]
- Bai, J.; Ramos, R.L.; Paramasivam, M.; Siddiqi, F.; Ackman, J.B.; LoTurco, J.J. The role of dcx and lis1 in migration through the lateral cortical stream of developing forebrain. Dev. Neurosci. 2008, 30, 144–156. [Google Scholar]
- Ocbina, P.J.; Dizon, M.L.; Shin, L.; Szele, F.G. Doublecortin is necessary for the migration of adult subventricular zone cells from neurospheres. Mol. Cell. Neurosci. 2006, 33, 126–135. [Google Scholar]
- Cohen, D.; Segal, M.; Reiner, O. Doublecortin supports the development of dendritic arbors in primary hippocampal neurons. Dev. Neurosci. 2008, 30, 187–199. [Google Scholar]
- Shmueli, O.; Gdalyahu, A.; Sorokina, K.; Nevo, E.; Avivi, A.; Reiner, O. DCX in PC12 cells: CREB-mediated transcription and neurite outgrowth. Hum. Mol. Genet. 2001, 10, 1061–1070. [Google Scholar]
- Burgess, H.A.; Martinez, S.; Reiner, O. Kiaa0369, doublecortin-like kinase, is expressed during brain development. J. Neurosci. Res. 1999, 58, 567–575. [Google Scholar] [CrossRef]
- Shu, T.; Tseng, H.C.; Sapir, T.; Stern, P.; Zhou, Y.; Sanada, K.; Fischer, A.; Coquelle, F.M.; Reiner, O.; Tsai, L.H. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and m phase progression. Neuron 2006, 49, 25–39. [Google Scholar]
- Deuel, T.A.; Liu, J.S.; Corbo, J.C.; Yoo, S.Y.; Rorke-Adams, L.B.; Walsh, C.A. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron 2006, 49, 41–53. [Google Scholar] [CrossRef]
- Verissimo, C.S.; Cheng, S.; Puigvert, J.C.; Qin, Y.; Vroon, A.; van Deutekom, J.; Price, L.S.; Danen, E.H.; van de Water, B.; Fitzsimons, C.P.; et al. Combining doublecortin-like kinase silencing and vinca alkaloids results in a synergistic apoptotic effect in neuroblastoma cells. J. Pharmacol. Exp. Ther. 2012, 342, 119–130. [Google Scholar] [CrossRef]
- Verissimo, C.S.; Molenaar, J.J.; Meerman, J.; Puigvert, J.C.; Lamers, F.; Koster, J.; Danen, E.H.; van de Water, B.; Versteeg, R.; Fitzsimons, C.P.; et al. Silencing of the microtubule-associated proteins doublecortin-like and doublecortin-like kinase-long induces apoptosis in neuroblastoma cells. Endoc. Relat. Cancer 2010, 17, 399–414. [Google Scholar]
- Schenk, G.J.; Vreugdenhil, E.; Hubens, C.J.; Veldhuisen, B.; de Kloet, E.R.; Oitzl, M.S. Hippocampal carp over-expression solidifies consolidation of contextual fear memories. Physiol. Behav. 2011, 102, 323–331. [Google Scholar]
- Schenk, G.J.; Werkman, T.; Wadman, W.; Veldhuisen, B.; Dijkmans, T.F.; Blaas, E.; Kegel, L.; de Kloet, E.R.; Vreugdenhil, E. Over-expression of the dclk gene transcript carp decreases CA3/CA1 network excitability. Brain Res. 2010, 1352, 21–34. [Google Scholar] [CrossRef]
- Schenk, G.J.; Veldhuisen, B.; Wedemeier, O.; McGown, C.C.; Schouten, T.G.; Oitzl, M.; de Kloet, E.R.; Vreugdenhil, E. Over-expression of deltac-dclk-short in mouse brain results in a more anxious behavioral phenotype. Physiol. Behav. 2010, 101, 541–548. [Google Scholar]
- Edelman, A.M.; Kim, W.Y.; Higgins, D.; Goldstein, E.G.; Oberdoerster, M.; Sigurdson, W. Doublecortin kinase-2, a novel doublecortin-related protein kinase associated with terminal segments of axons and dendrites. J. Biol. Chem. 2005, 280, 8531–8543. [Google Scholar]
- Tuy, F.P.; Saillour, Y.; Kappeler, C.; Chelly, J.; Francis, F. Alternative transcripts of Dclk1 and Dclk2 and their expression in doublecortin knockout mice. Dev. Neurosci. 2008, 30, 171–186. [Google Scholar] [CrossRef]
- Kerjan, G.; Koizumi, H.; Han, E.B.; Dube, C.M.; Djakovic, S.N.; Patrick, G.N.; Baram, T.Z.; Heinemann, S.F.; Gleeson, J.G. Mice lacking doublecortin and doublecortin-like kinase 2 display altered hippocampal neuronal maturation and spontaneous seizures. Proc. Natl. Acad. Sci. USA 2009, 106, 6766–6771. [Google Scholar]
- Ohmae, S.; Takemoto-Kimura, S.; Okamura, M.; Adachi-Morishima, A.; Nonaka, M.; Fuse, T.; Kida, S.; Tanji, M.; Furuyashiki, T.; Arakawa, Y.; et al. Molecular identification and characterization of a family of kinases with homology to camki/camkiv. J. Biol. Chem. 2006, 281, 20427–20439. [Google Scholar]
- Liu, Q.; Zuo, J.; Pierce, E.A. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J. Neurosci. 2004, 24, 6427–6436. [Google Scholar]
- Gao, J.; Cheon, K.; Nusinowitz, S.; Liu, Q.; Bei, D.; Atkins, K.; Azimi, A.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; et al. Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocalization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc. Natl. Acad. Sci. USA 2002, 99, 5698–5703. [Google Scholar]
- Massinen, S.; Hokkanen, M.E.; Matsson, H.; Tammimies, K.; Tapia-Paez, I.; Dahlstrom-Heuser, V.; Kuja-Panula, J.; Burghoorn, J.; Jeppsson, K.E.; Swoboda, P.; et al. Increased expression of the dyslexia candidate gene DCDC2 affects length and signaling of primary cilia in neurons. PLoS One 2011, 6, e20580. [Google Scholar]
- Kaplan, A.; Reiner, O. Linking cytoplasmic dynein and transport of rab8 vesicles to the midbody during cytokinesis by the doublecortin domain-containing 5 protein. J. Cell. Sci. 2011, 124, 3989–4000. [Google Scholar]
- Corbo, J.C.; Deuel, T.A.; Long, J.M.; LaPorte, P.; Tsai, E.; Wynshaw-Boris, A.; Walsh, C.A. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J. Neurosci. 2002, 22, 7548–7557. [Google Scholar]
- Ramos, R.L.; Bai, J.; LoTurco, J.J. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of dcx. Cereb. Cortex 2006, 16, 1323–1331. [Google Scholar]
- Koizumi, H.; Tanaka, T.; Gleeson, J.G. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron 2006, 49, 55–66. [Google Scholar] [CrossRef]
- Koizumi, H.; Higginbotham, H.; Poon, T.; Tanaka, T.; Brinkman, B.C.; Gleeson, J.G. Doublecortin maintains bipolar shape and nuclear translocation during migration in the adult forebrain. Nat. Neurosci. 2006, 9, 779–786. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, X.; Rosen, G.; Gabel, L.; Guadiana, S.M.; Sarkisian, M.R.; Galaburda, A.M.; Loturco, J.J. DCDC2 knockout mice display exacerbated developmental disruptions following knockdown of doublecortin. Neuroscience 2011, 190, 398–408. [Google Scholar]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar]
- Cullen, B.R. Enhancing and confirming the specificity of RNAi experiments. Nat. Methods 2006, 3, 677–681. [Google Scholar]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]
- Hüttenhofer, A.; Schattner, P.; Hall, J.; Mattick, J.S.; Brummelkamp, T.R.; Bernards, R.; Martienssen, R.A. Whither RNAi? Nat. Cell. Biol. 2003, 5, 489–490. [Google Scholar] [CrossRef]
- Sapir, T.; Sapoznik, S.; Levy, T.; Finkelshtein, D.; Shmueli, A.; Timm, T.; Mandelkow, E.M.; Reiner, O. Accurate balance of the polarity kinase MARK2/Par-1 is required for proper cortical neuronal migration. J. Neurosci. 2008, 28, 5710–5720. [Google Scholar]
- LoTurco, J.J.; Bai, J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006, 29, 407–413. [Google Scholar]
- Manent, J.B.; Wang, Y.; Chang, Y.; Paramasivam, M.; LoTurco, J.J. Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nat. Med. 2009, 15, 84–90. [Google Scholar] [CrossRef]
- LoTurco, J.; Manent, J.B.; Sidiqi, F. New and improved tools for in utero electroporation studies of developing cerebral cortex. Cereb. Cortex 2009, 19, i120–i125. [Google Scholar]
- Schaar, B.T.; Kinoshita, K.; McConnell, S.K. Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron 2004, 41, 203–213. [Google Scholar] [CrossRef]
- Sapir, T.; Shmueli, A.; Levy, T.; Timm, T.; Elbaum, M.; Mandelkow, E.M.; Reiner, O. Antagonistic effects of doublecortin and MARK2/Par-1 in the developing cerebral cortex. J. Neurosci. 2008, 28, 13008–13013. [Google Scholar]
- Tabata, H.; Nakajima, K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience 2001, 103, 865–872. [Google Scholar] [CrossRef]
- Saito, T.; Nakatsuji, N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001, 240, 237–246. [Google Scholar] [CrossRef]
- Borrell, V.; Yoshimura, Y.; Callaway, E.M. Targeted gene delivery to telencephalic inhibitory neurons by directional in utero electroporation. J. Neurosci. Methods 2005, 143, 151–158. [Google Scholar] [CrossRef]
- Navarro-Quiroga, I.; Chittajallu, R.; Gallo, V.; Haydar, T.F. Long-term, selective gene expression in developing and adult hippocampal pyramidal neurons using focal in utero electroporation. J. Neurosci. 2007, 27, 5007–5011. [Google Scholar] [CrossRef]
- Angevine, J.B.; Sidman, R.L. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature 1961, 192, 766–768. [Google Scholar] [CrossRef]
- Langevin, L.M.; Mattar, P.; Scardigli, R.; Roussigne, M.; Logan, C.; Blader, P.; Schuurmans, C. Validating in utero electroporation for the rapid analysis of gene regulatory elements in the murine telencephalon. Dev. Dyn. 2007, 236, 1273–1286. [Google Scholar] [CrossRef]
- Matsuda, T.; Cepko, C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA 2007, 104, 1027–1032. [Google Scholar] [CrossRef]
- Konno, D.; Shioi, G.; Shitamukai, A.; Mori, A.; Kiyonari, H.; Miyata, T.; Matsuzaki, F. Neuroepithelial progenitors undergo lgn-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nat. Cell Biol. 2008, 10, 93–101. [Google Scholar]
- Morin, X.; Jaouen, F.; Durbec, P. Control of planar divisions by the g-protein regulator lgn maintains progenitors in the chick neuroepithelium. Nat. Neurosci. 2007, 10, 1440–1448. [Google Scholar]
- Wiznerowicz, M.; Szulc, J.; Trono, D. Tuning silence: Conditional systems for RNA interference. Nat. Methods 2006, 3, 682–688. [Google Scholar] [CrossRef]
- Gal, J.S.; Morozov, Y.M.; Ayoub, A.E.; Chatterjee, M.; Rakic, P.; Haydar, T.F. Molecular and morphological heterogeneity of neural precursors in the mouse neocortical proliferative zones. J. Neurosci. 2006, 26, 1045–1056. [Google Scholar]
- Malatesta, P.; Hack, M.A.; Hartfuss, E.; Kettenmann, H.; Klinkert, W.; Kirchhoff, F.; Gotz, M. Neuronal or glial progeny: Regional differences in radial glia fate. Neuron 2003, 37, 751–764. [Google Scholar] [CrossRef]
- Feil, R.; Brocard, J.; Mascrez, B.; LeMeur, M.; Metzger, D.; Chambon, P. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10887–10890. [Google Scholar] [CrossRef]
- Hayashi, S.; McMahon, A.P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 2002, 244, 305–318. [Google Scholar]
- Stegmeier, F.; Hu, G.; Rickles, R.J.; Hannon, G.J.; Elledge, S.J. A lentiviral microRNA-based system for single-copy polymerase ii-regulated RNA interference in mammalian cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13212–13217. [Google Scholar] [CrossRef]
- Wiznerowicz, M.; Trono, D. Conditional suppression of cellular genes: Lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 2003, 77, 8957–8961. [Google Scholar]
- Ohkawa, J.; Taira, K. Control of the functional activity of an antisense RNA by a tetracycline-responsive derivative of the human u6 snRNA promoter. Hum. Gene Ther. 2000, 11, 577–585. [Google Scholar] [CrossRef]
- Hippenmeyer, S.; Youn, Y.H.; Moon, H.M.; Miyamichi, K.; Zong, H.; Wynshaw-Boris, A.; Luo, L. Genetic mosaic dissection of lis1 and ndel1 in neuronal migration. Neuron 2010, 68, 695–709. [Google Scholar]
- Zong, H.; Espinosa, J.S.; Su, H.H.; Muzumdar, M.D.; Luo, L. Mosaic analysis with double markers in mice. Cell 2005, 121, 479–492. [Google Scholar]
- Reiner, O.; Sapoznik, S.; Sapir, T. Lissencephaly 1 linking to multiple diseases: Mental retardation, neurodegeneration, schizophrenia, male sterility, and more. Neuromol. Med. 2006, 8, 547–566. [Google Scholar] [CrossRef]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar]
- O'Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.H.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K.; et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef]
- Kirov, G.; Grozeva, D.; Norton, N.; Ivanov, D.; Mantripragada, K.K.; Holmans, P.; Craddock, N.; Owen, M.J.; O’Donovan, M.C. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum. Mol. Genet. 2009, 18, 1497–1503. [Google Scholar]
- Elia, J.; Gai, X.; Xie, H.M.; Perin, J.C.; Geiger, E.; Glessner, J.T.; D’Arcy, M.; deBerardinis, R.; Frackelton, E.; Kim, C.; et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 2010, 15, 637–646. [Google Scholar] [CrossRef]
- Xu, B.; Roos, J.L.; Levy, S.; van Rensburg, E.J.; Gogos, J.A.; Karayiorgou, M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008, 40, 880–885. [Google Scholar]
- Walsh, T.; McClellan, J.M.; McCarthy, S.E.; Addington, A.M.; Pierce, S.B.; Cooper, G.M.; Nord, A.S.; Kusenda, M.; Malhotra, D.; Bhandari, A.; et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008, 320, 539–543. [Google Scholar]
- Stone, J.L.; O’Donovan, M.C.; Gurling, H.; Kirov, G.K.; Blackwood, D.H.; Corvin, A.; Craddock, N.J.; Gill, M.; Hultman, C.M.; Lichtenstein, P.; et al. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008, 455, 237–241. [Google Scholar]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietilainen, O.P.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reiner, O.; Gorelik, A.; Greenman, R. Use of RNA Interference by In Utero Electroporation to Study Cortical Development: The Example of the Doublecortin Superfamily. Genes 2012, 3, 759-778. https://doi.org/10.3390/genes3040759
Reiner O, Gorelik A, Greenman R. Use of RNA Interference by In Utero Electroporation to Study Cortical Development: The Example of the Doublecortin Superfamily. Genes. 2012; 3(4):759-778. https://doi.org/10.3390/genes3040759
Chicago/Turabian StyleReiner, Orly, Anna Gorelik, and Raanan Greenman. 2012. "Use of RNA Interference by In Utero Electroporation to Study Cortical Development: The Example of the Doublecortin Superfamily" Genes 3, no. 4: 759-778. https://doi.org/10.3390/genes3040759