Breaking the Gingival Epithelial Barrier: Role of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin in Oral Infectious Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
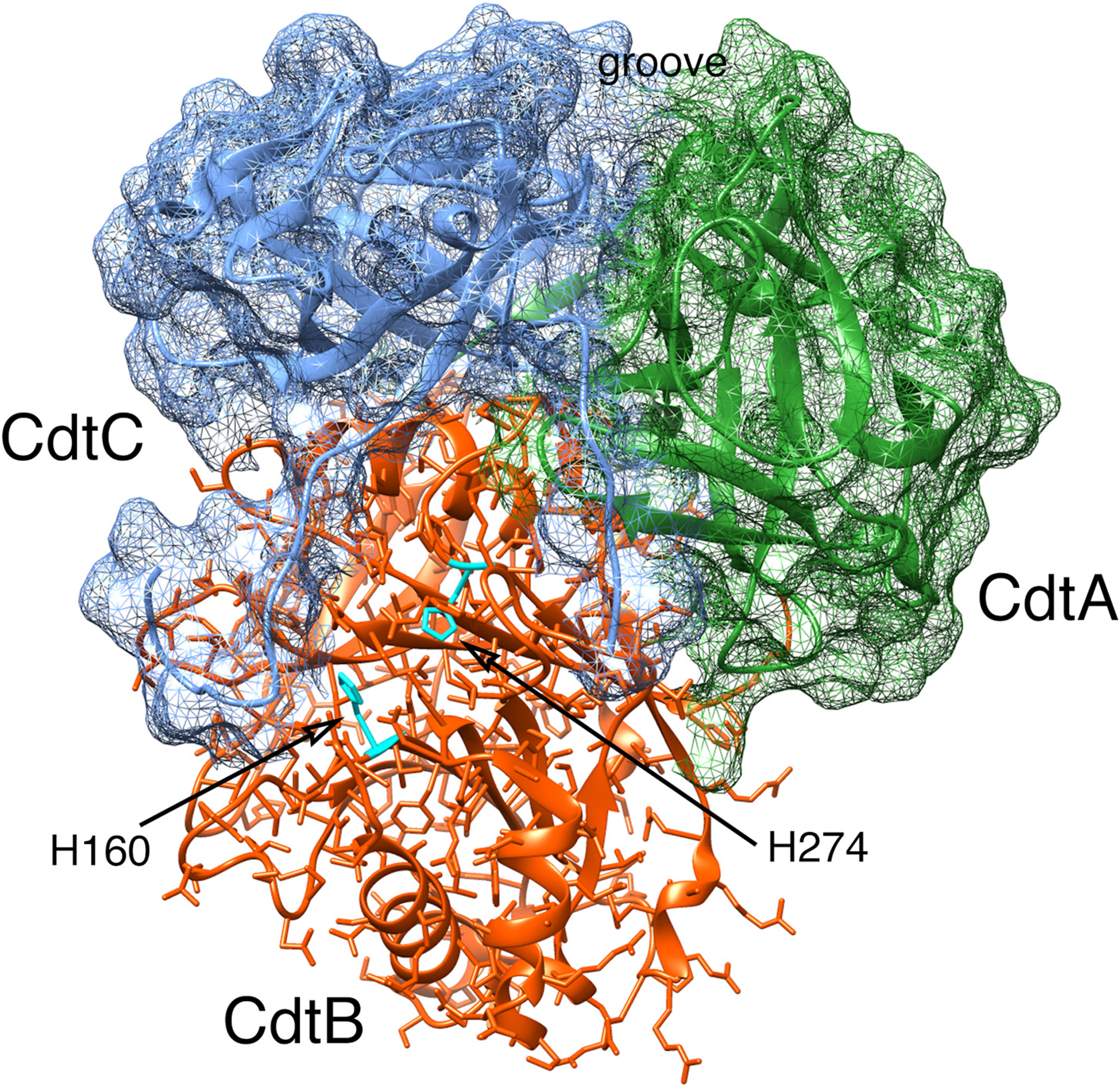
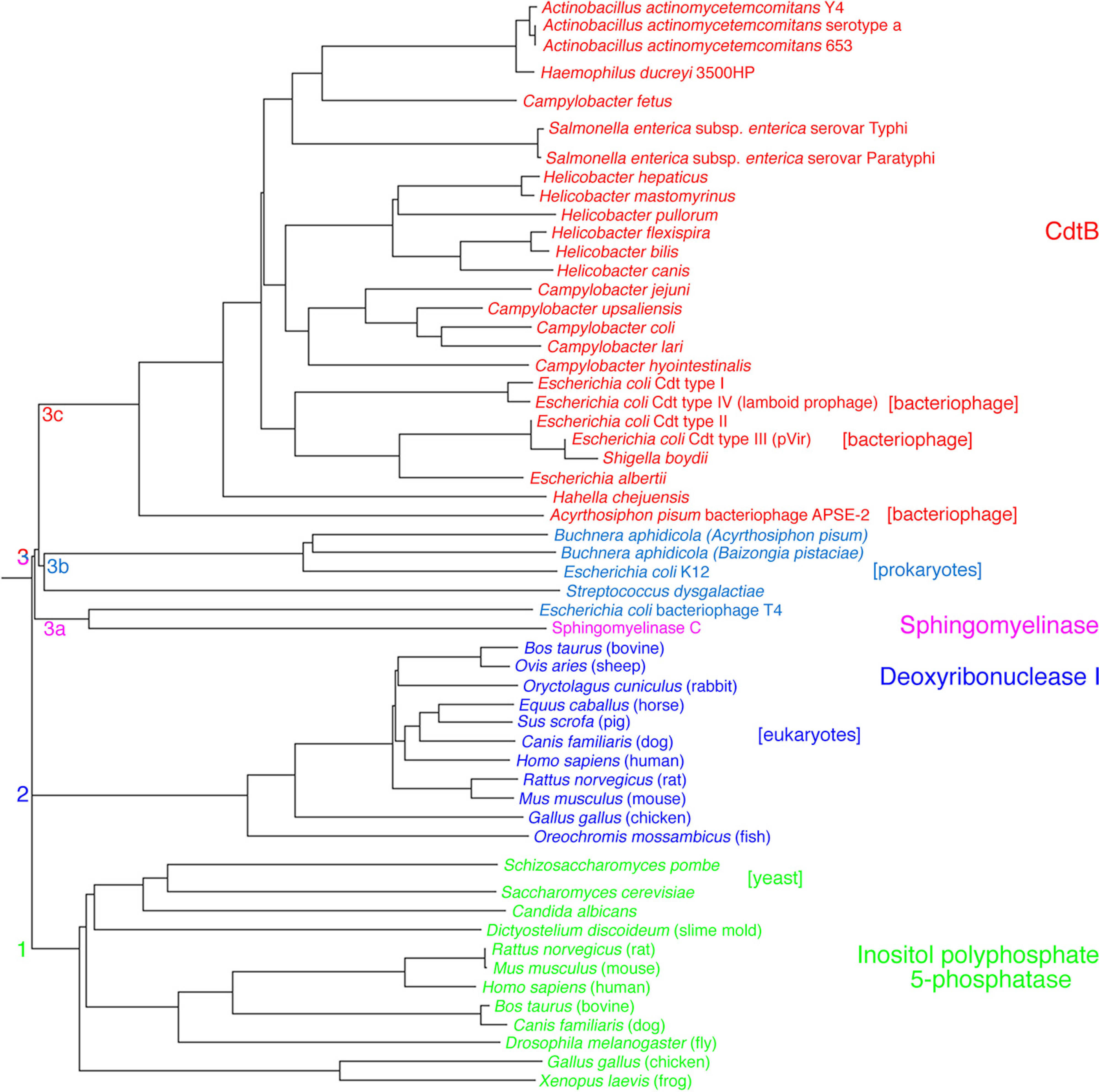
2. Aggregatibacter actinomycetemcomitans Cdt Structure and Function
2.1. Cell Surface Recognition
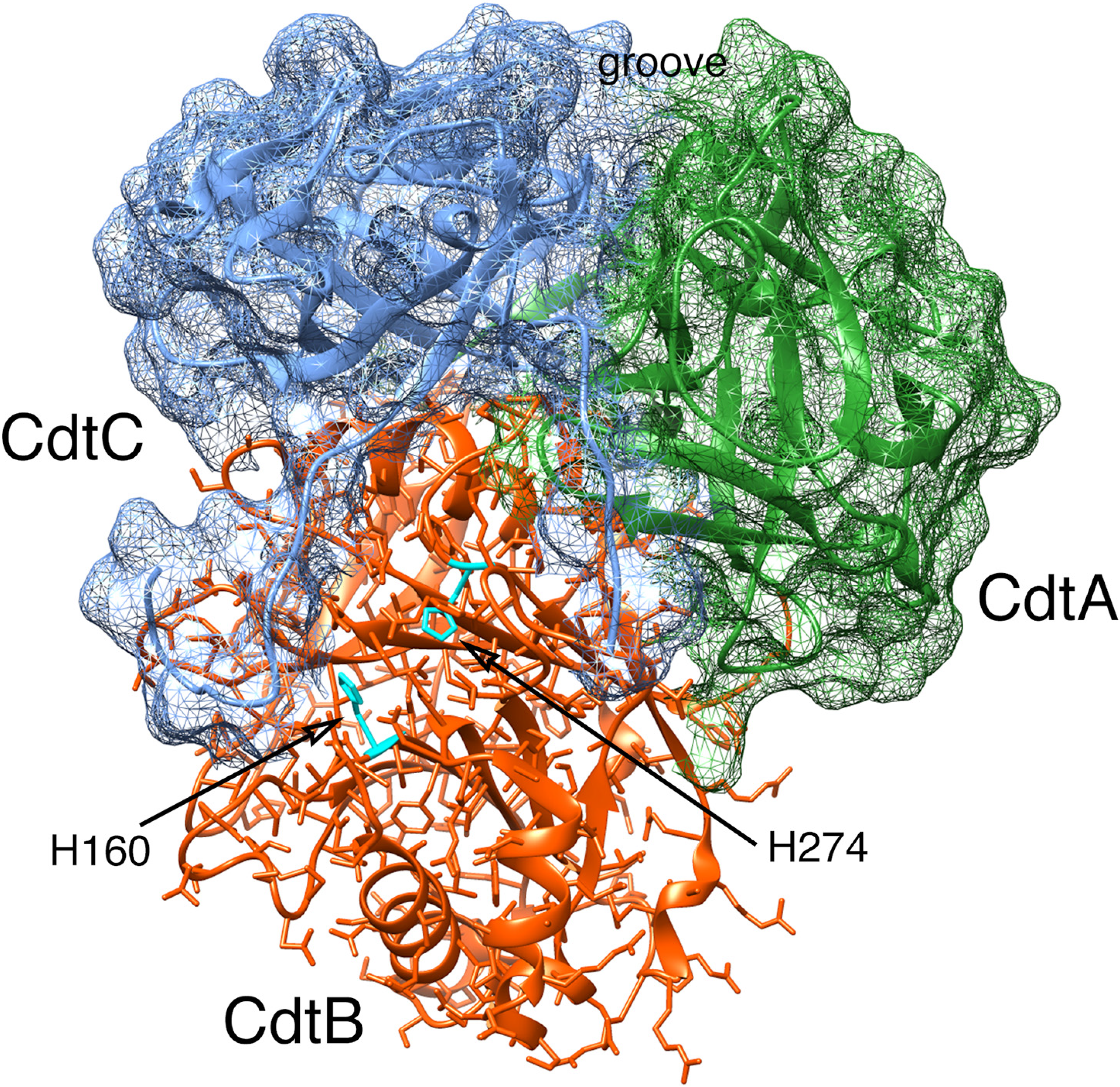
2.2. Cytotoxicity
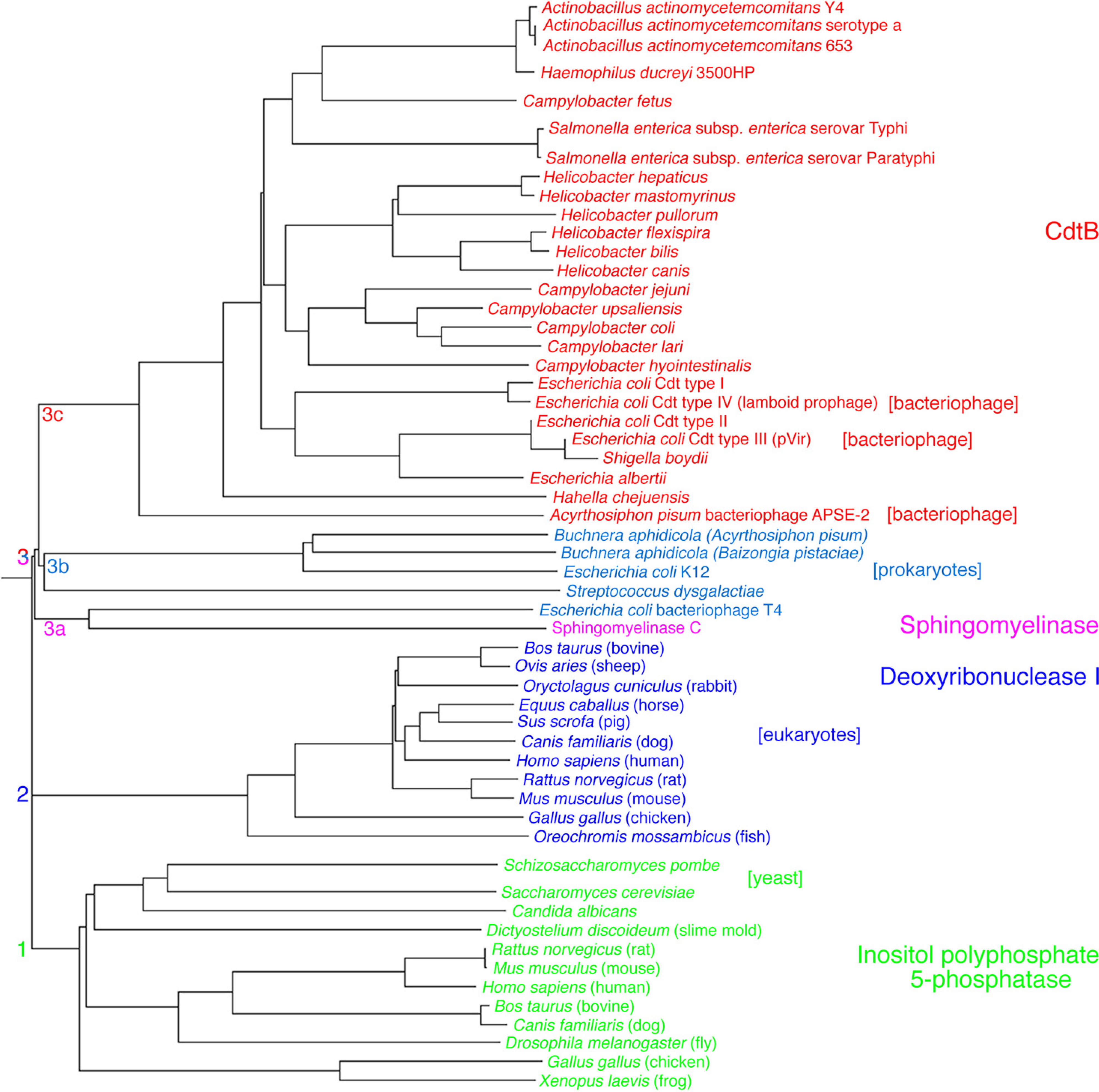
2.2.1. Primary Activities Associated with CdtB
2.2.2. Cell Signaling Activities
2.2.3. Cell Cycle Arrest as a Result of DNA Damage

3. Breakdown of the Gingival Epithelial Barrier
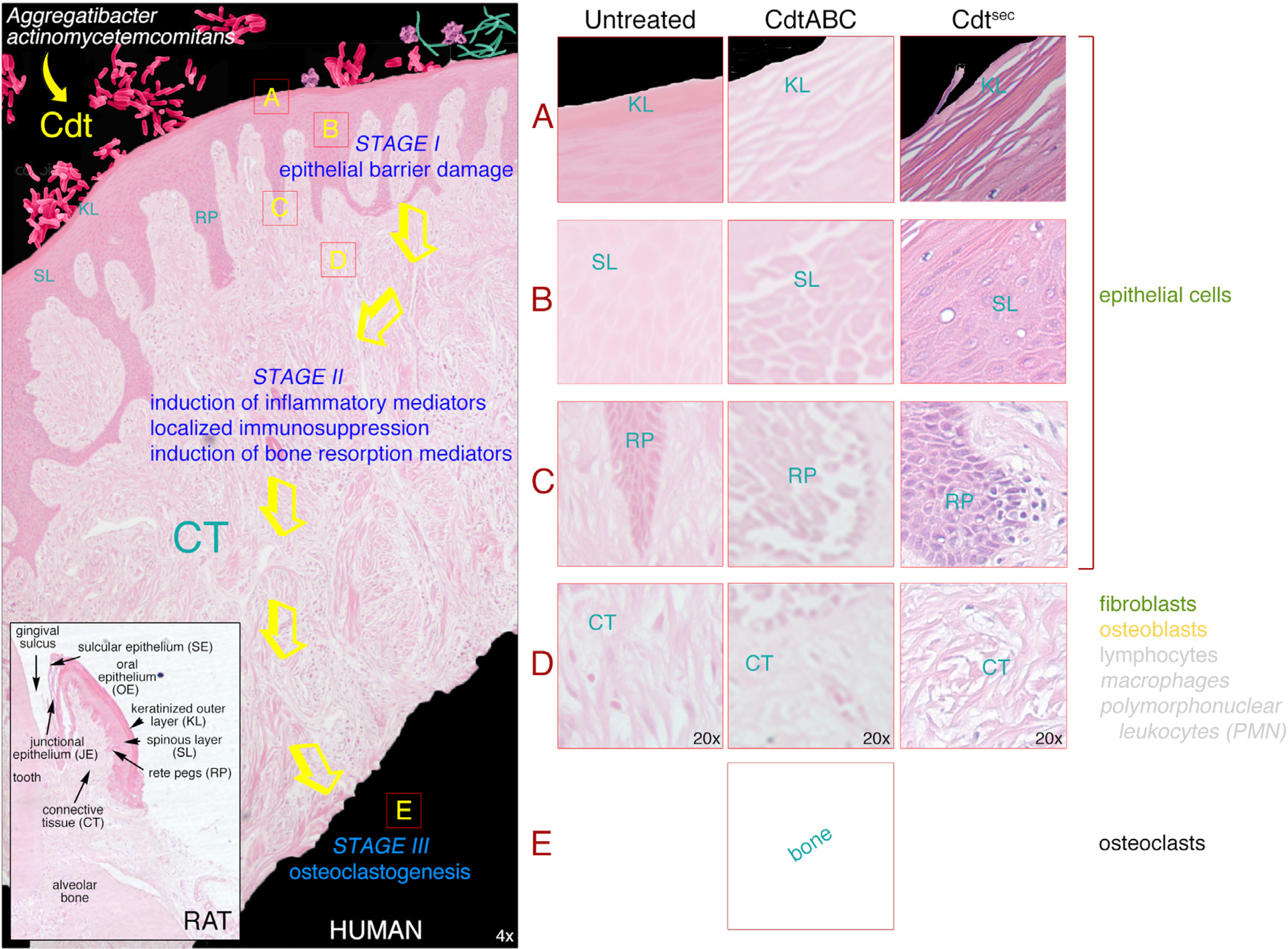
3.1. Gingival Tissue
3.2. Damaging Effects of Cdt on Human Gingival Tissue
3.2.1. Changes in Tissue Morphology and Cellular Organization
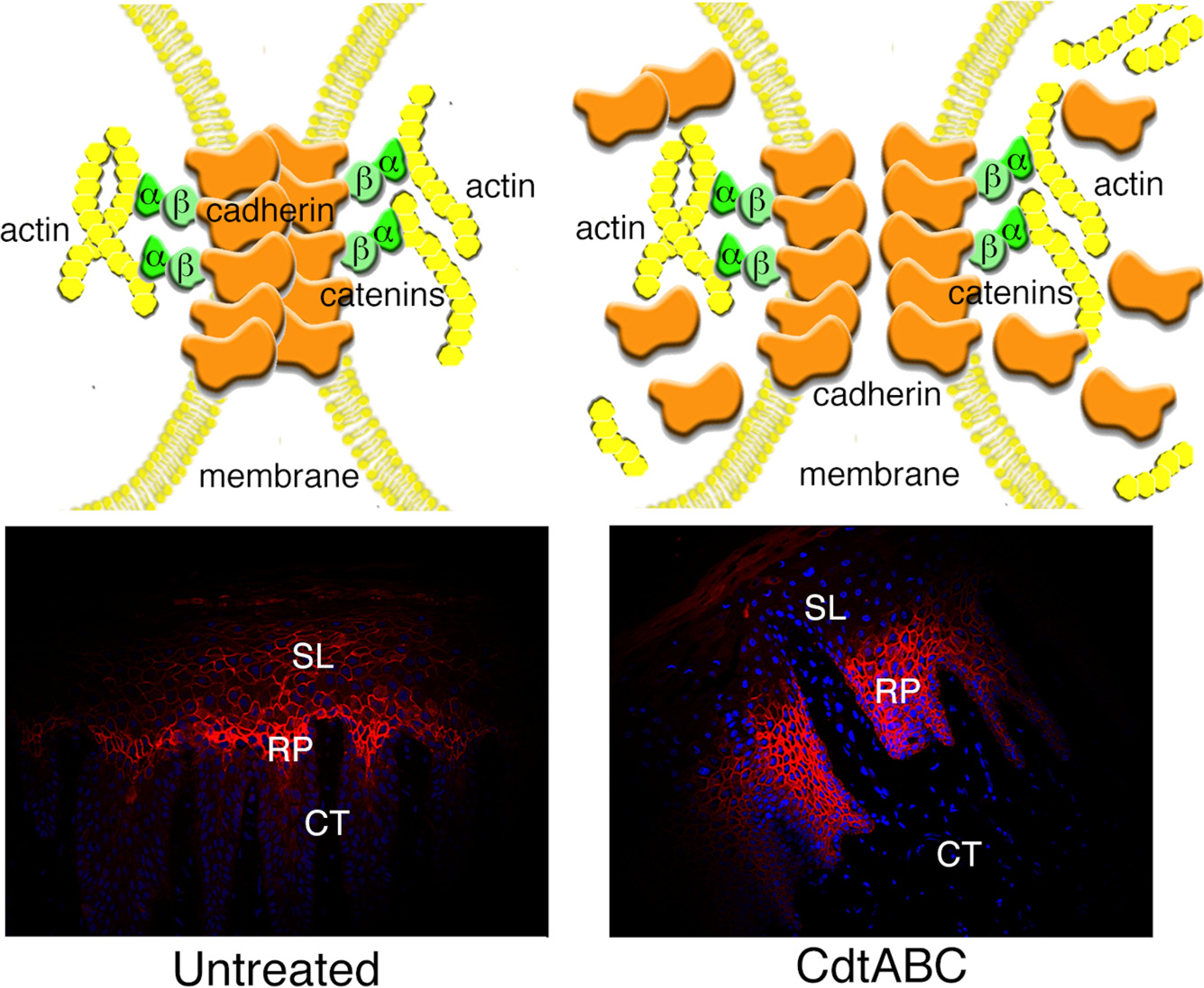
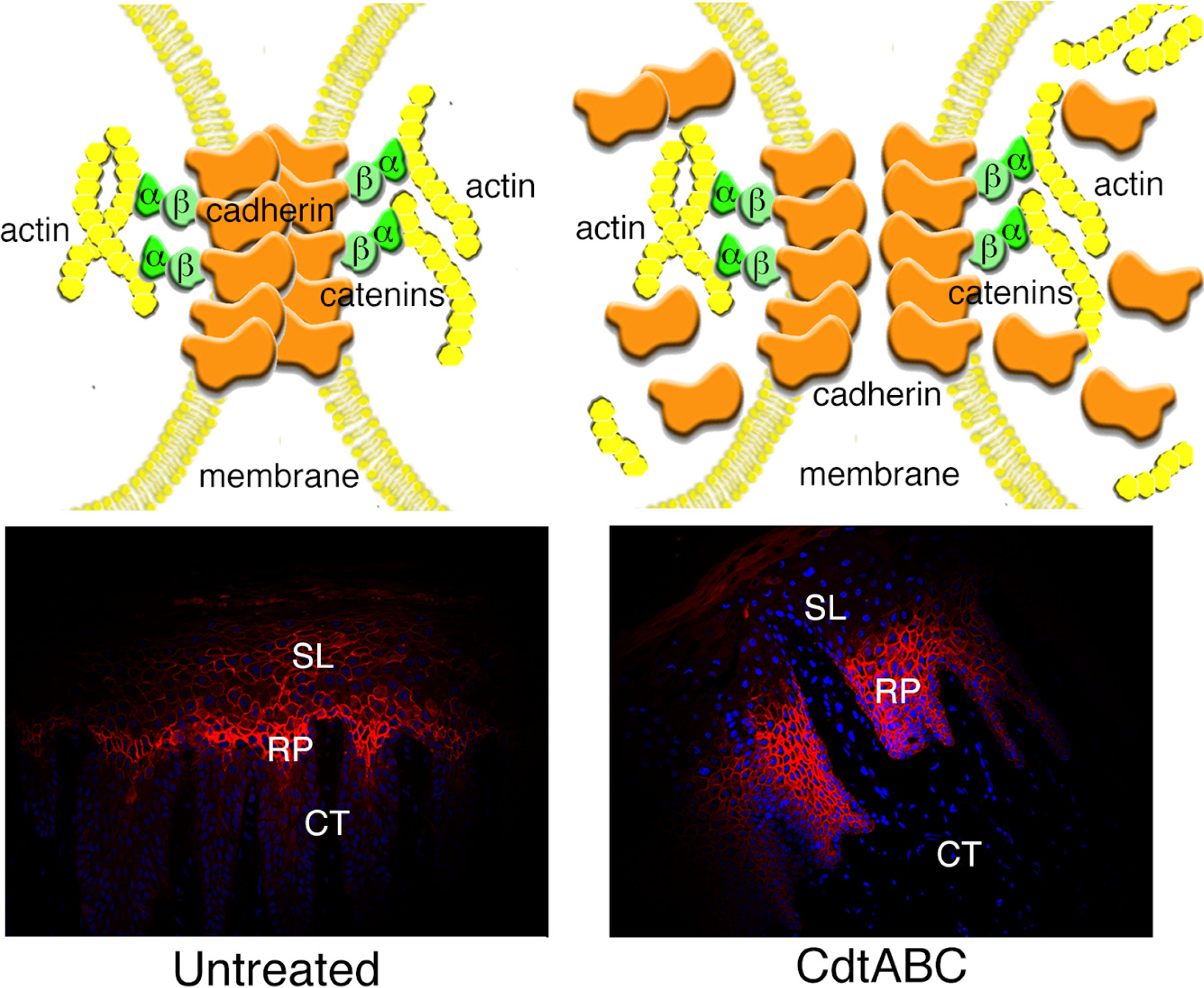
3.2.2. Disruption of Cell Junctions
3.2.3. Changes in Expression of Intracellular Scaffolding Proteins
3.3. Gingival Breakdown Model

4. Concluding Statements
Acknowledgments
Conflict of Interest
References
- Norskov-Lauritsen, N.; Kilian, M. Reclassification of Actinobacillus actinomycetemcomitans, Haemophilus aphrophilus, Haemophilus paraphrophilus; Haemophilus segnis as Aggregatibacter actinomycetemcomitans gen. nov., comb. nov., Aggregatibacter aphrophilus comb. nov. and Aggregatibacter segnis comb. nov., and emended description of Aggregatibacter aphrophilus to include V factor-dependent and V factor-independent isolates. Int. J. Syst. Evol. Microbiol. 2006, 56, 2135–2146. [Google Scholar] [CrossRef]
- Feder, H.M.J.; Roberts, J.C.; Salazar, J.; Leopold, H.B.; Toro-Salazar, O. HACEK endocarditis in infants and children: Two cases and a literature review. Pediatr. Infect. Dis. J. 2003, 22, 557–562. [Google Scholar]
- Armitage, G.C. Comparison of the microbiological features of chronic and aggressive periodontitis. Periodontology 2010, 53, 70–88. [Google Scholar] [CrossRef]
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef]
- Taubman, M.A.; Valverde, P.; Han, X.; Kawai, T. Immune response: The key to bone resorption in periodontal disease. J. Periodontol. 2005, 76, 2033–2041. [Google Scholar] [CrossRef]
- Lally, E.T.; Golub, E.E.; Kieba, I.R.; Taichman, N.S.; Rosenbloom, J.; Rosenbloom, J.C.; Gibson, C.W.; Demuth, D.R. Analysis of the Actinobacillus actinomycetemcomitans leukotoxin gene. Delineation of unique features and comparison to homologous toxins. J. Biol. Chem. 1989, 264, 15451–15456. [Google Scholar]
- Mayer, M.P.; Bueno, L.C.; Hansen, E.J.; DiRienzo, J.M. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect. Immun. 1999, 67, 1227–1237. [Google Scholar]
- Feng, Z.; Weinberg, A. Role of bacteria in health and disease of periodontal tissues. Periodontol 2000, 40, 50–76. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. Cellular internalization of cytolethal distending toxin from Haemophilus ducreyi. Infect. Immun. 2000, 68, 6903–6911. [Google Scholar] [CrossRef]
- Aberg, C.H.; Sjodin, B.; Lakio, L.; Pussinen, P.J.; Johansson, A.; Claesson, R. Presence of Aggregatibacter actinomycetemcomitans in young individuals: A 16-year clinical and microbiological follow-up study. J. Clin. Periodontol. 2009, 36, 815–822. [Google Scholar] [CrossRef]
- Ahmed, H.J.; Svensson, L.A.; Cope, L.D.; Latimer, J.L.; Hansen, E.J.; Ahlman, K.; Bayat-Turk, J.; Klamer, D.; Lagergård, T. Prevalence of cdtABC genes encoding cytolethal distending toxin among Haemophilus ducreyi and Actinobacillus actinomycetemcomitans strains. J. Med. Microbiol. 2001, 50, 860–864. [Google Scholar]
- Bandhaya, P.; Saraithong, P.; Likittanasombat, K.; Hengprasith, B.; Torrungruang, K. Aggregatibacter actinomycetemcomitans serotypes, the JP2 clone and cytolethal distending toxin genes in a Thai population. J. Clin. Periodontol. 2012, 39, 519–525. [Google Scholar] [CrossRef]
- Fabris, A.S.; DiRienzo, J.M.; Wikstrom, M.; Mayer, M.P. Detection of cytolethal distending toxin activity and cdt genes in Actinobacillus actinomycetemcomitans isolates from geographically diverse populations. Oral Microbiol. Immunol. 2002, 17, 231–238. [Google Scholar] [CrossRef]
- Nishikubo, S.; Ohara, M.; Ikura, M.; Katayanagi, K.; Fujiwara, T.; Komatsuzawa, H.; Kurihara, H.; Sugai, M. Single nucleotide polymorphism in the cytolethal distending toxin B gene confers heterogeneity in the cytotoxicity of Actinobacillus actinomycetemcomitans. Infect. Immun. 2006, 74, 7014–7020. [Google Scholar] [CrossRef]
- Tan, K.S.; Song, K.P.; Ong, G. Cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Occurrence and association with periodontal disease. J. Periodontal. Res. 2002, 37, 268–272. [Google Scholar]
- Yamano, R.; Ohara, M.; Nishikubo, S.; Fujiwara, T.; Kawamoto, T.; Ueno, Y.; Komatsuzawa, H.; Okuda, K.; Kurihara, H.; Suginaka, H.; et al. Prevalence of cytolethal distending toxin production in periodontopathogenic bacteria. J. Clin. Microbiol. 2003, 41, 1391–1398. [Google Scholar] [CrossRef]
- Ando, E.S.; De-Gennaro, L.A.; Faveri, M.; Feres, M.; DiRienzo, J.M.; Mayer, M.P. Immune response to cytolethal distending toxin of Aggregatibacter actinomycetemcomitans in periodontitis patients. J. Periodontal. Res. 2010, 45, 471–480. [Google Scholar]
- Johansson, A.; Buhlin, K.; Koski, R.; Gustafsson, A. The immunoreactivity of systemic antibodies to Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in adult periodontitis. Eur. J. Oral Sci. 2005, 113, 197–202. [Google Scholar]
- Xynogala, I.; Volgina, A.; DiRienzo, J.M.; Korostoff, J. Evaluation of the humoral immune response to the cytolethal distending toxin of Aggregatibacter actinomycetemcomitans Y4 in subjects with localized aggressive periodontitis. Oral Microbiol. Immunol. 2009, 24, 116–123. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Slots, J.; Sixou, M.; Sol, M.A.; Harmon, R.; McKay, T.L. Specific genetic variants of Actinobacillus actinomycetemcomitans correlate with disease and health in a regional population of families with localized juvenile periodontitis. Infect. Immun. 1994, 62, 3058–3065. [Google Scholar]
- DiRienzo, J.M.; McKay, T.L. Identification and characterization of genetic cluster groups of Actinobacillus actinomycetemcomitans isolated from the human oral cavity. J. Clin. Microbiol. 1994, 32, 75–81. [Google Scholar]
- Bueno, L.C.; Mayer, M.P.; DiRienzo, J.M. Relationship between conversion of localized juvenile periodontitis-susceptible children from health to disease and Actinobacillus actinomycetemcomitans leukotoxin promoter structure. J. Periodontol. 1998, 69, 998–1007. [Google Scholar] [CrossRef]
- Hoglund Aberg, C.; Antonoglou, G.; Haubek, D.; Kwamin, F.; Claesson, R.; Johansson, A. Cytolethal distending toxin in isolates of Aggregatibacter actinomycetemcomitans from Ghanaian adolescents and association with serotype and disease progression. PLoS One 2013, 8, e65781. [Google Scholar]
- Wang, X.; Li, L.; Yang, M.; Geng, Y.; Chen, H.; Xu, Y.; Sun, Y. Prevalence and distribution of Aggregatibacter actinomycetemcomitans and its cdtB gene in subgingival plaque of Chinese periodontitis patients. BMC Oral Health 2014, 14, 37. [Google Scholar] [CrossRef]
- Gargi, A.; Reno, M.; Blanke, S.R. Bacterial toxin modulation of the eukaryotic cell cycle: Are all cytolethal distending toxins created equally? Front. Cell. Infect. Microbiol. 2012, 2, 124. [Google Scholar]
- Akifusa, S.; Poole, S.; Lewthwaite, J.; Henderson, B.; Nair, S.P. Recombinant Actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect. Immun. 2001, 69, 5925–5930. [Google Scholar] [CrossRef]
- Mao, X.; DiRienzo, J.M. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell. Microbiol. 2002, 4, 245–255. [Google Scholar]
- Saiki, K.; Konishi, K.; Gomi, T.; Nishihara, T.; Yoshikawa, M. Reconstitution and purification of cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiol. Immunol. 2001, 45, 497–506. [Google Scholar] [CrossRef]
- Yamada, T.; Komoto, J.; Saiki, K.; Konishi, K.; Takusagawa, F. Variation of loop sequence alters stability of cytolethal distending toxin (CDT): Crystal structure of CDT from Actinobacillus actinomycetemcomitans. Protein Sci. 2006, 15, 362–372. [Google Scholar] [CrossRef]
- McSweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060. [Google Scholar] [CrossRef]
- Kanno, F.; Korostoff, J.; Volgina, A.; DiRienzo, J.M. Resistance of human periodontal ligament fibroblasts to the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. J. Periodontol. 2005, 76, 1189–1201. [Google Scholar] [CrossRef]
- Cao, L.; Volgina, A.; Huang, C.M.; Korostoff, J.; DiRienzo, J.M. Characterization of point mutations in the cdtA gene of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Mol. Microbiol. 2005, 58, 1303–1321. [Google Scholar] [CrossRef]
- Lee, R.B.; Hassane, D.C.; Cottle, D.L.; Pickett, C.L. Interactions of Campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 2003, 71, 4883–4890. [Google Scholar] [CrossRef]
- Akifusa, S.; Heywood, W.; Nair, S.P.; Stenbeck, G.; Henderson, B. Mechanism of internalization of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiology 2005, 151, 1395–1402. [Google Scholar]
- Mise, K.; Akifusa, S.; Watarai, S.; Ansai, T.; Nishihara, T.; Takehara, T. Involvement of ganglioside GM3 in G2/M cell cycle arrest of human monocytic cells induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2005, 73, 4846–4852. [Google Scholar] [CrossRef]
- Prokazova, N.V.; Samovilova, N.N.; Gracheva, E.V.; Golovanova, N.K. Ganglioside GM3 and its biological functions. Biochemistry (Moscow) 2009, 74, 235–249. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K.; Brown, A.; Walker, L.; Besack, D.; Zekavat, A.; Wrenn, S.; Krummenacher, C.; Shenker, B.J. Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J. Biol. Chem. 2009, 284, 10650–10658. [Google Scholar] [CrossRef]
- Eshraghi, A.; Maldonado-Arocho, F.J.; Gargi, A.; Cardwell, M.M.; Prouty, M.G.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. J. Biol. Chem. 2010, 285, 18199–18207. [Google Scholar]
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235. [Google Scholar] [CrossRef]
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 5895–5900. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736. [Google Scholar] [CrossRef]
- Frisan, T.; Cortes-Bratti, X.; Chaves-Olarte, E.; Stenerlow, B.; Thelestam, M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707. [Google Scholar] [CrossRef]
- Dassanayake, R.P.; Griep, M.A.; Duhamel, G.E. The cytolethal distending toxin B sub-unit of Helicobacter hepaticus is a Ca2+- and Mg2+-dependent neutral nuclease. FEMS Microbiol. Lett. 2005, 251, 219–225. [Google Scholar] [CrossRef]
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Cao, L.; Volgina, A.; Bandelac, G.; Korostoff, J. Functional and structural characterization of chimeras of a bacterial genotoxin and human type I DNAse. FEMS Microbiol. Lett. 2009, 291, 222–231. [Google Scholar] [CrossRef]
- Hu, X.; Nesic, D.; Stebbins, C.E. Comparative structure-function analysis of cytolethal distending toxins. Proteins 2006, 62, 421–434. [Google Scholar]
- Lara-Tejero, M.; Galán, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef]
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681. [Google Scholar]
- Dlakic, M. Is CdtB a nuclease or a phosphatase? Science 2001, 291, 547. [Google Scholar] [CrossRef]
- Shenker, B.J.; Dlakic, M.; Walker, L.P.; Besack, D.; Jaffe, E.; Labelle, E.; Boesze-Battaglia, K. A novel mode of action for a microbial-derived immunotoxin: the cytolethal distending toxin subunit B exhibits phosphatidylinositol 3,4,5-triphosphate phosphatase activity. J. Immunol. 2007, 178, 5099–5108. [Google Scholar]
- Shenker, B.J.; Walker, L.P.; Zekavat, A.; Dlakic, M.; Boesze-Battaglia, K. Blockade of the PI-3K signaling pathway by the Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces macrophages to synthesize and secrete pro-inflammatory cytokines. Cell. Microbiol. 2014. [Google Scholar] [CrossRef]
- Hickey, T.E.; McVeigh, A.L.; Scott, D.A.; Michielutti, R.E.; Bixby, A.; Carroll, S.A.; Bourgeois, A.L.; Guerry, P. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect. Immun. 2000, 68, 6535–6541. [Google Scholar] [CrossRef]
- Ando-Suguimoto, E.S.; da Silva, M.P.; Kawamoto, D.; Chen, C.; DiRienzo, J.M.; Mayer, M.P. The cytolethal distending toxin of Aggregatibacter actinomycetemcomitans inhibits macrophage phagocytosis and subverts cytokine production. Cytokine 2014, 66, 46–53. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Johansson, A.; Wang, Y.; Chen, C.; Kalfas, S.; Lerner, U.H. The cytolethal distending toxin induces receptor activator of NF-κB ligand expression in human gingival fibroblasts and periodontal ligament cells. Infect. Immun. 2005, 73, 342–351. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Johansson, A.; Wang, Y.; Chen, C.; Lagergård, T.; Kalfas, S.; Lerner, U.H. Cytokine responses of human gingival fibroblasts to Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cytokine 2005, 30, 56–63. [Google Scholar] [CrossRef]
- Uchida, Y.; Shiba, H.; Komatsuzawa, H.; Takemoto, T.; Sakata, M.; Fujita, T.; Kawaguchi, H.; Sugai, M.; Kurihara, H. Expression of IL-1β and IL-8 by human gingival epithelial cells in response to Actinobacillus actinomycetemcomitans. Cytokine 2001, 14, 152–161. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Brage, M.; Lagergård, T.; Johansson, A. Cytolethal distending toxin upregulates RANKL expression in Jurkat T-cells. Apmis 2008, 116, 499–506. [Google Scholar] [CrossRef]
- Hsu, H.; Lacey, D.L.; Dunstan, C.R.; Solovyev, I.; Colombero, A.; Timms, E.; Tan, H.L.; Elliott, G.; Kelley, M.J.; Sarosi, I.; et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3540–3545. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Liu, D.; Xu, J.K.; Figliomeni, L.; Huang, L.; Pavlos, N.J.; Rogers, M.; Tan, A.; Price, P.; Zheng, M.H. Expression of RANKL and OPG mRNA in periodontal disease: Possible involvement in bone destruction. Int. J. Mol. Med. 2003, 11, 17–21. [Google Scholar]
- Mogi, M.; Ozeki, N.; Nakamura, H.; Togari, A. Dual roles for NF-κB activation in osteoblastic cells by serum deprivation: Osteoblastic apoptosis and cell-cycle arrest. Bone 2004, 35, 507–516. [Google Scholar] [CrossRef]
- Escalas, N.; Davezac, N.; de Rycke, J.; Baldin, V.; Mazars, R.; Ducommun, B. Study of the cytolethal distending toxin-induced cell cycle arrest in HeLa cells: Involvement of the CDC25 phosphatase. Exp. Cell Res. 2000, 257, 206–212. [Google Scholar] [CrossRef]
- Johnson, W.M.; Lior, H. A new heat-labile cytolethal distending toxin (CLDT) produced by Campylobacter spp. Microb. Pathog. 1988, 4, 115–126. [Google Scholar] [CrossRef]
- Pérès, S.Y.; Marches, O.; Daigle, F.; Nougayrede, J.P.; Herault, F.; Tasca, C.; de Rycke, J.; Oswald, E. A new cytolethal distending toxin (CDT) from Escherichia coli producing CNF2 blocks HeLa cell division in G2/M phase. Mol. Microbiol. 1997, 24, 1095–1107. [Google Scholar]
- Sato, T.; Koseki, T.; Yamato, K.; Saiki, K.; Konishi, K.; Yoshikawa, M.; Ishikawa, I.; Nishihara, T. p53-independent expression of p21(CIP1/WAF1) in plasmacytic cells during G2 cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2002, 70, 528–534. [Google Scholar] [CrossRef]
- Sugai, M.; Kawamoto, T.; Pérès, S.Y.; Ueno, Y.; Komatsuzawa, H.; Fujiwara, T.; Kurihara, H.; Suginaka, H.; Oswald, E. The cell cycle-specific growth-inhibitory factor produced by Actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect. Immun. 1998, 66, 5008–5019. [Google Scholar]
- Whitehouse, C.A.; Balbo, P.B.; Pesci, E.C.; Cottle, D.L.; Mirabito, P.M.; Pickett, C.L. Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect. Immun. 1998, 66, 1934–1940. [Google Scholar]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. The cytolethal distending toxin from the chancroid bacterium Haemophilus ducreyi induces cell-cycle arrest in the G2 phase. J. Clin. Invest. 1999, 103, 107–115. [Google Scholar] [CrossRef]
- Stevens, M.K.; Latimer, J.L.; Lumbley, S.R.; Ward, C.K.; Cope, L.D.; Lagergård, T.; Hansen, E.J. Characterization of a Haemophilus ducreyi mutant deficient in expression of cytolethal distending toxin. Infect. Immun. 1999, 67, 3900–3908. [Google Scholar]
- Smith, J.L.; Bayles, D.O. The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit. Rev. Microbiol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157, 1851–1875. [Google Scholar] [CrossRef]
- Oswald, E.; Nougayrede, J.P.; Taieb, F.; Sugai, M. Bacterial toxins that modulate host cell-cycle progression. Curr. Opin. Microbiol. 2005, 8, 83–91. [Google Scholar] [CrossRef]
- Comayras, C.; Tasca, C.; Pérès, S.Y.; Ducommun, B.; Oswald, E.; de Rycke, J. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect. Immun. 1997, 65, 5088–5095. [Google Scholar]
- Alaoui-El-Azher, M.; Mans, J.J.; Baker, H.V.; Chen, C.; Progulske-Fox, A.; Lamont, R.J.; Handfield, M. Role of the ATM-checkpoint kinase 2 pathway in CDT-mediated apoptosis of gingival epithelial cells. PLoS One 2010, 5, e11714. [Google Scholar]
- Nougayrede, J.P.; Taieb, F.; de Rycke, J.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Song, M.; Wan, L.S.; Ellen, R.P. Kinetics of KB and HEp-2 cell responses to an invasive, cytolethal distending toxin-producing strain of Actinobacillus actinomycetemcomitans. Oral Microbiol. Immunol. 2002, 17, 245–251. [Google Scholar] [CrossRef]
- Lepine, G.; Caudry, S.; DiRienzo, J.M.; Ellen, R.P. Epithelial cell invasion by Actinobacillus actinomycetemcomitans strains from restriction fragment-length polymorphism groups associated with juvenile periodontitis or carrier status. Oral Microbiol. Immunol. 1998, 13, 341–347. [Google Scholar] [CrossRef]
- Meyer, D.H.; Lippmann, J.E.; Fives-Taylor, P.M. Invasion of epithelial cells by Actinobacillus actinomycetemcomitans: A dynamic, multistep process. Infect. Immun. 1996, 64, 2988–2997. [Google Scholar]
- Meyer, D.H.; Sreenivasan, P.K.; Fives-Taylor, P.M. Evidence for invasion of a human oral cell line by Actinobacillus actinomycetemcomitans. Infect. Immun. 1991, 59, 2719–2726. [Google Scholar]
- Gilchrist, E.P.; Moyer, M.P.; Shillitoe, E.J.; Clare, N.; Murrah, V.A. Establishment of a human polyclonal oral epithelial cell line. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2000, 90, 340–347. [Google Scholar]
- Kang, P.; Korostoff, J.; Volgina, A.; Grzesik, W.; DiRienzo, J.M. Differential effect of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans on co-cultures of human oral cells. J. Med. Microbiol. 2005, 54, 785–794. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Haris, M.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Cytolethal distending toxin damages the oral epithelium of gingival explants. J. Dent. Res. 2011, 90, 874–879. [Google Scholar] [CrossRef]
- Belibasakis, G.; Johansson, A.; Wang, Y.; Claesson, R.; Chen, C.; Asikainen, S.; Kalfas, S. Inhibited proliferation of human periodontal ligament cells and gingival fibroblasts by Actinobacillus actinomycetemcomitans: involvement of the cytolethal distending toxin. Eur. J. Oral Sci. 2002, 110, 366–373. [Google Scholar] [CrossRef]
- Belibasakis, G.N.; Mattsson, A.; Wang, Y.; Chen, C.; Johansson, A. Cell cycle arrest of human gingival fibroblasts and periodontal ligament cells by Actinobacillus actinomycetemcomitans: involvement of the cytolethal distending toxin. Apmis 2004, 112, 674–685. [Google Scholar] [CrossRef]
- Bosshardt, D.D.; Lang, N.P. The junctional epithelium: From health to disease. J. Dent. Res. 2005, 84, 9–20. [Google Scholar] [CrossRef]
- Presland, R.B.; Dale, B.A. Epithelial structural proteins of the skin and oral cavity: Function in health and disease. Crit. Rev. Oral Biol. Med. 2000, 11, 383–408. [Google Scholar] [CrossRef]
- Bikle, D.D.; Oda, Y.; Xie, Z. Calcium and 1,25(OH)2D: Interacting drivers of epidermal differentiation. J. Steroid Biochem. Mol. Biol. 2004, 89, 355–360. [Google Scholar] [CrossRef]
- Christersson, L.A.; Albini, B.; Zambon, J.J.; Wikesjo, U.M.; Genco, R.J. Tissue localization of Actinobacillus actinomycetemcomitans in human periodontitis. I. Light, immunofluorescence and electron microscopic studies. J. Periodontol. 1987, 58, 529–539. [Google Scholar]
- Gibbons, R.J. Bacterial adhesion to oral tissues: A model for infectious diseases. J. Dent. Res. 1989, 68, 750–760. [Google Scholar] [CrossRef]
- Powell, R.N. Gingival tissue physiology in vitro. 3. Cell proliferation in gingival epithelium. J. Periodontal. Res. 1971, 6, 38–44. [Google Scholar] [CrossRef]
- Powell, R.N. Gingival tissue physiology in vitro. 1. Gingival organ culture. J. Periodontal. Res. 1967, 2, 290–296. [Google Scholar] [CrossRef]
- Ohara, M.; Miyauchi, M.; Tsuruda, K.; Takata, T.; Sugai, M. Topical application of Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces cell cycle arrest in the rat gingival epithelium in vivo. J. Periodontal. Res. 2011, 46, 389–395. [Google Scholar] [CrossRef]
- Franke, W.W. Discovering the molecular components of intercellular junctions-a historical view. Cold Spring Harb. Perspect. Biol. 2009, 1, a003061. [Google Scholar] [CrossRef]
- Cavey, M.; Lecuit, T. Molecular bases of cell-cell junctions stability and dynamics. Cold Spring Harb. Perspect. Biol. 2009, 1, a002998. [Google Scholar]
- Shen, L. Tight junctions on the move: Molecular mechanisms for epithelial barrier regulation. Ann. N.Y. Acad. Sci. 2012, 1258, 9–18. [Google Scholar]
- Miyaguchi, K. Ultrastructure of the zonula adherens revealed by rapid-freeze deep-etching. J. Struct. Biol. 2000, 132, 169–178. [Google Scholar] [CrossRef]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar]
- Kowalczyk, A.P.; Nanes, B.A. Adherens junction turnover: regulating adhesion through cadherin endocytosis, degradation, and recycling. Subcell. Biochem. 2012, 60, 197–222. [Google Scholar] [CrossRef]
- Hirokawa, N.; Heuser, J.E. Quick-freeze, deep-etch visualization of the cytoskeleton beneath surface differentiations of intestinal epithelial cells. J. Cell Biol. 1981, 91, 399–409. [Google Scholar] [CrossRef]
- Thilander, H.; Bloom, G.D. Cell contacts in oral epithelia. J. Periodontal. Res. 1968, 3, 96–110. [Google Scholar] [CrossRef]
- Barnett, M.L.; Szabo, G. Gap junctions in human gingival keratinized epithelium. J. Periodontal. Res. 1973, 8, 117–126. [Google Scholar] [CrossRef]
- Meyle, J.; Gultig, K.; Rascher, G.; Wolburg, H. Transepithelial electrical resistance and tight junctions of human gingival keratinocytes. J. Periodontal. Res. 1999, 34, 214–222. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Moffatt, C.E.; Hagerty, D.; Whitmore, S.E.; Brown, T.A.; Graves, D.T.; Lamont, R.J. Interaction of oral bacteria with gingival epithelial cell multilayers. Mol. Oral Microbiol. 2011, 26, 210–220. [Google Scholar] [CrossRef]
- Katz, J.; Sambandam, V.; Wu, J.H.; Michalek, S.M.; Balkovetz, D.F. Characterization of Porphyromonas gingivalis-induced degradation of epithelial cell junctional complexes. Infect. Immun. 2000, 68, 1441–1449. [Google Scholar] [CrossRef]
- Damek-Poprawa, M.; Korostoff, J.; Gill, R.; DiRienzo, J.M. Cell junction remodeling in gingival tissue exposed to a microbial toxin. J. Dent. Res. 2013, 92, 518–523. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Ye, P.; Chapple, C.C.; Kumar, R.K.; Hunter, N. Expression patterns of E-cadherin, involucrin, and connexin gap junction proteins in the lining epithelia of inflamed gingiva. J. Pathol. 2000, 192, 58–66. [Google Scholar] [CrossRef]
- Downer, C.S.; Speight, P.M. E-cadherin expression in normal, hyperplastic and malignant oral epithelium. Eur. J. Cancer B Oral Oncol. 1993, 29B, 303–305. [Google Scholar] [CrossRef]
- Drees, F.; Pokutta, S.; Yamada, S.; Nelson, W.J.; Weis, W.I. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 2005, 123, 903–915. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Knudsen, K.A. Cadherins and associated proteins. In Vivo 1991, 5, 505–513. [Google Scholar]
- Brieher, W.M.; Yap, A.S. Cadherin junctions and their cytoskeleton(s). Curr. Opin. Cell Biol. 2012, 25, 1–8. [Google Scholar]
- Yamada, S.; Pokutta, S.; Drees, F.; Weis, W.I.; Nelson, W.J. Deconstructing the cadherin-catenin-actin complex. Cell 2005, 123, 889–901. [Google Scholar] [CrossRef]
- Aragon, V.; Chao, K.; Dreyfus, L.A. Effect of cytolethal distending toxin on F-actin assembly and cell division in Chinese hamster ovary cells. Infect. Immun. 1997, 65, 3774–3780. [Google Scholar]
- Lagergard, T.; Keith, J. Cytolethal distending toxin as virulence factor, protective antigen, and target for vaccine development. Vaccine: Dev. Ther. 2012, 2, 51–60. [Google Scholar] [CrossRef]
- Ueno, Y.; Ohara, M.; Kawamoto, T.; Fujiwara, T.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Biogenesis of the Actinobacillus actinomycetemcomitans cytolethal distending toxin holotoxin. Infect. Immun. 2006, 74, 3480–3487. [Google Scholar] [CrossRef]
- Tan, K.S.; Ong, G.; Song, K.P. Introns in the cytolethal distending toxin gene of Actinobacillus actinomycetemcomitans. J. Bacteriol. 2005, 187, 567–575. [Google Scholar] [CrossRef]
- Jeon, B.; Itoh, K.; Ryu, S. Promoter analysis of cytolethal distending toxin genes (cdtA, B, and C) and effect of a luxS mutation on CDT production in Campylobacter jejuni. Microbiol. Immunol. 2005, 49, 599–603. [Google Scholar] [CrossRef]
- Coburn, J.; Leong, J.M. Microbiology. Arresting features of bacterial toxins. Science 2000, 290, 287–288. [Google Scholar] [CrossRef]
- Handfield, M.; Baker, H.V.; Lamont, R.J. Beyond good and evil in the oral cavity: Insights into host-microbe relationships derived from transcriptional profiling of gingival cells. J. Dent. Res. 2008, 87, 203–223. [Google Scholar] [CrossRef]
- Kagnoff, M.F.; Eckmann, L. Epithelial cells as sensors for microbial infection. J. Clin. Invest. 1997, 100, 6–10. [Google Scholar] [CrossRef]
- Nalbant, A.; Chen, C.; Wang, Y.; Zadeh, H.H. Induction of T-cell apoptosis by Actinobacillus actinomycetemcomitans mutants with deletion of ltxA and cdtABC genes: possible activity of GroEL-like molecule. Oral Microbiol. Immunol. 2003, 18, 339–349. [Google Scholar] [CrossRef]
- Shenker, B.J.; McKay, T.; Datar, S.; Miller, M.; Chowhan, R.; Demuth, D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 1999, 162, 4773–4780. [Google Scholar]
- Shenker, B.J.; Hoffmaster, R.H.; Zekavat, A.; Yamaguchi, N.; Lally, E.T.; Demuth, D.R. Induction of apoptosis in human T cells by Actinobacillus actinomycetemcomitans cytolethal distending toxin is a consequence of G2 arrest of the cell cycle. J. Immunol. 2001, 167, 435–441. [Google Scholar]
- Reed, J.C.; Meister, L.; Tanaka, S.; Cuddy, M.; Yum, S.; Geyer, C.; Pleasure, D. Differential expression of bcl2 protooncogene in neuroblastoma and other human tumor cell lines of neural origin. Cancer Res. 1991, 51, 6529–6538. [Google Scholar]
- Griffith, T.S.; Brunner, T.; Fletcher, S.M.; Green, D.R.; Ferguson, T.A. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995, 270, 1189–1192. [Google Scholar]
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Miyauchi, M.; Takata, T.; Sugai, M. Caspase-2 and caspase-7 are involved in cytolethal distending toxin-induced apoptosis in Jurkat and MOLT-4 T-cell lines. Infect. Immun. 2004, 72, 871–879. [Google Scholar] [CrossRef]
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Nakachi, K.; Fujiwara, T.; Komatsuzawa, H.; Sugai, M. Cytolethal distending toxin induces caspase-dependent and -independent cell death in MOLT-4 cells. Infect. Immun. 2008, 76, 4783–4791. [Google Scholar] [CrossRef]
- Rabin, S.D.; Flitton, J.G.; Demuth, D.R. Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces apoptosis in nonproliferating macrophages by a phosphatase-independent mechanism. Infect. Immun. 2009, 77, 3161–3169. [Google Scholar] [CrossRef]
- Fernandes, K.P.; Mayer, M.P.; Ando, E.S.; Ulbrich, A.G.; Amarente-Mendes, J.G.; Russo, M. Inhibition of interferon-gamma-induced nitric oxide production in endotoxin-activated macrophages by cytolethal distending toxin. Oral Microbiol. Immunol. 2008, 23, 360–366. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
DiRienzo, J.M. Breaking the Gingival Epithelial Barrier: Role of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin in Oral Infectious Disease. Cells 2014, 3, 476-499. https://doi.org/10.3390/cells3020476
DiRienzo JM. Breaking the Gingival Epithelial Barrier: Role of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin in Oral Infectious Disease. Cells. 2014; 3(2):476-499. https://doi.org/10.3390/cells3020476
Chicago/Turabian StyleDiRienzo, Joseph M. 2014. "Breaking the Gingival Epithelial Barrier: Role of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin in Oral Infectious Disease" Cells 3, no. 2: 476-499. https://doi.org/10.3390/cells3020476