Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
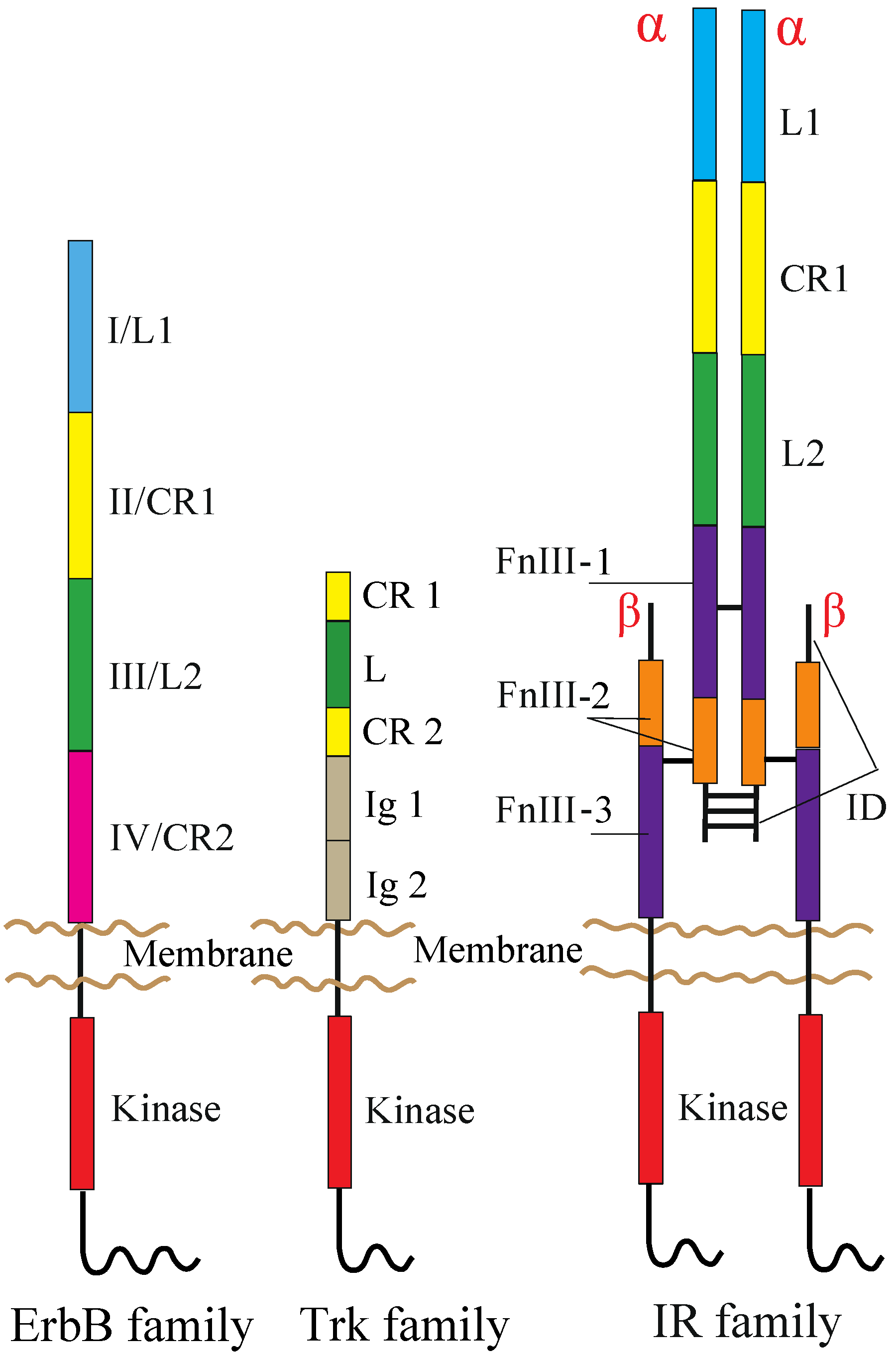
2. RTKs and Their Ligands
2.1. ErbB Family
2.2. NT Receptor Family
2.3. IR Family
3. Are TRKs Monomeric or Dimeric Prior to Ligand Binding?
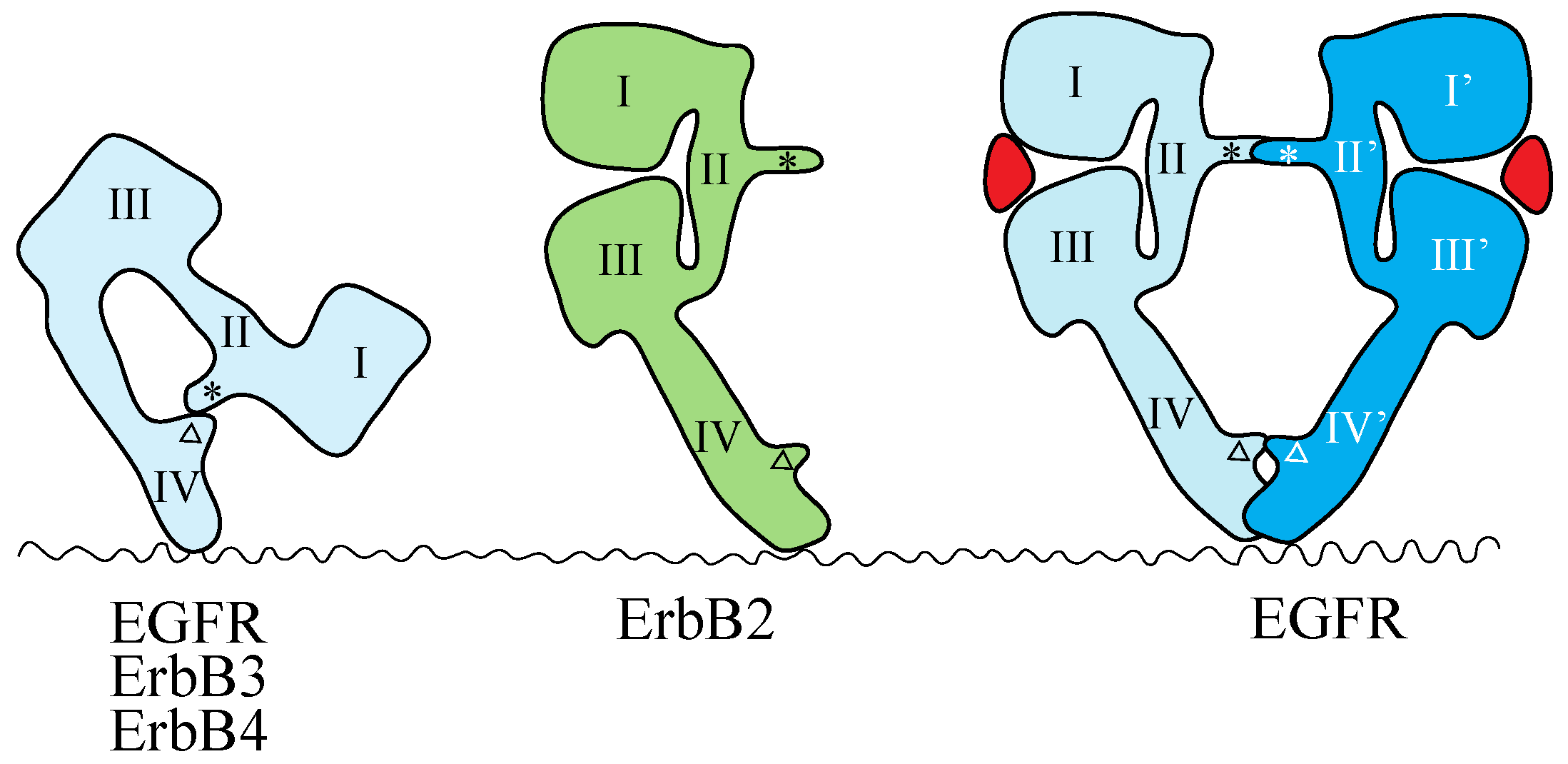
3.1. ErbB Family
3.2. NT Receptor Family
3.3. IR Family
4. Mechanisms of RTK Activation
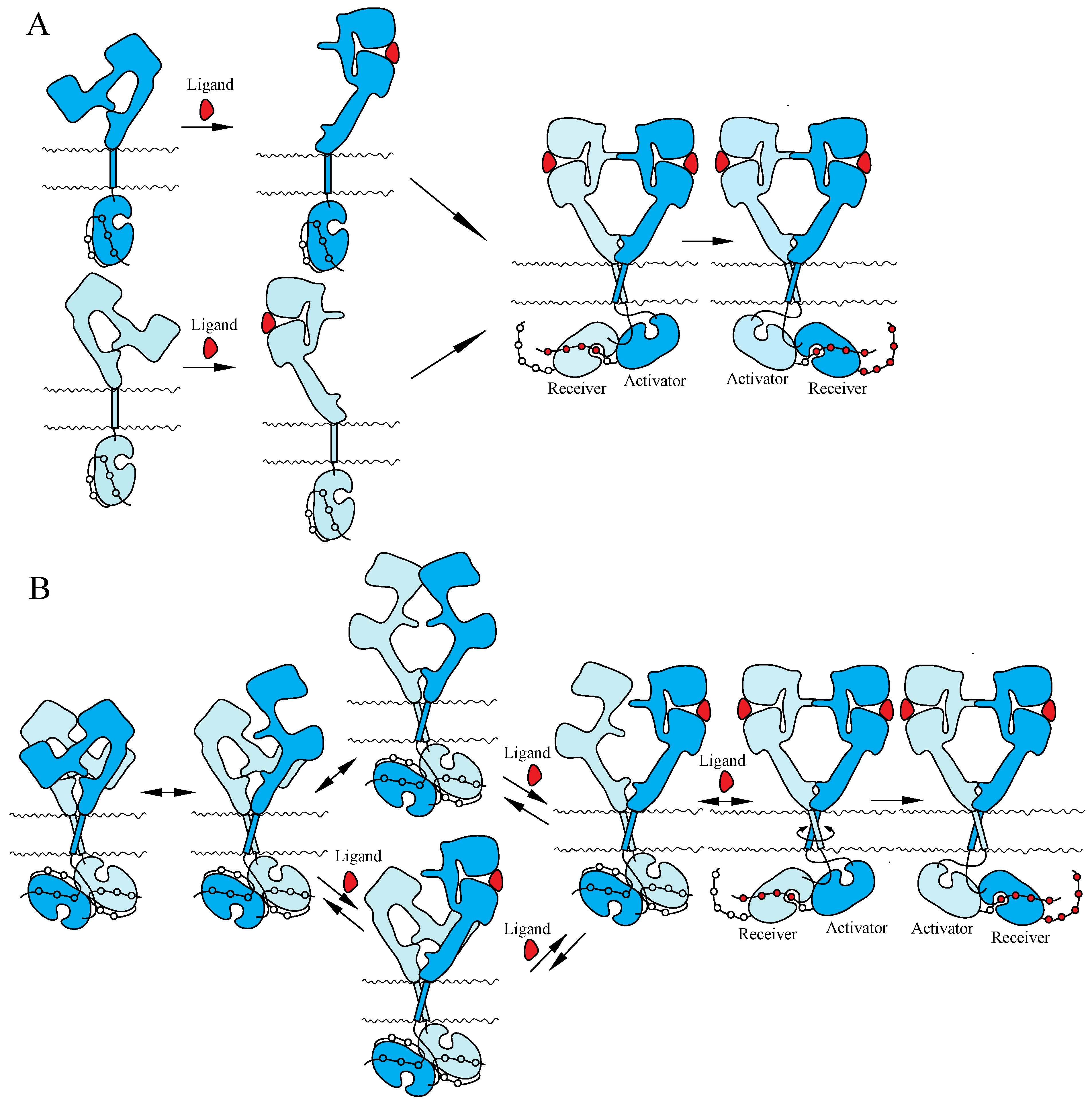
4.1. ErbB Family
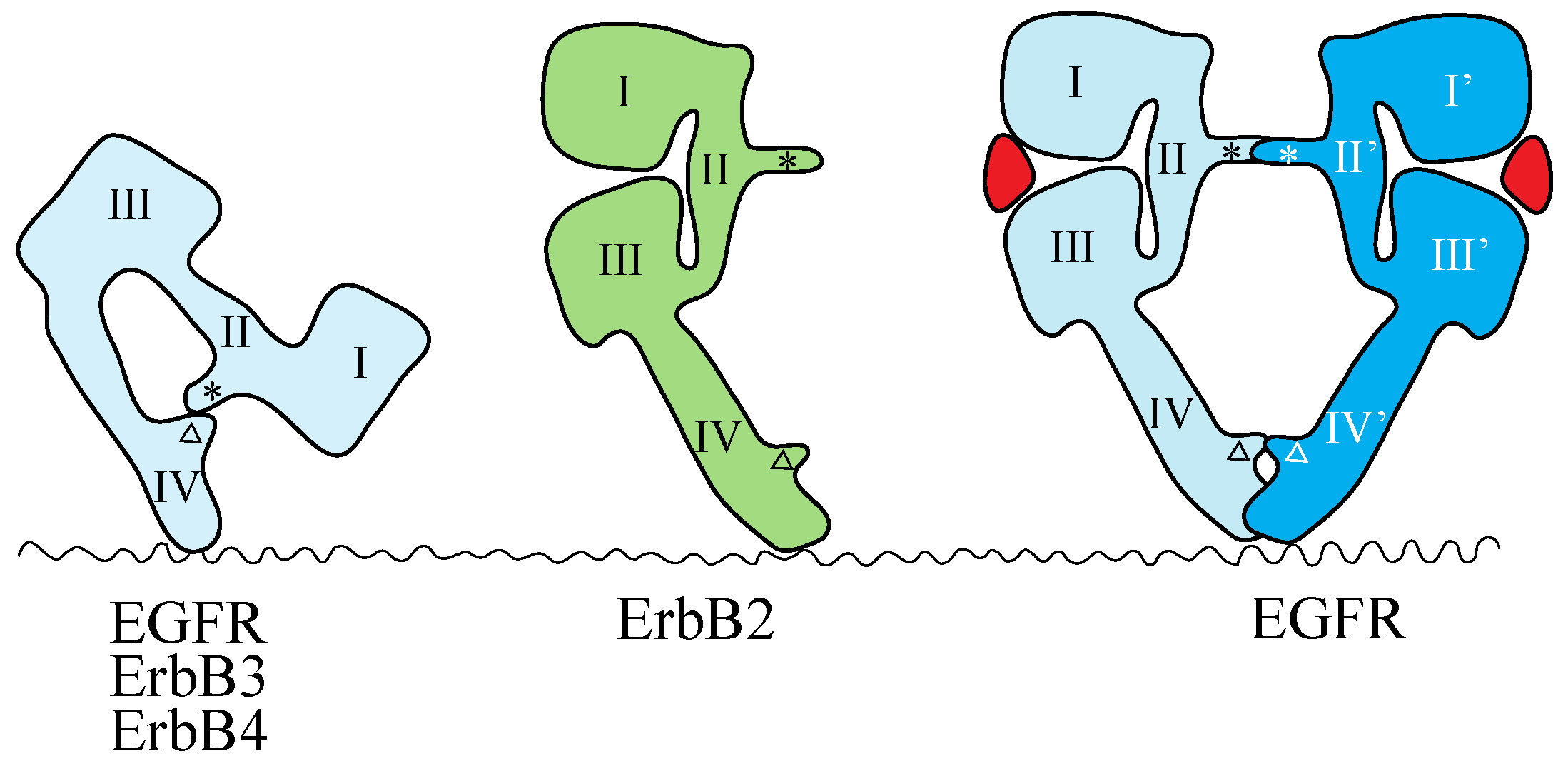
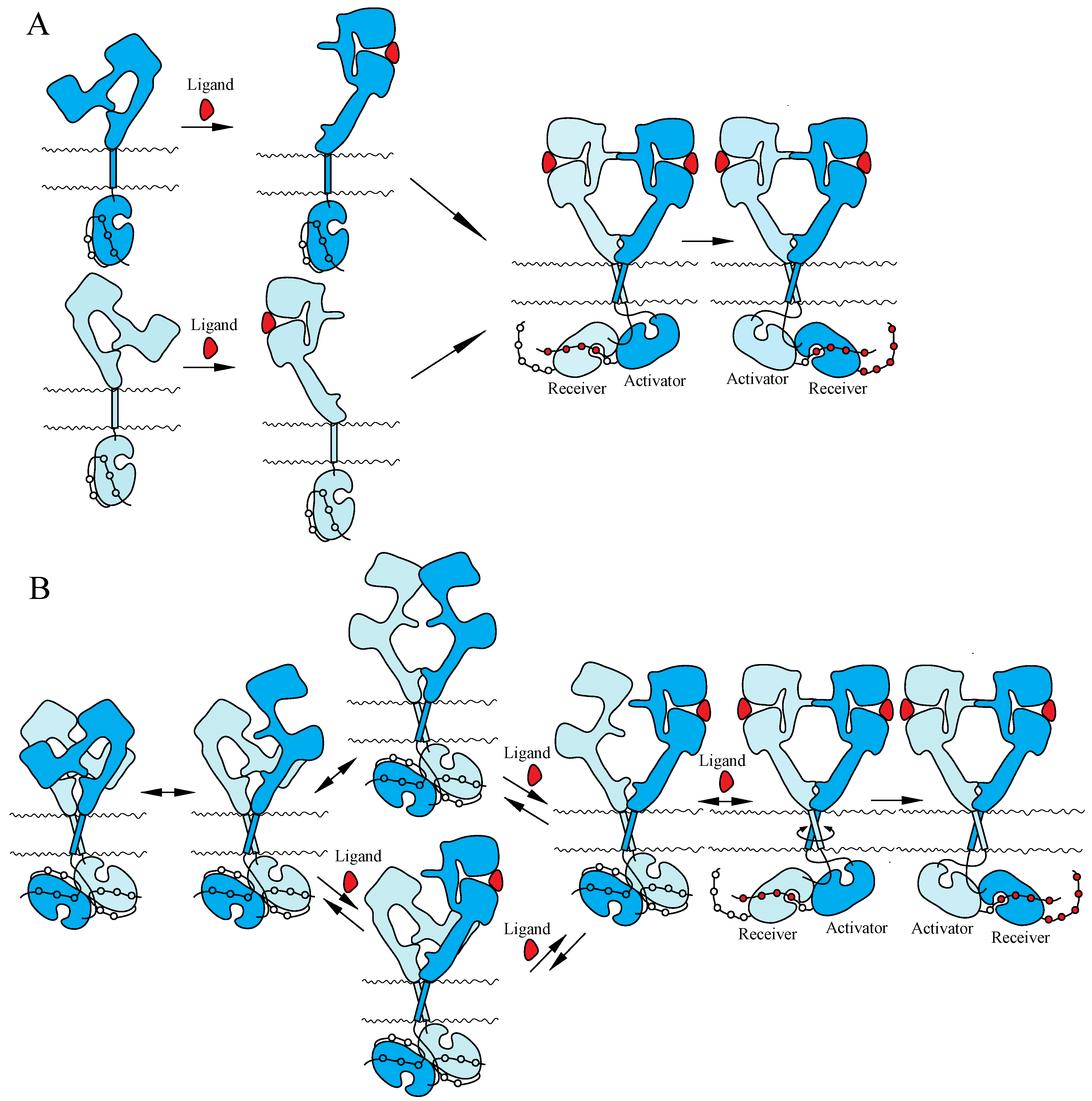
4.2. NT Receptor Family
4.3. IR Family
5. Cooperative Ligand Binding
5.1. ErbB Family
5.2. NT Receptor Family
5.3. IR Family
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signaling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Blagoev, B.; Ong, S.E.; Kratchmarova, I.; Mann, M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat. Biotechnol. 2004, 22, 1139–1145. [Google Scholar] [CrossRef]
- Bradshaw, R.A.; Chalkley, R.J.; Biarc, J.; Burlingame, A.L. Receptor tyrosine kinase signaling mechanisms: Devolving TrkA responses with phosphoproteomics. Adv. Biol. Regul. 2013, 53, 87–96. [Google Scholar] [CrossRef]
- Gotoh, N.; Tojo, A.; Hino, M.; Yazaki, Y.; Shibuya, M. A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 1992, 186, 768–774. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef]
- Pawson, T. Regulation and targets of receptor tyrosine kinases. Eur. J. Cancer 2002, 38, S3–S10. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Heldin, C.H. Dimerization of cell surface receptors in signal transduction. Cell 1995, 80, 213–223. [Google Scholar] [CrossRef]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Burgess, A.W.; Cho, H.S.; Eigenbrot, C.; Ferguson, K.M.; Garrett, T.P.; Leahy, D.J.; Lemmon, M.A.; Sliwkowski, M.X.; Ward, C.W.; Yokoyama, S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 2003, 12, 541–552. [Google Scholar] [CrossRef]
- Bublil, E.M.; Yarden, Y. The EGF receptor family: Spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 2007, 19, 124–134. [Google Scholar] [CrossRef]
- Endres, N.F.; Das, R.; Smith, A.W.; Arkhipov, A.; Kovacs, E.; Huang, Y.; Pelton, J.G.; Shan, Y.; Shaw, D.E.; Wemmer, D.E.; et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 2013, 152, 543–556. [Google Scholar]
- Moriki, T.; Maruyama, H.; Maruyama, I.N. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 2001, 311, 1011–1026. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.; Clarke, D.T.; Tobin, M.J.; Jones, S.V.; Jones, G.R. Preformed oligomeric epidermal growth factor receptors undergo an ectodomain structure change during signaling. Biophys. J. 2002, 82, 2415–2427. [Google Scholar] [CrossRef]
- Yu, X.; Sharma, K.D.; Takahashi, T.; Iwamoto, R.; Mekada, E. Ligand-independent dimer formation of epidermal growth factor receptor (EGFR) is a step separable from ligand-induced EGFR signaling. Mol. Biol. Cell 2002, 13, 2547–2557. [Google Scholar] [CrossRef]
- Clayton, A.H.; Walker, F.; Orchard, S.G.; Henderson, C.; Fuchs, D.; Rothacker, J.; Nice, E.C.; Burgess, A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor-A multidimensional microscopy analysis. J. Biol. Chem. 2005, 280, 30392–30399. [Google Scholar] [CrossRef]
- Teramura, Y.; Ichinose, J.; Takagi, H.; Nishida, K.; Yanagida, T.; Sako, Y. Single-molecule analysis of epidermal growth factor binding on the surface of living cells. EMBO J. 2006, 25, 4215–4222. [Google Scholar] [CrossRef]
- Liu, P.; Sudhaharan, T.; Koh, R.M.; Hwang, L.C.; Ahmed, S.; Maruyama, I.N.; Wohland, T. Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 2007, 93, 684–698. [Google Scholar] [CrossRef]
- Saffarian, S.; Li, Y.; Elson, E.L.; Pike, L.J. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 2007, 93, 1021–1031. [Google Scholar] [CrossRef]
- Tao, R.H.; Maruyama, I.N. All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 2008, 121, 3207–3217. [Google Scholar] [CrossRef]
- Bader, A.N.; Hofman, E.G.; Voortman, J.; En Henegouwen, P.M.; Gerritsen, H.C. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys. J. 2009, 97, 2613–2622. [Google Scholar] [CrossRef]
- Yang, K.S.; Ilagan, M.X.; Piwnica-Worms, D.; Pike, L.J. Luciferase fragment complementation imaging of conformational changes in the epidermal growth factor receptor. J. Biol. Chem. 2009, 284, 7474–7482. [Google Scholar] [CrossRef]
- Ma, X.; Ahmed, S.; Wohland, T. EGFR activation monitored by SW-FCCS in live cells. Front. Biosci. (Elite Ed.) 2011, 3, 22–32. [Google Scholar] [CrossRef]
- Shen, J.; Maruyama, I.N. Nerve growth factor receptor TrkA exists as a preformed, yet inactive, dimer in living cells. FEBS Lett. 2011, 585, 295–299. [Google Scholar] [CrossRef]
- Shen, J.; Maruyama, I.N. Brain-derived neurotrophic factor receptor TrkB exists as a preformed dimer in living cells. J. Mol. Signal. 2012, 7, 2. [Google Scholar] [CrossRef]
- Macdonald-Obermann, J.L.; Piwnica-Worms, D.; Pike, L.J. Mechanics of EGF receptor/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc. Natl. Acad. Sci. USA 2012, 109, 137–142. [Google Scholar] [CrossRef]
- Macdonald-Obermann, J.L.; Adak, S.; Landgraf, R.; Piwnica-Worms, D.; Pike, L.J. Dynamic analysis of the epidermal growth factor (EGF) receptor-ErbB2-ErbB3 protein network by luciferase fragment complementation imaging. J. Biol. Chem. 2013, 288, 30773–30784. [Google Scholar]
- Ward, C.W.; Lawrence, M.C.; Streltsov, V.A.; Adams, T.E.; McKern, N.M. The insulin and EGF receptor structures: New insights into ligand-induced receptor activation. Trends Biochem. Sci. 2007, 32, 129–137. [Google Scholar] [CrossRef]
- Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962, 237, 1555–1562. [Google Scholar]
- Carpenter, G.; Lembach, K.J.; Morrison, M.M.; Cohen, S. Characterization of the binding of 125-I-labeled epidermal growth factor to human fibroblasts. J. Biol. Chem. 1975, 250, 4297–4304. [Google Scholar]
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signaling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar] [CrossRef]
- Tidcombe, H.; Jackson-Fisher, A.; Mathers, K.; Stern, D.F.; Gassmann, M.; Golding, J.P. Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proc. Natl. Acad. Sci. USA 2003, 100, 8281–8286. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Lemmon, M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signaling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Ward, C.W.; Garrett, T.P.J. The relationship between the L1 and L2 domains of the insulin and epidermal growth factor receptors and leucine-rich repeat modules. BMC Bioinform. 2001, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.W.; Garrett, T.P.J.; McKern, N.M.; Lou, M.; Cosgrove, L.J.; Sparrow, L.G.; Frenkel, M.J.; Hoyne, P.A.; Elleman, T.C.; Adams, T.E.; et al. The three dimensional structure of the type I insulin-like growth factor receptor. Mol. Pathol. 2001, 54, 125–132. [Google Scholar] [CrossRef]
- Wilson, K.J.; Gilmore, J.L.; Foley, J.; Lemmon, M.A.; Riese, D.J., 2nd. Functional selectivity of EGF family peptide growth factors: Implications for cancer. Pharmacol. Ther. 2009, 122, 1–8. [Google Scholar]
- Karunagaran, D.; Tzahar, E.; Beerli, R.R.; Chen, X.; Graus-Porta, D.; Ratzkin, B.J.; Seger, R.; Hynes, N.E.; Yarden, Y. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: Implications for breast cancer. EMBO J. 1996, 15, 254–264. [Google Scholar]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287. [Google Scholar]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef]
- Guy, P.M.; Platko, J.V.; Cantley, L.C.; Cerione, R.A.; Carraway, K.L., 3rd. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8132–8136. [Google Scholar]
- Sierke, S.L.; Cheng, K.; Kim, H.H.; Koland, J.G. Biochemical characterization of the protein tyrosine kinase homology domain of the ErbB3 (HER3) receptor protein. Biochem. J. 1997, 322, 757–763. [Google Scholar]
- Prigent, S.A.; Gullick, W.J. Identification of c-erbB-3 binding sites for phosphatidylinositol 3'-kinase and SHC using an EGF receptor/c-erbB-3 chimera. EMBO J. 1994, 13, 2831–2841. [Google Scholar]
- Wallasch, C.; Weiss, F.U.; Niederfellner, G.; Jallal, B.; Issing, W.; Ullrich, A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995, 14, 4267–4275. [Google Scholar]
- Shi, F.; Telesco, S.E.; Liu, Y.; Radhakrishnan, R.; Lemmon, M.A. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 7692–7697. [Google Scholar] [CrossRef]
- Zhang, Q.; Park, E.; Kani, K.; Landgraf, R. Functional isolation of activated and unilaterally phosphorylated heterodimers of ERBB2 and ERBB3 as scaffolds in ligand-dependent signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 13237–13242. [Google Scholar] [CrossRef]
- Levi-Montalcini, R.; Angeletti, P.U. Essential role of the nerve growth factor in the survival and maintenance of dissociated sensory and sympathetic embryonic nerve cells in vitro. Dev. Biol. 1963, 7, 653–659. [Google Scholar] [CrossRef]
- Lai, K.O.; Fu, W.Y.; Ip, F.C.; Ip, N.Y. Cloning and expression of a novel neurotrophin, NT-7, from carp. Mol. Cell. Neurosci. 1998, 11, 64–76. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Hempstead, B.L.; Martin-Zanca, D.; Chao, M.V.; Parada, L.F. The trk proto-oncogene product: A signal transducing receptor for nerve growth factor. Science 1991, 252, 554–558. [Google Scholar]
- Ip, N.Y.; Ibanez, C.F.; Nye, S.H.; McClain, J.; Jones, P.F.; Gies, D.R.; Belluscio, L.; Le Beau, M.M.; Espinosa, R., 3rd; Squinto, S.P.; et al. Mammalian neurotrophin-4: Structure, chromosomal localization, tissue distribution, and receptor specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 3060–3064. [Google Scholar] [CrossRef]
- Klein, R.; Nanduri, V.; Jing, S.A.; Lamballe, F.; Tapley, P.; Bryant, S.; Cordon-Cardo, C.; Jones, K.R.; Reichardt, L.F.; Barbacid, M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 1991, 66, 395–403. [Google Scholar] [CrossRef]
- Lamballe, F.; Klein, R.; Barbacid, M. trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell 1991, 66, 967–979. [Google Scholar] [CrossRef]
- Soppet, D.; Escandon, E.; Maragos, J.; Middlemas, D.S.; Reid, S.W.; Blair, J.; Burton, L.E.; Stanton, B.R.; Kaplan, D.R.; Hunter, T.; et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell 1991, 65, 895–903. [Google Scholar] [CrossRef]
- Squinto, S.P.; Stitt, T.N.; Aldrich, T.H.; Davis, S.; Bianco, S.M.; Radziejewski, C.; Glass, D.J.; Masiakowski, P.; Furth, M.E.; Valenzuela, D.M.; et al. trkB encodes a functional receptor for brain-derived neurotrophic factor and nejurotrophin-3 but not nerve growth factor. Cell 1991, 65, 885–893. [Google Scholar] [CrossRef]
- Rodriguez-Tebar, A.; Dechant, G.; Gotz, R.; Barde, Y.A. Binding of neurotrophin-3 to its neuronal receptors and interactions with nerve growth factor and brain-derived neurotrophic factor. EMBO J. 1992, 11, 917–922. [Google Scholar]
- Barbacid, M. The Trk family of neurotrophin receptors. J. Neurobiol. 1994, 25, 1386–1403. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef]
- Lessmann, V.; Brigadski, T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: An update. Neurosci. Res. 2009, 65, 11–22. [Google Scholar] [CrossRef]
- Lessmann, V.; Gottmann, K.; Malcangio, M. Neurotrophin secretion: Current facts and future prospects. Prog. Neurobiol. 2003, 69, 341–374. [Google Scholar] [CrossRef]
- Chao, M.V.; Rajagopal, R.; Lee, F.S. Neurotrophin signalling in health and disease. Clin. Sci. (Lond.) 2006, 110, 167–173. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Martin-Zanca, D.; Parada, L.F. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 1991, 350, 158–160. [Google Scholar] [CrossRef]
- Hempstead, B.L.; Martin-Zanca, D.; Kaplan, D.R.; Parada, L.F.; Chao, M.V. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 1991, 350, 678–683. [Google Scholar] [CrossRef]
- Klein, R.; Lamballe, F.; Bryant, S.; Barbacid, M. The trkB tyrosine protein kinase is a receptor for neurotrophin-4. Neuron 1992, 8, 947–956. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; Tapley, P.; Jing, S.Q.; Nanduri, V.; O’Rourke, E.; Lambelle, F.; Kovary, K.; Klein, R.; Jones, K.R.; Reichardt, L.F.; et al. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell 1991, 66, 173–183. [Google Scholar] [CrossRef]
- Simi, A.; Ibáñez, C.F. Assembly and activation of neurotrophic factor receptor complexes. Dev. Neurobiol. 2010, 70, 323–331. [Google Scholar]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signaling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Lyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Schneider, R.; Schweiger, M. A novel modular mosaic of cell adhesion motifs in the extracellular domains of the neurogenic trk and trkB tyrosine kinase receptors. Oncogene 1991, 6, 1807–1811. [Google Scholar]
- Bertrand, T.; Kothe, M.; Liu, J.; Dupuy, A.; Rak, A.; Berne, P.F.; Davis, S.; Gladysheva, T.; Valtre, C.; et al. The crystal structures of TrkA and TrkB suggest key regions for achieving selective inhibition. J. Mol. Biol. 2012, 423, 439–453. [Google Scholar] [CrossRef]
- Holden, P.H.; Asopa, V.; Robertson, A.G.; Clarke, A.R.; Tyler, S.; Bennett, G.S.; Brain, S.D.; Wilcock, G.K.; Allen, S.J.; Smith, S.K.; et al. Immunogloblin-like domains define the nerve growth factor binding site of the TrkA receptor. Nat. Biotechnol. 1997, 15, 668–672. [Google Scholar] [CrossRef]
- Perez, P.; Coll, P.M.; Hempstead, B.L.; Martin-Zanca, D.; Chao, M.V. NGF binding to the trk tyrosine kinase receptor requires the extracellular immunoglobulin-like domains. Mol. Cell. Neurosci. 1995, 6, 97–105. [Google Scholar] [CrossRef]
- Urfer, R.; Tsoulfas, P.; O’Connell, L.; Presta, L.G. Specificity determinants in neurotrophin-3 and design of nerve growth factor-based trkC agonists by changing central β-strand bundle residues of their neurotrophin-3 analogs. Biochemistry 1997, 36, 4775–4781. [Google Scholar] [CrossRef]
- Ultsch, M.H.; Wiesmann, C.; Simmons, L.C.; Henrich, J.; Yang, M.; Reilly, D.; Bass, S.H.; de Vos, A.M. Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J. Mol. Biol. 1999, 290, 149–159. [Google Scholar] [CrossRef]
- Wiesmann, C.; Ultsch, M.H.; Bass, S.H.; de Vos, A.M. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 1999, 401, 184–188. [Google Scholar] [CrossRef]
- Steiner, D.F. Insulin Today. Diabetes 1976, 26, 322–340. [Google Scholar] [CrossRef]
- Levine, R.; Goldstein, M.; Klein, S.; Huddlestun, B. The action of insulin on the distribution of galactose in eviscerated nephrectomized dogs. J. Biol. Chem. 1949, 179, 985–986. [Google Scholar]
- Renehan, A.G.; Brennan, B.M. Acromegaly, growth hormone and cancer risk. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 639–657. [Google Scholar] [CrossRef]
- Walenkamp, M.J.E.; Wit, J.M. Genetic disorders in the growth hormone – insulin-like growth factor-I axis. Horm. Res. 2006, 66, 221–230. [Google Scholar] [CrossRef]
- Gicquel, C.; Le Bouc, Y. Hormonal regulation of fetal growth. Horm. Res. 2006, 65, 28–33. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 2010, 21, 610–618. [Google Scholar] [CrossRef]
- Freychet, P.; Roth, J.; Neville, D.M., Jr. Insulin receptors in the liver: Specific binding of [125I]insulin to the plasma membrane and its relation to insulin bioactivity. Proc. Natl. Acad. Sci. USA 1971, 68, 1833–1837. [Google Scholar] [CrossRef]
- Denley, A.; Wallace, J.C.; Cosgrove, L.J.; Forbes, B.E. The insulin receptor isoform exon 11- (IR-A) in cancer and other diseases: A review. Horm. Metab. Res. 2003, 35, 778–785. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar]
- Denley, A.; Bonython, E.R.; Booker, G.W.; Cosgrove, L.J.; Forbes, B.E.; Ward, C.W.; Wallace, J.C. Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 2004, 18, 2502–2512. [Google Scholar] [CrossRef]
- Slaaby, R.; Schaffer, L.; Lautrup-Larsen, I.; Andersen, A.S.; Shaw, A.C.; Mathiasen, I.S.; Brandt, J. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J. Biol. Chem. 2006, 281, 25869–25874. [Google Scholar] [CrossRef]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of insulin/IGF hybrid receptors: Contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007, 403, 603–613. [Google Scholar] [CrossRef]
- De Meyts, P. Insulin and insulin-like growth factors: The paradox of signaling specificity. Growth Horm. IGF Res. 2002, 12, 81–83. [Google Scholar] [CrossRef]
- Dupont, J.; LeRoith, D. Insulin and insulin-like growth factor I receptors: Similarities and differences in signal transduction. Horm. Res. 2001, 55, 22–26. [Google Scholar]
- Jui, H.Y.; Suzuki, Y.; Accili, D.; Taylor, S.I. Expression of a cDNA encoding the human insulin receptor-related receptor. J. Biol. Chem. 1994, 269, 22446–22452. [Google Scholar]
- Zhang, B.; Roth, R.A. The insulin receptor-related receptor. Tissue expression, ligand binding specificity, and signaling capabilities. J. Biol. Chem. 1992, 267, 18320–18328. [Google Scholar]
- Hirayama, I.; Tamemoto, H.; Yokota, H.; Kubo, S.K.; Wang, J.; Kuwano, H.; Nagamachi, Y.; Takeuchi, T.; Izumi, T. Insulin receptor-related receptor is expressed in pancreatic beta-cells and stimulates tyrosine phosphorylation of insulin receptor substrate-1 and -2. Diabetes 1999, 48, 1237–1244. [Google Scholar] [CrossRef]
- Mathi, S.K.; Chan, J.; Watt, V.M. Insulin receptor-related receptor messenger ribonucleic acid: Quantitative distribution and localization to subpopulations of epithelial cells in stomach and kidney. Endocrinology 1995, 136, 4125–4132. [Google Scholar] [CrossRef]
- Deyev, I.E.; Sohet, F.; Vassilenko, K.P.; Serova, O.V.; Popova, N.V.; Zozulya, S.A.; Burova, E.B.; Houillier, P.; Rzhevsky, D.I.; Berchatova, A.A.; et al. Insulin receptor-related receptor as an extracellular alkali sensor. Cell Metab. 2011, 13, 679–689. [Google Scholar] [CrossRef]
- Nef, S.; Verma-Kurvari, S.; Merenmies, J.; Vassalli, J.D.; Efstratiadis, A.; Accili, D.; Parada, L.F. Testis determination requires insulin receptor family function in mice. Nature 2003, 426, 291–295. [Google Scholar] [CrossRef]
- Adams, T.E.; Epa, V.C.; Garrett, T.P.; Ward, C.W. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. 2000, 57, 1050–1093. [Google Scholar] [CrossRef]
- Ward, C.W.; Garrett, T.P. Structural relationships between the insulin receptor and epidermal growth factor receptor families and other proteins. Curr. Opin. Drug Discov. Dev. 2004, 7, 630–638. [Google Scholar]
- Hubbard, S.R. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 464–471. [Google Scholar] [CrossRef]
- Cheng, I.; Stram, D.O.; Penney, K.L.; Pike, M.; Le Marchand, L.; Kolonel, L.N.; Hirschhorn, J.; Altshuler, D.; Henderson, B.E.; Freedman, M.L. Common genetic variation in IGF1 and prostate cance risk in the multiethnic cohort. J. Natl. Cancer Inst. 2006, 98, 123–134. [Google Scholar] [CrossRef]
- Cohen, P. The twentieth centrury struggle to decipher insulin signaling. Nat. Rev. Mol. Cell Biol. 2006, 7, 867–873. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signaling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Zaid, H.; Antonescu, C.N.; Randhawa, V.K.; Klip, A. Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem. J. 2008, 413, 201–215. [Google Scholar] [CrossRef]
- Yarden, Y.; Schlessinger, J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 1987, 26, 1443–1451. [Google Scholar] [CrossRef]
- Boni-Schnetzler, M.; Pilch, P.F. Mechanism of epidermal growth factor receptor autophosphorylation and high-affinity binding. Proc. Natl. Acad. Sci. USA 1987, 84, 7832–7836. [Google Scholar] [CrossRef]
- Weiss, F.U.; Daub, H.; Ullrich, A. Novel mechanisms of RTK signal generation. Curr. Opin. Genet. Dev. 1997, 7, 80–86. [Google Scholar] [CrossRef]
- Canals, F. Signal transmission by epidermal growth factor receptor: Coincidence of activation and dimerization. Biochemistry 1992, 31, 4493–4501. [Google Scholar] [CrossRef]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef]
- Knebel, A.; Rahmsdorf, H.J.; Ullrich, A.; Herrlich, P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996, 15, 5314–5325. [Google Scholar]
- Nagy, P.; Claus, J.; Jovin, T.M.; Arndt-Jovin, D.J. Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc. Natl. Acad. Sci. USA 2010, 107, 16524–16529. [Google Scholar] [CrossRef]
- Sawano, A.; Takayama, S.; Matsuda, M.; Miyawaki, A. Lateral propagation of EGF signaling after local stimulation is dependent on receptor density. Dev. Cell 2002, 3, 245–257. [Google Scholar] [CrossRef]
- Arevalo, J.C.; Conde, B.; Hempstead, B.L.; Chao, M.V.; Martin-Zanca, D.; Perez, P. TrkA immunoglobulin-like ligand binding domains inhibit spontaneous activation of the receptor. Mol. Cell. Biol. 2000, 20, 5908–5916. [Google Scholar] [CrossRef]
- Ohira, K.; Shimizu, K.; Hayashi, M. TrkB dimerization during development of the prefrontal cortex of the macaque. J. Neurosci. Res. 2001, 65, 463–469. [Google Scholar] [CrossRef]
- Mischel, P.S.; Umbach, J.A.; Eskandari, S.; Smith, S.G.; Gundersen, C.B.; Zampighi, G.A. Nerve growth factor signals via preexisting TrkA receptor oligomers. Biophys. J. 2002, 83, 968–976. [Google Scholar] [CrossRef]
- Vilar, M.; Charalampopoulos, I.; Kenchappa, R.S.; Simi, A.; Karaca, E.; Reversi, A.; Choi, S.; Bothwell, M.; Mingarro, I.; Friedman, W.J.; Schiavo, G.; Bastiaens, P.I.; Verveer, P.J.; Carter, B.D.; Ibanez, C.F. Activation of the p75 neurotrophin receptor through conformational rearrangement of disulphide-linked receptor dimers. Neuron 2009, 62, 72–83. [Google Scholar] [CrossRef]
- Marchetti, L.; Callegari, A.; Luin, S.; Signore, G.; Viegi, A.; Beltram, F.; Cattaneo, A. Ligand signature in the membrane dynamics of single TrkA receptor molecules. J. Cell Sci. 2013, 126, 4445–4456. [Google Scholar] [CrossRef]
- He, X.L.; Garcia, K.C. Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 2004, 304, 870–875. [Google Scholar] [CrossRef]
- Wehrman, T.; He, X.; Raab, B.; Dukipatti, A.; Blau, H.; Garcia, K.C. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 2007, 53, 25–38. [Google Scholar] [CrossRef]
- Iacaruso, M.F.; Galli, S.; Marti, M.; Villalta, J.I.; Estrin, D.A.; Jares-Erijman, E.A.; Pietrasanta, L.I. Structural model for p75(NTR)-TrkA intracellular domain interaction: A combined FRET and bioinformatics study. J. Mol. Biol. 2011, 414, 681–698. [Google Scholar] [CrossRef]
- Matusica, D.; Skeldal, S.; Sykes, A.M.; Palstra, N.; Sharma, A.; Coulson, E.J. An intracellular domain fragment of the p75 neurotrophin receptor (p75(NTR)) enhances tropomyosin receptor kinase A (TrkA) receptor function. J. Biol. Chem. 2013, 288, 11144–11154. [Google Scholar]
- Massague, J.; Pilch, P.F.; Czech, M.P. Electrophoretic resolution of three major insulin receptor structures with unique subunit stoichiometries. Proc. Natl. Acad. Sci. USA 1980, 77, 7137–7141. [Google Scholar] [CrossRef]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.H.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signaling. Cell 1985, 40, 747–58. [Google Scholar] [CrossRef]
- Moxham, C.P.; Duronio, V.; Jacobs, S. Insulin-like growth factor I receptor beta-subunit heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I and insulin receptor heterodimers. J. Biol. Chem. 1989, 264, 13238–13244. [Google Scholar]
- Soos, M.A.; Siddle, K. Immunological relationships between receptors for insulin and insulin-like growth factor I. Evidence for structural heterogeneity of insulin-like growth factor I receptors involving hybrids with insulin receptors. Biochem. J. 1989, 263, 553–563. [Google Scholar]
- Federici, M.; Porzio, O.; Zucaro, L.; Giovannone, B.; Borboni, P.; Marini, M.A.; Lauro, D.; Sesti, G. Increased abundance of insulin/IGF-I hybrid receptors in adipose tissue from NIDDM patients. Mol. Cell Endocrinol. 1997, 135, 41–47. [Google Scholar] [CrossRef]
- Bailyes, E.M.; Nave, B.T.; Soos, M.A.; Orr, S.R.; Hayward, A.C.; Siddle, K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: Quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem. J. 1997, 327, 209–215. [Google Scholar]
- Sandhu, M.S.; Dunger, D.B.; Giovannucci, E.L. Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J. Natl. Cancer Inst. 2002, 94, 972–980. [Google Scholar] [CrossRef]
- Cho, H.S.; Leahy, D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Moll. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Bouyain, S.; Longo, P.A.; Li, S.; Ferguson, K.M.; Leahy, D.J. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 15024–15029. [Google Scholar] [CrossRef]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Berezov, A.; Chen, J.; Liu, Q.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling receptor ensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 2002, 277, 28330–28339. [Google Scholar]
- Walker, F.; Orchard, S.G.; Jorissen, R.N.; Hall, N.E.; Zhang, H.H.; Hoyne, P.A.; Adams, T.E.; Johns, T.G.; Ward, C.; Garrett, T.P.; et al. CR1/CR2 interactions modulate the functions of the cell surface epidermal growth factor receptor. J. Biol. Chem. 2004, 279, 22387–22398. [Google Scholar] [CrossRef]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef]
- Jura, N.; Endres, N.F.; Engel, K.; Deindl, S.; Das, R.; Lamers, M.H.; Wemmer, D.E.; Zhang, X.; Kuriyan, J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009, 137, 1293–1307. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massague, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef]
- Red Brewer, M.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell 2009, 34, 641–651. [Google Scholar] [CrossRef]
- Jing, S.; Tapley, P.; Barbacid, M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron 1992, 9, 1067–1079. [Google Scholar] [CrossRef]
- Bothwell, M.A.; Shooter, E.M. Dissociation equilibrium constant of beta nerve growth factor. J. Biol. Chem. 1977, 252, 8532–8536. [Google Scholar]
- McDonald, N.Q.; Lapatto, R.; Murray-Rust, J.; Gunning, J.; Wlodawer, A.; Blundell, T.L. New protein fold revealed by a 2.3-Å resolution crystal structure of nerve growth factor. Nature 1991, 354, 411–414. [Google Scholar] [CrossRef]
- Radziejewski, C.; Robinson, R.C.; DiStefano, P.S.; Taylor, J.W. Dimeric structure and conformational stability of brain-derived neurotrophic factor and neurotrophin-3. Biochemistry 1992, 31, 4431–4436. [Google Scholar] [CrossRef]
- Ibanez, C.F.; Ilag, L.L.; Murray-Rust, J.; Persson, H. An extended surface of binding to Trk tyrosine kinase receptors in NGF and BDNF allows the engineering of a multifunctional pan-neurotrophin. EMBO J. 1993, 12, 2281–2293. [Google Scholar]
- Artim, S.C.; Mendrola, J.M.; Lemmon, M.A. Assessing the range of kinase autoinhibition mechanisms in the insulin receptor family. Biochem. J. 2012, 448, 213–220. [Google Scholar] [CrossRef]
- Vilar, M.; Charalampopoulos, I.; Kenchappa, R.S.; Reversi, A.; Klos-Applequist, J.M.; Karaca, E.; Simi, A.; Spuch, C.; Choi, S.; Friedman, W.J.; et al. Ligand-independent signaling by disulfide-crosslinked dimers of the p75 neurotrophin receptor. J. Cell Sci. 2009, 122, 3351–3357. [Google Scholar] [CrossRef]
- Li, S.; Covino, N.D.; Stein, E.G.; Till, J.H.; Hubbard, S.R. Structural and biochemical evidence for an autoinhibitory role for tyrosine 984 in the juxtamembrane region of the insulin receptor. J. Biol. Chem. 2003, 278, 26007–26014. [Google Scholar]
- Hubbard, S.R. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef]
- Munshi, S.; Kornienko, M.; Hall, D.L.; Reid, J.C.; Waxman, L.; Stirdivant, S.M.; Darke, P.L.; Kuo, L.C. Crystal structure of the Apo, unactivated insulin-like growth factor-1 receptor kinase. Implication for inhibitor specificity. J. Biol. Chem. 2002, 277, 38797–38802. [Google Scholar] [CrossRef]
- Menting, J.G.; Whittaker, J.; Margetts, M.B.; Whittaker, L.J.; Kong, G.K.; Smith, B.J.; Watson, C.J.; Zakova, L.; Kletvikova, E.; Jiracek, J.; et al. How insulin engages its primary binding site on the insulin receptor. Nature 2013, 493, 241–245. [Google Scholar] [CrossRef]
- Schlessinger, J. Allosteric regulation of the epidermal growth factor receptor kinase. J. Cell Biol. 1986, 103, 2067–2072. [Google Scholar] [CrossRef]
- Macdonald, J.L.; Pike, L.J. Heterogeneity in EGF-binding affinities arises from negative cooperativity in an aggregating system. Proc. Natl. Acad. Sci. USA 2008, 105, 112–117. [Google Scholar] [CrossRef]
- Macdonald-Obermann, J.L.; Pike, L.J. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. J. Biol. Chem. 2009, 284, 13570–13576. [Google Scholar] [CrossRef]
- De Meyts, P.; Roth, J.; Neville, D.M., Jr.; Gavin, J.R., III; Lesniak, M.A. Insulin interactions with its receptors: Experimental evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 1973, 55, 154–161. [Google Scholar] [CrossRef]
- Shoyab, M.; De Larco, J.E.; Todaro, G.J. Biologically active phorbol esters specifically alter affinity of epidermal growth factor membrane receptors. Nature 1979, 279, 387–391. [Google Scholar] [CrossRef]
- Magun, B.E.; Matrisian, L.M.; Bowden, G.T. Epidermal growth factor. Ability of tumor promoter to alter its degradation, receptor affinity and receptor number. J. Biol. Chem. 1980, 255, 6373–6381. [Google Scholar]
- King, A.C.; Cuatrecasas, P. Resolution of high and low affinity epidermal growth factor receptors. Inhibition of high affinity component by low temperature, cycloheximide, and phorbol esters. J. Biol. Chem. 1982, 257, 3053–3060. [Google Scholar]
- Kawamoto, T.; Sato, J.D.; Le, A.; Polikoff, J.; Sato, G.H.; Mendelsohn, J. Growth stimulation of A431 cells by epidermal growth factor: Identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc. Natl. Acad. Sci. USA 1983, 80, 1337–1341. [Google Scholar] [CrossRef]
- Defize, L.H.; Boonstra, J.; Meisenhelder, J.; Kruijer, W.; Tertoolen, L.G.; Tilly, B.C.; Hunter, T.; van Bergen en Henegouwen, P.M.; Moolenaar, W.H.; de Laat, S.W. Signal transduction by epidermal growth factor occurs through the subclass of high affinity receptors. J. Cell Biol. 1989, 109, 2495–2507. [Google Scholar] [CrossRef]
- Bellot, F.; Moolenaar, W.; Kris, R.; Mirakhur, B.; Verlaan, I.; Ullrich, A.; Schlessinger, J.; Felder, S. High-affinity epidermal growth factor binding is specifically reduced by a monoclonal antibody, and appears necessary for early responses. J. Cell Biol. 1990, 110, 491–502. [Google Scholar] [CrossRef]
- Hunter, T.; Ling, N.; Cooper, J.A. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature 1984, 311, 480–483. [Google Scholar] [CrossRef]
- Özcan, F.; Klein, P.; Lemmon, M.A.; Lax, I.; Schlessinger, J. On the nature of low- and high-affinity EGF receptors on living cells. Proc. Natl. Acad. Sci. USA 2006, 103, 5735–5740. [Google Scholar] [CrossRef]
- Livneh, E.; Prywes, R.; Kashles, O.; Reiss, N.; Sasson, I.; Mory, Y.; Ullrich, A.; Schlessinger, J. Reconstitution of human epidermal growth factor receptors and its deletion mutants in cultured hamster cells. J. Biol. Chem. 1986, 261, 12490–12497. [Google Scholar]
- Wofsy, C.; Goldstein, B.; Lund, K.; Wiley, H.S. Implications of epidermal growth factor (EGF) induced egf receptor aggregation. Biophys. J. 1992, 63, 98–110. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Bu, Z.; Ladbury, J.E.; Zhou, M.; Pinchasi, D.; Lax, I.; Engelman, D.M.; Schlessinger, J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997, 16, 281–294. [Google Scholar] [CrossRef]
- Klein, P.; Mattoon, D.; Lemmon, M.A.; Schlessinger, J. A structure-based model for ligand binding and dimerization of EGF receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 929–934. [Google Scholar] [CrossRef]
- Holbrook, M.R.; Slakey, L.L.; Gross, D.J. Thermodynamic mixing of molecular states of the epidermal growth factor receptor modulates macroscopic ligand binding affinity. Biochem. J. 2000, 352, 99–108. [Google Scholar] [CrossRef]
- Mayawala, K.; Vlachos, D.G.; Edwards, J.S. Heterogeneities in EGF receptor density at the cell surface can lead to concave up scatchard plot of EGF binding. FEBS Lett. 2005, 579, 3043–3047. [Google Scholar] [CrossRef]
- Alvarado, D.; Klein, D.E.; Lemmon, M.A. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell 2010, 142, 568–579. [Google Scholar] [CrossRef]
- Liu, P.; Cleveland, T.E., 4th; Bouyain, S.; Byrne, P.O.; Longo, P.A.; Leahy, D.J. A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. USA 2012, 109, 10861–10866. [Google Scholar]
- Sako, Y.; Minoghchi, S.; Yanagida, T. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2000, 2, 168–172. [Google Scholar] [CrossRef]
- Lu, C.; Mi, L.Z.; Schurpf, T.; Walz, T.; Springer, T.A. Mechanisms for kinase-mediated dimerization of the epidermal growth factor receptor. J. Biol. Chem. 2012, 287, 38244–38253. [Google Scholar] [CrossRef]
- Adak, S.; DeAndrade, D.; Pike, L.J. The tethering arm of the EGF receptor is required for negative cooperativity and signal transduction. J. Biol. Chem. 2011, 286, 1545–1555. [Google Scholar] [CrossRef]
- Adak, S.; Yang, K.S.; Macdonald-Obermann, J.; Pike, L.J. The membrane-proximal intracellular domain of the epidermal growth factor receptor underlies negative cooperativity in ligand binding. J. Biol. Chem. 2011, 286, 45146–45155. [Google Scholar]
- Downward, J.; Waterfield, M.D.; Parker, P.J. Autophosphorylation and protein kinase C phosphorylation of the epidermal growth factor receptor. Effect on tyrosine kinase activity and ligand binding affinity. J. Biol. Chem. 1985, 260, 14538–14546. [Google Scholar]
- Mahadeo, D.; Kaplan, L.; Chao, M.V.; Hempstead, B.L. High affinity nerve growth factor binding displays a faster rate of association than p140trk binding. Implications for multi-subunit polypeptide receptors. J. Biol. Chem. 1994, 269, 6884–6891. [Google Scholar]
- Bothwell, M. Keeping track of neurotrophin receptors. Cell 1991, 65, 915–918. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophin receptors: A window into neuronal differentiation. Neuron 1992, 9, 583–593. [Google Scholar] [CrossRef]
- Vesa, J.; Kruttgen, A.; Shooter, E.M. p75 reduces TrkB tyrosine autophosphorylation in response to brain-derived neurotrophic factor and neurotrophin 4/5. J. Biol. Chem. 2000, 275, 24414–24420. [Google Scholar] [CrossRef]
- Barker, P.A. High affinity not in the vicinity? Neuron 2007, 53, 1–4. [Google Scholar] [CrossRef]
- Esposito, D.; Patel, P.; Stephens, R.M.; Perez, P.; Chao, M.V.; Kaplan, D.R.; Hempstead, B.L. The cytoplasmic and transmembrane domains of the p75 and Trk A receptors regulate high affinity binding to nerve growth factor. J. Biol. Chem. 2001, 276, 32687–32695. [Google Scholar]
- Frattali, A.L.; Treadway, J.L.; Pessin, J.E. Transmembrane signaling by the human insulin receptor kinase. Relationship between intramolecular beta subunit trans- and cis-autophosphorylation and substrate kinase activation. J. Biol. Chem. 1992, 267, 19521–19528. [Google Scholar]
- De Meyts, P.; Whittaker, J. Structural biology of insulin and IGF1 receptors: Implication for drug design. Nat. Rev. Drug Discov. 2002, 1, 769–783. [Google Scholar] [CrossRef]
- De Meyts, P. The structural basis of insulin and insulin-like growth factor-I receptor binding and negative co-operativity, and its relevance to mitogenic versus metabolic signaling. Diabetologia 1994, 37, S135–S148. [Google Scholar] [CrossRef]
- De Meyts, P. The insulin receptor: A prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 2008, 33, 376–384. [Google Scholar] [CrossRef]
- Kiselyov, V.V.; Versteyhe, S.; Gauguin, L.; De Meyts, P. Harmonic oscillator model of the insulin and IGF1 receptors’ allosteric binding and activation. Mol. Syst. Biol. 2009, 5, 243. [Google Scholar]
- Knudsen, L.; De Meyts, P.; Kiselyov, V.V. Insight into the molecular basis for the kinetic differences between the two insulin receptor isoforms. Biochem. J. 2011, 440, 397–403. [Google Scholar] [CrossRef]
- Boni-Schnetzler, M.; Scott, W.; Waugh, S.M.; DiBella, E.; Pilch, P.F. The insulin receptor. Structural basis for high affinity ligand binding. J. Biol. Chem. 1987, 262, 8395–401. [Google Scholar]
- Sweet, L.J.; Morrison, B.D.; Pessin, J.E. Isolation of functional alpha beta heterodimers from the purified human placental alpha 2 beta 2 heterotetrameric insulin receptor complex. A structural basis for insulin binding. J. Biol. Chem. 1987, 262, 6939–6942. [Google Scholar]
- Pang, D.T.; Shafer, J.A. Stoichiometry for the binding of insulin to insulin receptors in adipocyte membranes. J. Biol. Chem. 1983, 258, 2514–2518. [Google Scholar]
- Pang, D.T.; Shafer, J.A. Evidence that insulin receptor from human placenta has a high affinity for only one molecule of insulin. J. Biol. Chem. 1984, 259, 8589–8596. [Google Scholar]
- Hoyne, P.A.; Elleman, T.C.; Adams, T.E.; Richards, K.M.; Ward, C.W. Properties of an insulin receptor with an IGF-1 receptor loop exchange in the cysteine-rich region. FEBS Lett. 2000, 469, 57–60. [Google Scholar]
- Marsh, J.W.; Westley, J.; Steiner, D.F. Insulin-receptor interactions. Presence of a positive cooperative effect. J. Biol. Chem. 1984, 259, 6641–6649. [Google Scholar]
- Riedel, H.; Dull, T.J.; Schlessinger, J.; Ullrich, A. A chimaeric receptor allows insulin to stimulate tyrosine kinase activity of epidermal growth factor receptor. Nature 1986, 324, 68–70. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maruyama, I.N. Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers. Cells 2014, 3, 304-330. https://doi.org/10.3390/cells3020304
Maruyama IN. Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers. Cells. 2014; 3(2):304-330. https://doi.org/10.3390/cells3020304
Chicago/Turabian StyleMaruyama, Ichiro N. 2014. "Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers" Cells 3, no. 2: 304-330. https://doi.org/10.3390/cells3020304