3.1. VEGFR2 Recycles to the Plasma Membrane via Rab4a-Containing Endosomes
Previous work has shown that the VEGFR2 exhibits both plasma membrane and endosomal localisation [
6,
12,
15,
21]. To test whether VEGFR2 undergoes a ligand-independent endosome-to-plasma membrane recycling step in quiescent primary human endothelial cells, monensin can be used, as this agent inhibits cell surface receptor recycling [
22,
23,
24,
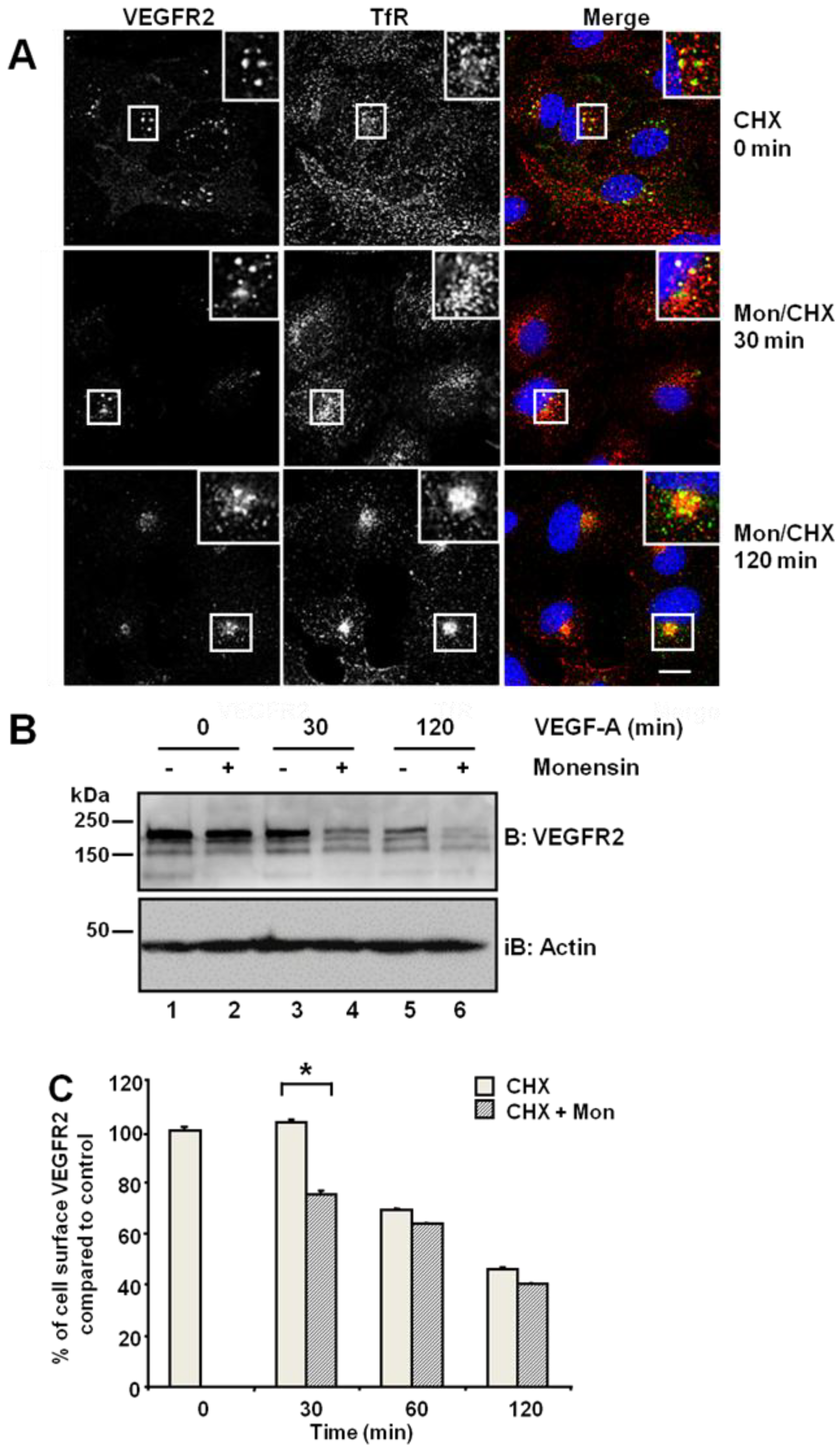
25]. Primary endothelial cells were first subjected to a block in new protein synthesis using cycloheximide: we detected a stable and distal pool of VEGFR2, which showed partial co-distribution with the transferrin receptor (TfR) present in early endosomes (
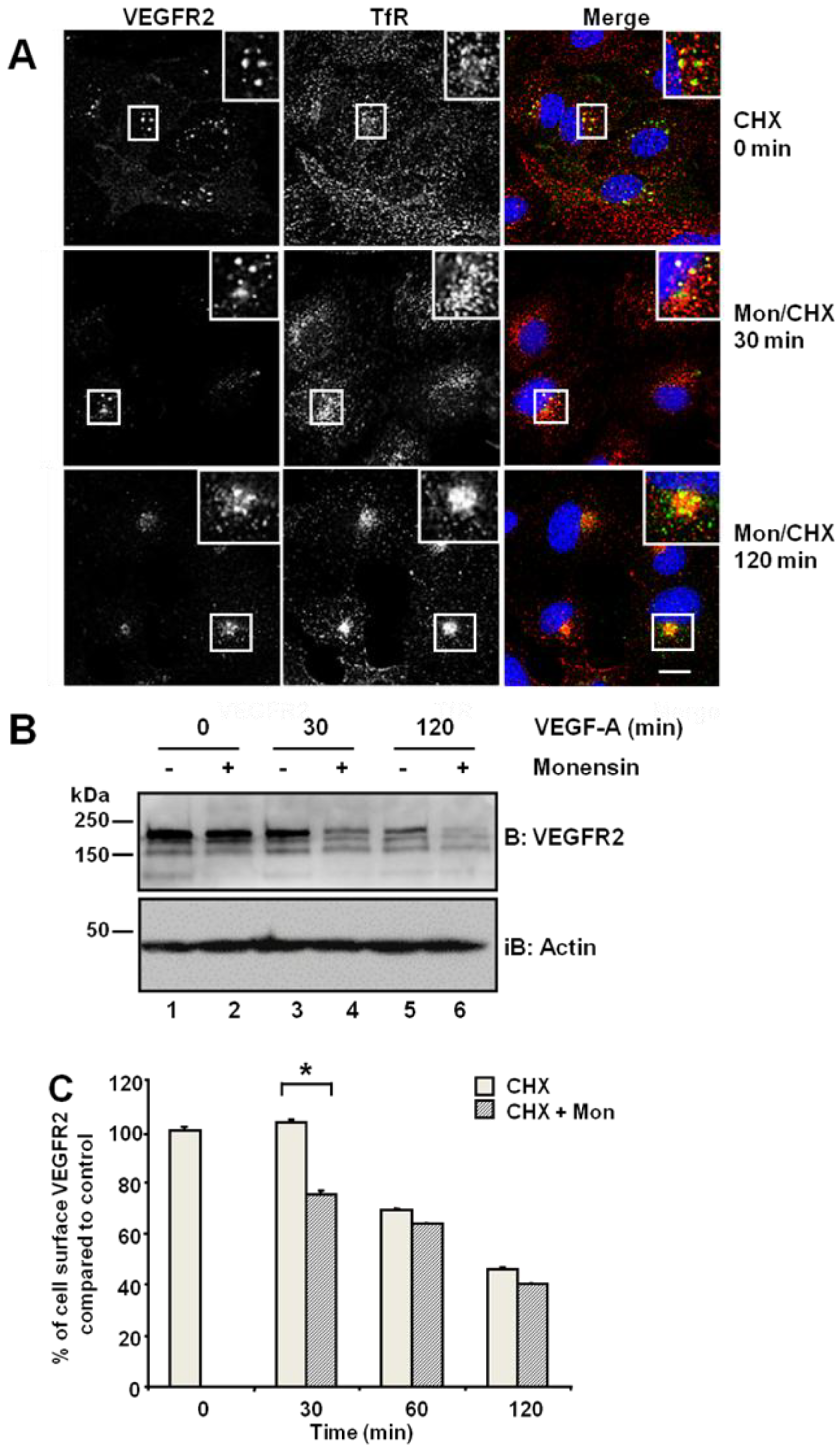
Figure 1A, upper panels). Simultaneous treatment with monensin and cycloheximide for 30 min increased VEGFR2 co-distribution with TfR in early endosomes (
Figure 1A, insets). Following simultaneous VEGF-A and cycloheximide treatment, VEGFR2 levels markedly decreased in the absence of new protein synthesis (
Figure 1B). Monensin further potentiated this VEGF-A-stimulated decrease in VEGFR2 levels (
Figure 1B). Inhibition of endosome-to-plasma membrane recycling would be thus predicted to decrease cell surface VEGFR2 levels. Flow cytometry analysis showed a ~25% decrease in cell surface VEGFR2 levels after 30-min exposure to monensin, compared with cycloheximide-treated cells, which showed no significant change in VEGFR2 levels (
Figure 1C). Thus, both quiescent and activated VEGFR2 undergoes endosome-to-plasma membrane recycling.
Figure 1.
Inhibition of endosome-to-plasma membrane recycling restricts VEGFR2 to a perinuclear compartment. (A) Human umbilical vein endothelial cells (HUVECs) were pre-treated for 2 h with cycloheximide (CHX) prior to treatment with 20 μM monensin for zero, 30 or 120 min. Cells were then fixed, permeabilised and labelled with goat anti-VEGFR2 extracellular domain (green) and mouse anti-TfR (red). Primary antibodies were visualized using AlexaFluor-conjugated species-specific secondary antibodies, and the nucleus was labelled with DAPI (blue). Bar: 10 μm. Insets show a two-fold magnification of the indicated regions. (B) Serum-starved HUVECs were pre-treated with 20 μM monensin prior to VEGF-A stimulation for zero, 30 or 120 min. Cell lysates were immunoblotted with antibodies specific for VEGFR2 or actin. The data shown is representative of three independent experiments. (C) Flow cytometry analysis of HUVECs treated with either 20 μM monensin or 50 μg/mL cycloheximide (CHX) for zero, 30, 60 or 120 min. Error bars denote ± SEM (n = 3); *, p < 0.05.
Figure 1.
Inhibition of endosome-to-plasma membrane recycling restricts VEGFR2 to a perinuclear compartment. (A) Human umbilical vein endothelial cells (HUVECs) were pre-treated for 2 h with cycloheximide (CHX) prior to treatment with 20 μM monensin for zero, 30 or 120 min. Cells were then fixed, permeabilised and labelled with goat anti-VEGFR2 extracellular domain (green) and mouse anti-TfR (red). Primary antibodies were visualized using AlexaFluor-conjugated species-specific secondary antibodies, and the nucleus was labelled with DAPI (blue). Bar: 10 μm. Insets show a two-fold magnification of the indicated regions. (B) Serum-starved HUVECs were pre-treated with 20 μM monensin prior to VEGF-A stimulation for zero, 30 or 120 min. Cell lysates were immunoblotted with antibodies specific for VEGFR2 or actin. The data shown is representative of three independent experiments. (C) Flow cytometry analysis of HUVECs treated with either 20 μM monensin or 50 μg/mL cycloheximide (CHX) for zero, 30, 60 or 120 min. Error bars denote ± SEM (n = 3); *, p < 0.05.
![Cells 03 00363 g001]()
Rab GTPases, such as Rab4a or Rab11a, regulate different endosome-to-plasma membrane recycling routes [
26,
27,
28,
29,
30,
31,
32] and are functionally linked to VEGFRs in endothelial cells [
6,
21,
33]. To differentiate between these two endosome-linked recycling pathways, we overexpressed GFP-tagged wild-type Rab4a or Rab11a in endothelial cells and assessed co-distribution with VEGFR2 using quantitative microscopy (
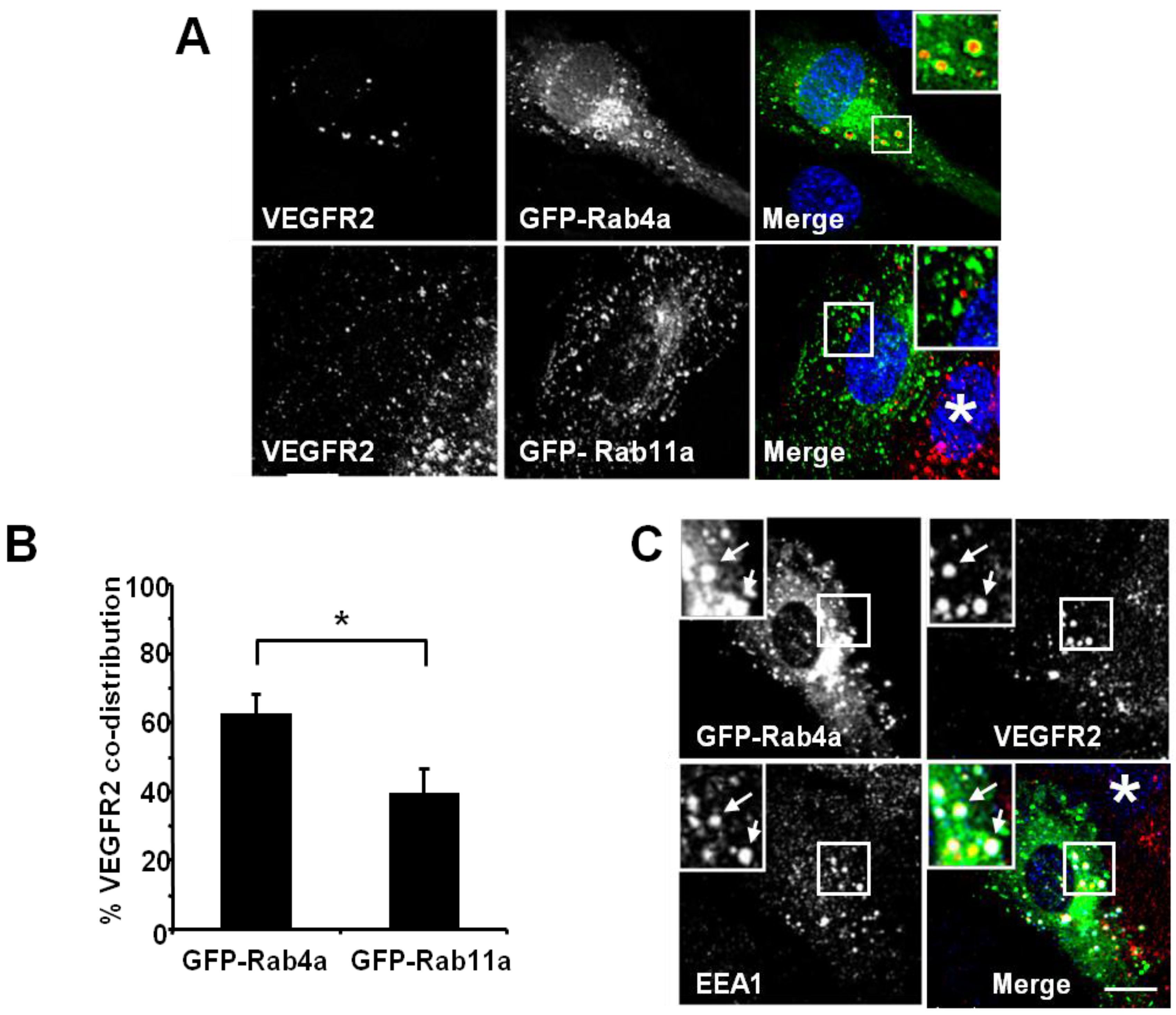
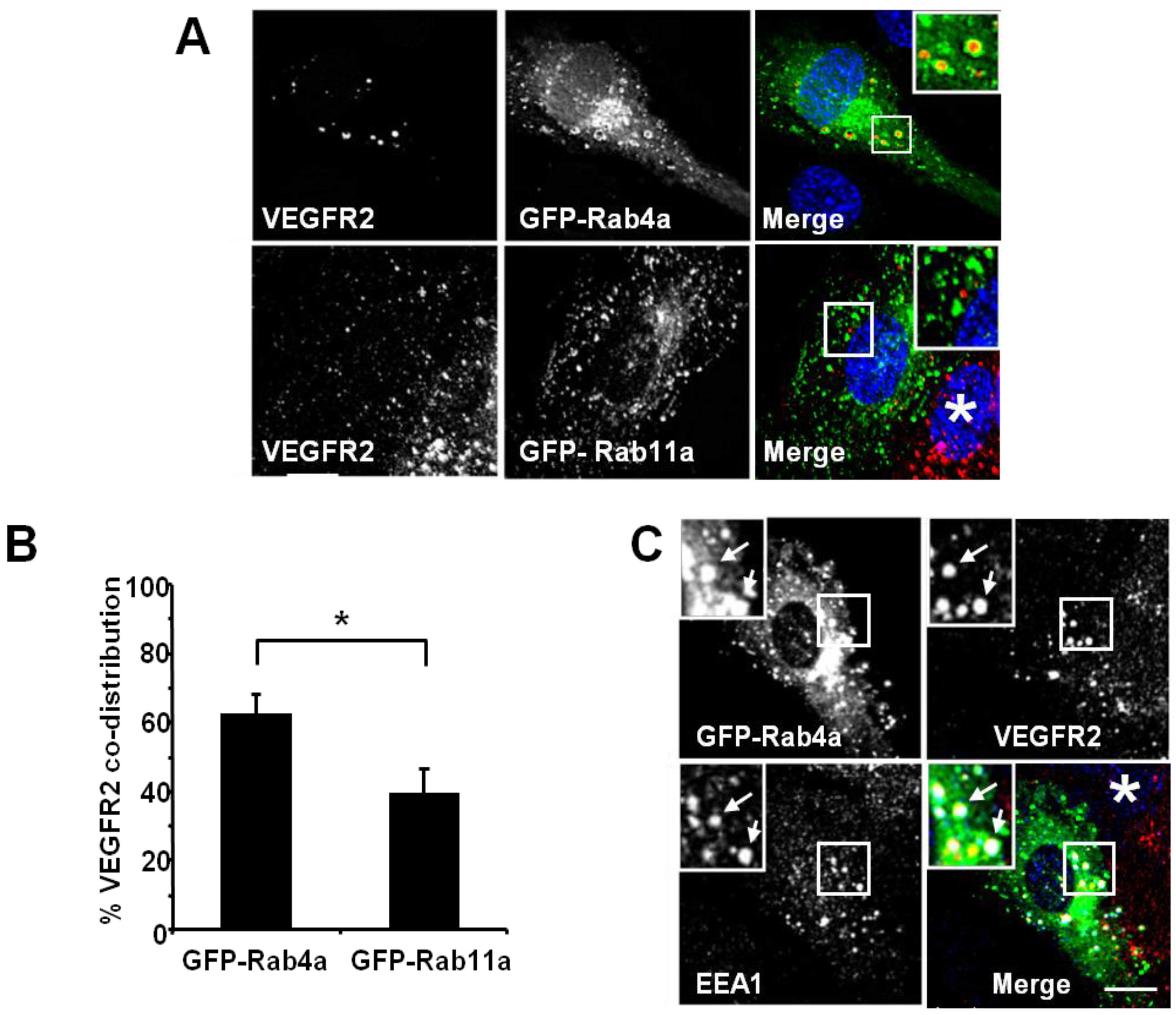
Figure 2). Endothelial cell transfection and expression of GFP-Rab4a caused the accumulation of punctate structures with Rab4a-enriched membranes and a central core of ‘trapped’ VEGFR2 within the internal lumen (
Figure 2A, upper panels and inset). In contrast, transfection and expression of GFP-Rab11a showed non-overlapping distribution of VEGFR2 and Rab11a to different vesicles (
Figure 2A, lower panels and inset). Quantification of VEGFR2 co-distribution of either Rab4a or Rab11a in such experiments revealed a significant difference between these two GTPases (
Figure 2B). To further assess the link between early endosomes and VEGFR2 dynamics, we compared the co-distribution of early endosomal antigen 1 (EEA1), GFP-Rab4a and VEGFR2 (
Figure 2C). These experiments showed that a proportion of EEA1-positive endosomes also contained Rab4a and VEGFR2 (
Figure 2C, arrows).
Figure 2.
VEGFR2 trafficking in early endosomes is closely associated with the Rab4a GTPase. (A) HUVECs were transfected with either GFP-Rab4a or GFP-Rab11a (green), fixed and probed with goat antibodies to the VEGFR2 extracellular domain, followed by AlexaFluor-labelled secondary antibodies (red). The nucleus was visualised with DAPI (blue). (B) Quantification of VEGFR2 co-localisation with GFP-Rab4a or GFP-Rab11a (see Materials and Methods). Error bars denote SEM (n = 45); * p < 0.05. (C) VEGFR2 is present in Rab4a-positive early endosomes. HUVECs were transiently transfected to express GFP-Rab4a (green), and cells were fixed, permeabilised and labelled with goat anti-VEGFR2 (red) and rabbit anti-EEA1 (blue). Bound primary antibodies were visualised with AlexaFluor-conjugated secondary antibodies. Insets show a two-fold magnification of the highlighted region. Bar: 10 μm.
Figure 2.
VEGFR2 trafficking in early endosomes is closely associated with the Rab4a GTPase. (A) HUVECs were transfected with either GFP-Rab4a or GFP-Rab11a (green), fixed and probed with goat antibodies to the VEGFR2 extracellular domain, followed by AlexaFluor-labelled secondary antibodies (red). The nucleus was visualised with DAPI (blue). (B) Quantification of VEGFR2 co-localisation with GFP-Rab4a or GFP-Rab11a (see Materials and Methods). Error bars denote SEM (n = 45); * p < 0.05. (C) VEGFR2 is present in Rab4a-positive early endosomes. HUVECs were transiently transfected to express GFP-Rab4a (green), and cells were fixed, permeabilised and labelled with goat anti-VEGFR2 (red) and rabbit anti-EEA1 (blue). Bound primary antibodies were visualised with AlexaFluor-conjugated secondary antibodies. Insets show a two-fold magnification of the highlighted region. Bar: 10 μm.
3.2. VEGFR2 Endosome-to-Plasma Membrane Recycling Depends on Rab4a GTPase Activity
VEGF-A-activated VEGFR2 undergoes ubiquitination and proteolysis within the endosome-lysosome system within 2 h, even when new protein synthesis is blocked [
12]. To address whether Rab4a influences VEGFR2 trafficking and degradation, we analysed VEGF-A-stimulated VEGFR2 activation and internalisation in GFP-Rab4a-expressing endothelial cells (
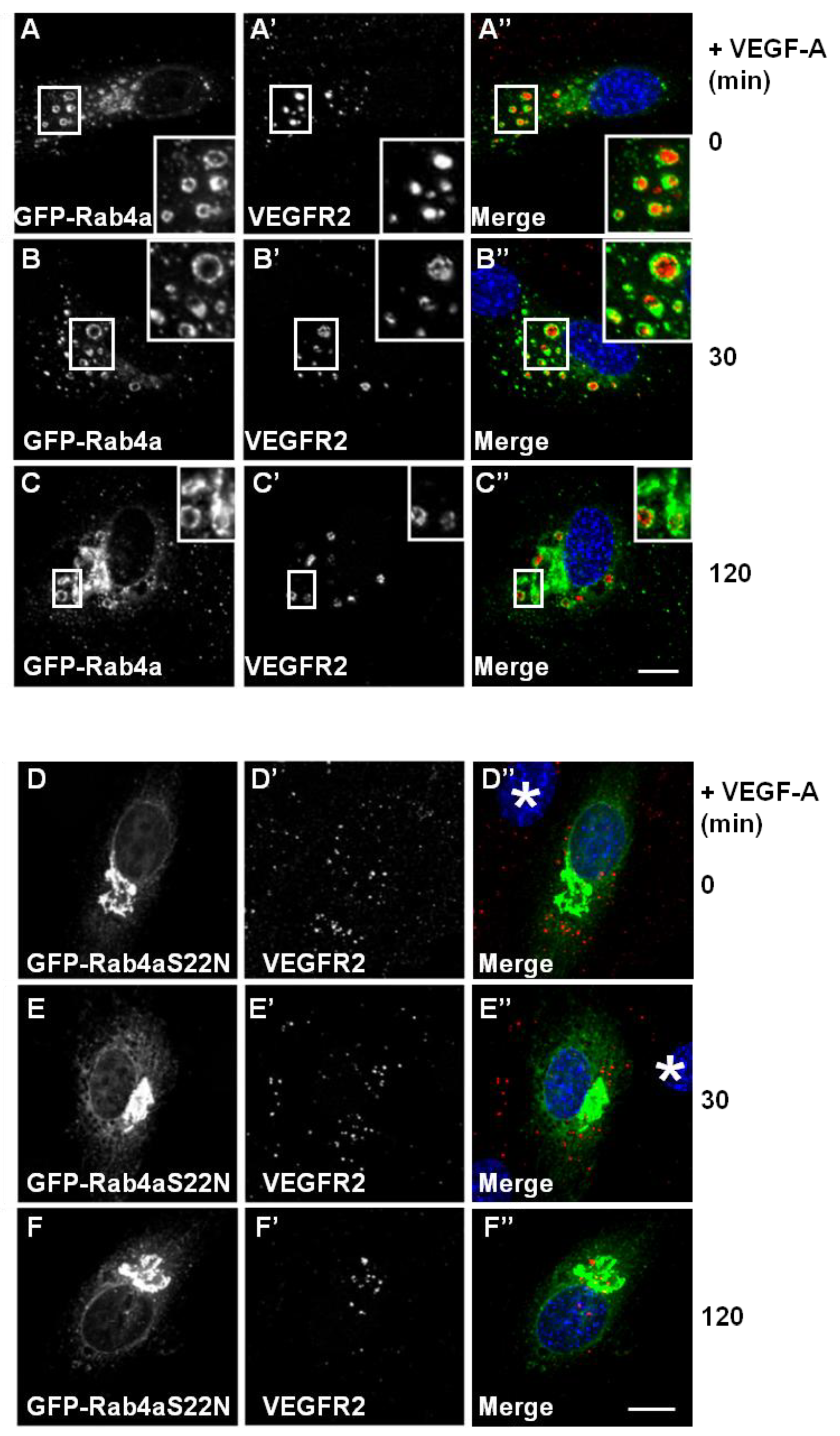
Figure 3A–C). VEGFR2-positive/Rab4a-positive endosomes were prominent even 2 h after ligand stimulation in the presence of cycloheximide to inhibit further protein synthesis (
Figure 3C’, insets). This delayed VEGFR2 trafficking effect was linked to increased Rab4a expression; and therefore, may be regulated by the Rab4a guanosine triphosphate
(GTP)/guanosine diphosphate (GDP)-bound state. To test this, we overexpressed a GDP-bound dominant-negative Rab4a mutant (GFP-Rab4a-S22N) in endothelial cells and evaluated VEGFR2 trafficking and distribution following VEGF-A stimulation using microscopy (
Figure 3D–F). This GDP-bound Rab4a mutant may be expected to prevent the recycling of VEGFR2 and, therefore, enhances the proportion of the receptor pool that is subject to lysosomal degradation. GDP-bound Rab4a-S22N localised to a juxtanuclear membrane compartment clearly distinct from VEGFR2 localization (
Figure 3D). Endothelial cells expressing juxtanuclear Rab4a-S22N did not show co-distribution with endosomal VEGFR2; however, in these cells, endosomal VEGFR2 levels persisted (
Figure 3D’–F’). Interestingly, VEGF-A-stimulation still exhibited increased VEGFR2 retention within endosomes and reduced VEGFR2 degradation (
Figure 3F’) in comparison to control non-transfected cells.
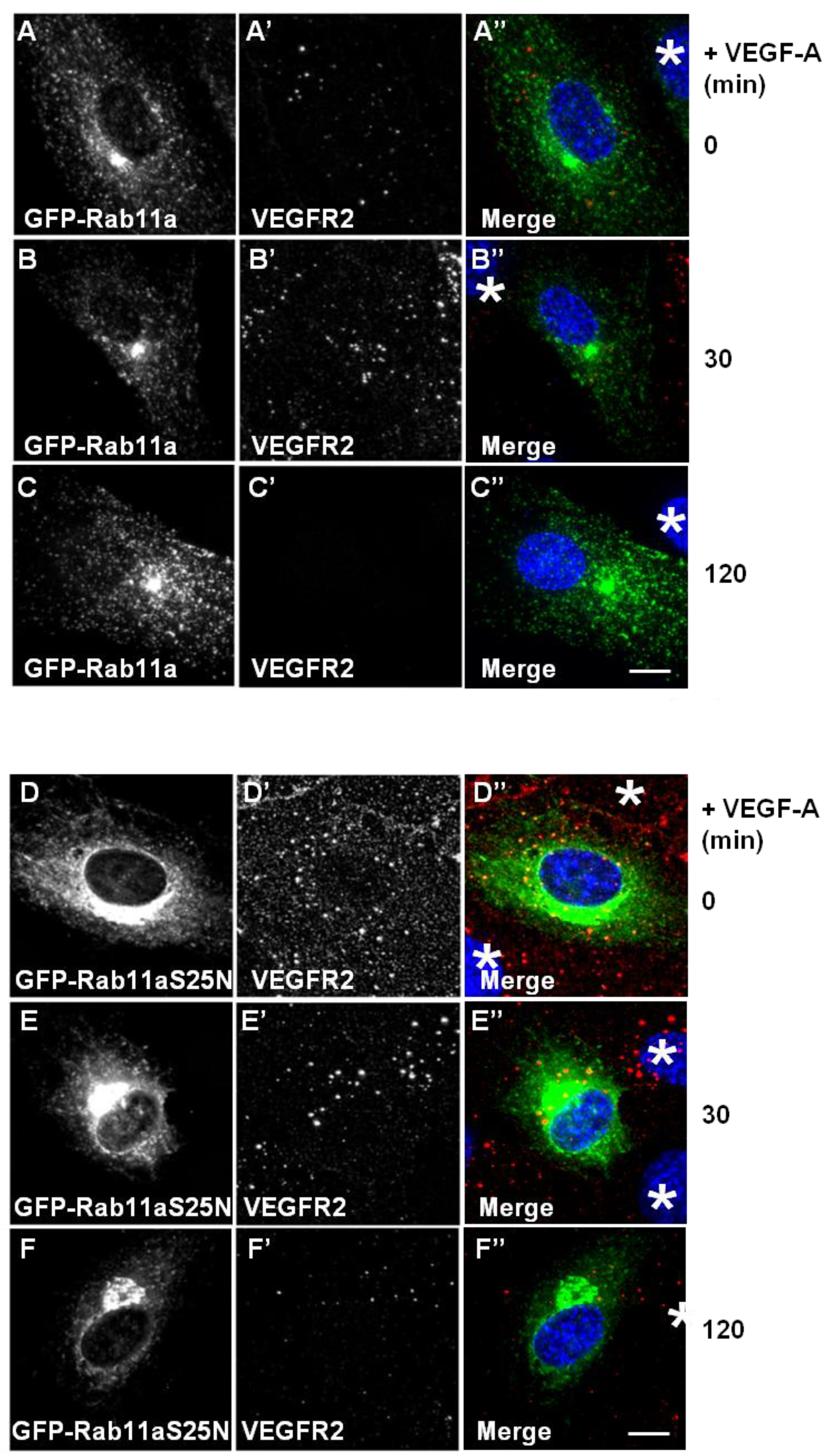
Different membrane receptors recycle from the endosome-to-plasma membrane via Rab4a- or Rab11a-dependent pathways [
26,
29]. To further test for Rab11a involvement in the regulation of VEGFR2 endosome-to-plasma membrane recycling, we overexpressed either wild-type or dominant-negative (S25N) Rab11a proteins in endothelial cells and stimulated with VEGF-A (
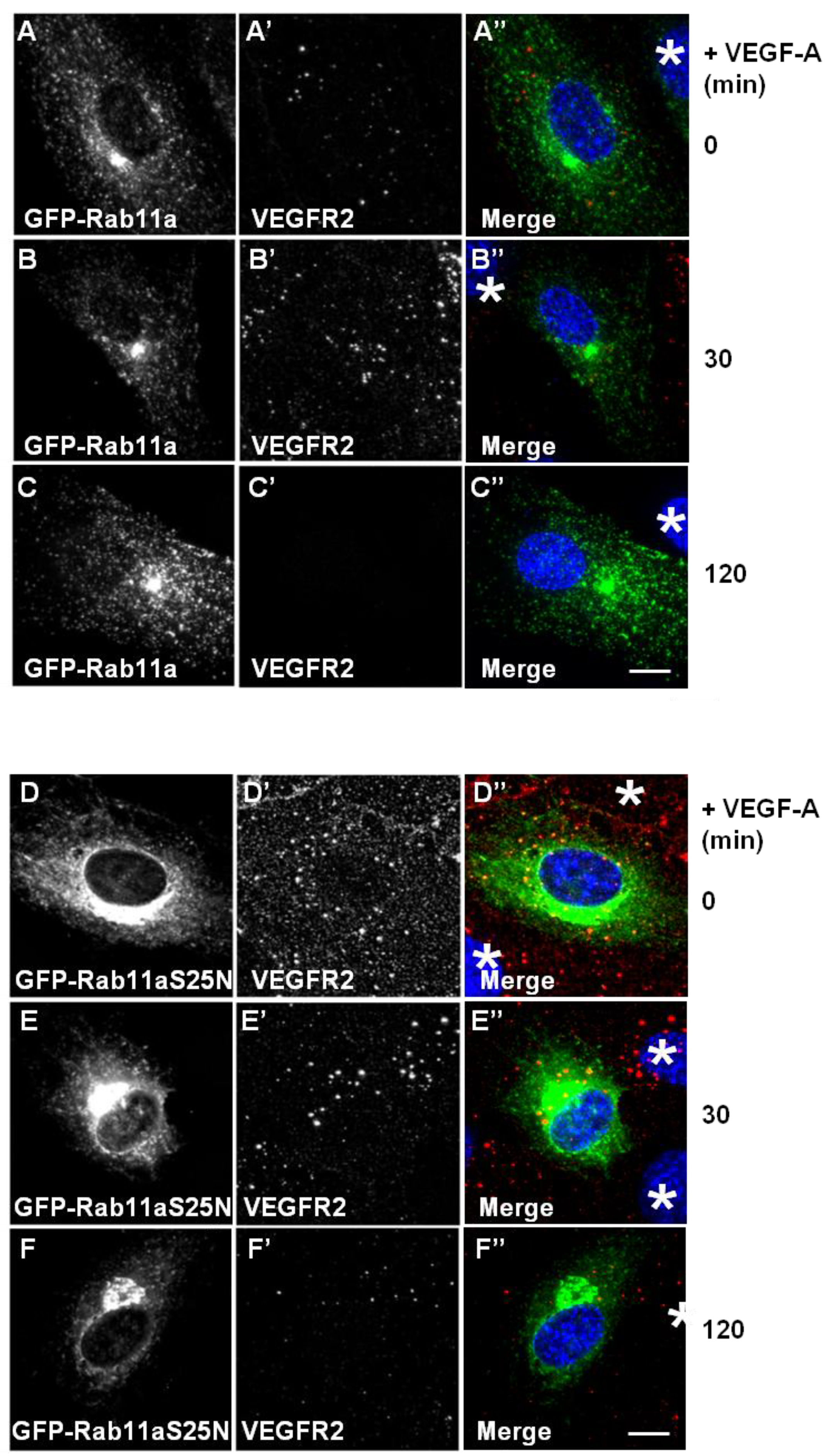
Figure 4). Expression of GFP-Rab11a in endothelial cells followed by VEGF-A stimulation revealed two findings. Firstly, there was little or no co-distribution between VEGFR2 and Rab11a (
Figure 4A,B). Secondly, VEGF-A-stimulated VEGFR2 activation for 2 h caused noticeable VEGFR2 degradation (
Figure 4C’) in cells overexpressing Rab11a (
Figure 4C), thus resembling control non-transfected endothelial cells (see
Figure 1A). We also overexpressed a GDP-bound dominant-negative Rab11a mutant (GFP-Rab11a-S25N) and evaluated VEGFR2 trafficking and distribution in transfected endothelial cells (
Figure 4D–F). Interestingly, GDP-bound Rab11a-S25N also localised to a juxtanuclear membrane compartment in transfected endothelial cells (
Figure 4D), and this was distinct from VEGFR2 distribution (
Figure 4D’). In contrast to previous experiments with Rab4a-S22N, Rab11a-S25N expression did not significantly affect VEGF-A-stimulated VEGFR2 activation and degradation via the endosome-lysosome system (
Figure 4F’).
Figure 3.
Wild-type and mutant Rab4a-S22N blocks VEGF-A-stimulated VEGFR2 degradation. (A–C) HUVECs were transfected with GFP-Rab4a (green) and cells were then stimulated with VEGF-A for (A) 0 min, (B) 30 min or (C) 120 min in the presence of CHX. Cells were subsequently fixed, permeabilised and labelled with goat anti-VEGFR2 followed by AlexaFluor-conjugated secondary antibody (red). The nucleus is visualized using DAPI (blue). Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm. (D–F) HUVECs were transiently transfected to express dominant-negative GDP-bound GFP-Rab4a-S22N (green) and then stimulated with VEGF-A for (D) 0 min, € 30 min or (F) 120 min in the presence of cycloheximide (CHX) and processed for immunofluorescence microscopy. VEGFR2 was detected using goat anti-VEGFR2 followed by AlexaFluor-conjugated secondary antibody (red), whilst the nuclear DNA was labelled with DAPI (blue). The images shown are representative of three independent experiments. Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm.
Figure 3.
Wild-type and mutant Rab4a-S22N blocks VEGF-A-stimulated VEGFR2 degradation. (A–C) HUVECs were transfected with GFP-Rab4a (green) and cells were then stimulated with VEGF-A for (A) 0 min, (B) 30 min or (C) 120 min in the presence of CHX. Cells were subsequently fixed, permeabilised and labelled with goat anti-VEGFR2 followed by AlexaFluor-conjugated secondary antibody (red). The nucleus is visualized using DAPI (blue). Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm. (D–F) HUVECs were transiently transfected to express dominant-negative GDP-bound GFP-Rab4a-S22N (green) and then stimulated with VEGF-A for (D) 0 min, € 30 min or (F) 120 min in the presence of cycloheximide (CHX) and processed for immunofluorescence microscopy. VEGFR2 was detected using goat anti-VEGFR2 followed by AlexaFluor-conjugated secondary antibody (red), whilst the nuclear DNA was labelled with DAPI (blue). The images shown are representative of three independent experiments. Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm.
![Cells 03 00363 g003]()
Figure 4.
Rab11a perturbation does not affect VEGF-A stimulated VEGFR2 degradation. (A–C) HUVECs were transiently transfected to express GFP-Rab11a (green) and then stimulated with VEGF-A for (A) 0 min, (B) 30 min or (C) 120 min in the presence of cycloheximide (CHX) and processed for immunofluorescence microscopy. VEGFR2 was detected using goat anti-VEGFR2 antibody followed by AlexaFluor-conjugated secondary antibody (red), whilst the nuclear DNA was labelled with DAPI (blue). The images shown are representative of three independent experiments. Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm. (D–F) HUVECs were transiently transfected to express dominant-negative GDP-bound GFP-Rab11a-S25N (green) and then stimulated with VEGF-A for (D) 0 min, (E) 30 min or (F) 120 min in the presence of cycloheximide (CHX) and processed for immunofluorescence microscopy. VEGFR2 was detected using goat anti-VEGFR2 followed by AlexaFluor-conjugated secondary antibody (red), whilst the nuclear DNA was labelled with DAPI (blue). The images shown are representative of three independent experiments. Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm.
Figure 4.
Rab11a perturbation does not affect VEGF-A stimulated VEGFR2 degradation. (A–C) HUVECs were transiently transfected to express GFP-Rab11a (green) and then stimulated with VEGF-A for (A) 0 min, (B) 30 min or (C) 120 min in the presence of cycloheximide (CHX) and processed for immunofluorescence microscopy. VEGFR2 was detected using goat anti-VEGFR2 antibody followed by AlexaFluor-conjugated secondary antibody (red), whilst the nuclear DNA was labelled with DAPI (blue). The images shown are representative of three independent experiments. Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm. (D–F) HUVECs were transiently transfected to express dominant-negative GDP-bound GFP-Rab11a-S25N (green) and then stimulated with VEGF-A for (D) 0 min, (E) 30 min or (F) 120 min in the presence of cycloheximide (CHX) and processed for immunofluorescence microscopy. VEGFR2 was detected using goat anti-VEGFR2 followed by AlexaFluor-conjugated secondary antibody (red), whilst the nuclear DNA was labelled with DAPI (blue). The images shown are representative of three independent experiments. Inset panels show a two-fold magnification of boxed highlighted regions. Bar: 10 μm.
![Cells 03 00363 g004]()
3.3. The VEGFR2 and TfR Recycling Pathways Are Distinct and Modulate Signal Transduction
A number of previous studies have established that VEGFR2 undergoes endosome-to-plasma membrane recycling [
6,
15,
21]. We used the previously described antibody-based recycling assay to monitor VEGFR2 and TfR recycling [
15]. Chloroquine (CHQ) is an inhibitor of endosomal compartment acidification and, therefore, blocks endocytosis [
34]. Upon CHQ treatment of endothelial cells, both recycling VEGFR2 and TfR accumulated in enlarged endosomal structures (
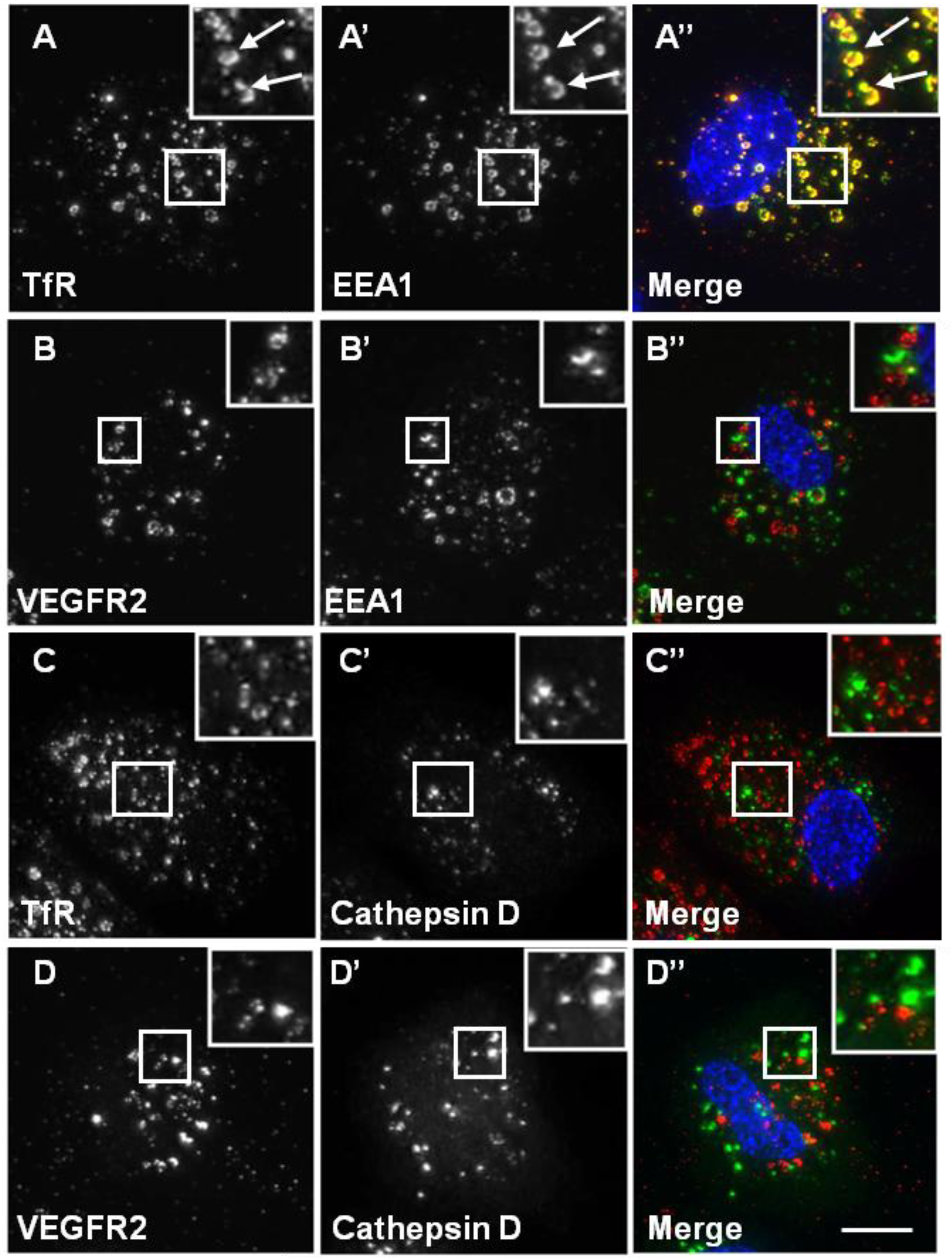
Figure 5). Intriguingly, whilst TfR showed a high level of co-localisation with EEA1 (
Figure 5A), VEGFR2 showed little co-localisation with EEA1 under similar conditions (
Figure 5B). These data indicate differences in VEGFR2 and TfR recycling. Neither VEGFR2 nor TfR showed any co-localisation with cathepsin D in the presence of CHQ (
Figure 5C and D).
Figure 5.
Chloroquine causes TfR, but not VEGFR2, accumulation in endosomes. A direct recycling assay was performed using the goat anti-VEGFR2 extracellular domain or mouse anti-TfR (red). Samples were fixed, permeabilised and labelled with either rabbit anti-EEA1 (green) or rabbit anti-cathepsin D (green). Primary antibodies were visualised using either FITC anti-rabbit IgG or AlexaFluor-488-conjugated anti-rabbit IgG, and the nucleus was seen with DAPI (blue). The images are 2D projections of a stacked series of 15–30 μm optical sections taken using a wide field deconvolution microscopy system. Insets show a two-fold magnification of the indicated region. Bar: 10 μm.
Figure 5.
Chloroquine causes TfR, but not VEGFR2, accumulation in endosomes. A direct recycling assay was performed using the goat anti-VEGFR2 extracellular domain or mouse anti-TfR (red). Samples were fixed, permeabilised and labelled with either rabbit anti-EEA1 (green) or rabbit anti-cathepsin D (green). Primary antibodies were visualised using either FITC anti-rabbit IgG or AlexaFluor-488-conjugated anti-rabbit IgG, and the nucleus was seen with DAPI (blue). The images are 2D projections of a stacked series of 15–30 μm optical sections taken using a wide field deconvolution microscopy system. Insets show a two-fold magnification of the indicated region. Bar: 10 μm.
Does Rab4a GTPase activity and regulation of endosome-to-plasma membrane recycling influence VEGF-A-stimulated intracellular signalling in endothelial cells? We have previously used RNA interference (RNAi) to evaluate Rab5a and Rab7a GTPase regulation of VEGFR2 function linked to endosome-lysosome trafficking and VEGF-A-stimulated intracellular signalling in primary endothelial cells [
17]. We used this approach to check whether Rab4a or Rab11a depletion modulated VEGF-A-stimulated short-term intracellular signalling, therefore altering longer term cellular responses, such as endothelial cell migration, proliferation and endothelial tube formation,
i.e., tubulogenesis.
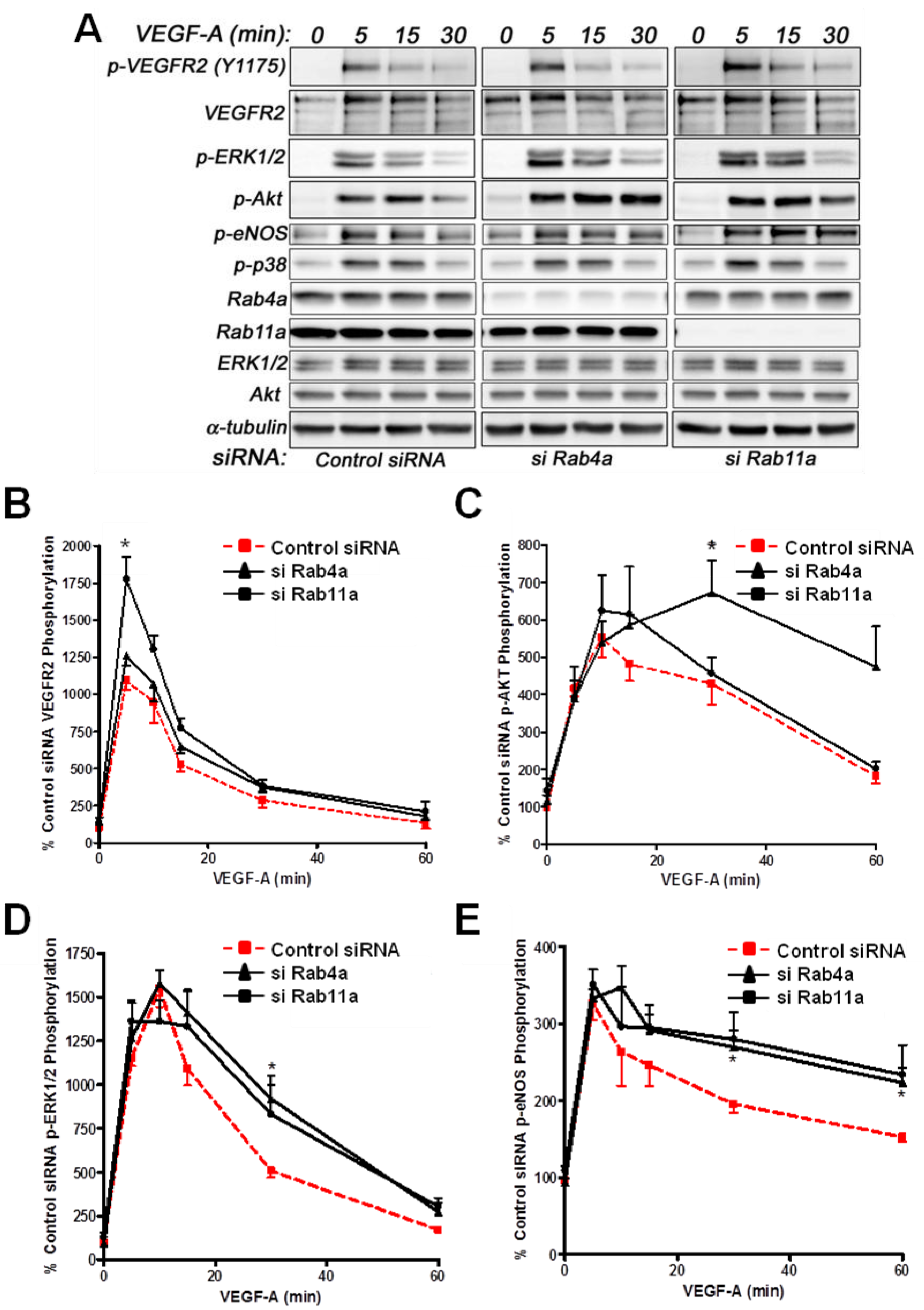
Analysis of VEGF-A-stimulated VEGFR2 activation revealed subtle differences in VEGFR2 activation and downstream signal transduction (
Figure 6A). Quantification showed a nearly two-fold increase in VEGFR2-pY1175 levels upon Rab11a depletion, whereas Rab4a depletion did not significantly affect VEGFR2-pY1175 levels (
Figure 6B). Furthermore, VEGF-A-stimulated Akt activation was prolonged upon Rab4a depletion compared to controls and Rab11a-depleted cells, although peak phospho-Akt levels were not significantly altered (
Figure 6C). However, the effects on p42/44 MAPK (ERK1/2) activation were less pronounced (
Figure 6D). Rab4a or Rab11a depletion did not affect peak phospho-p42/44 MAPK levels, but the duration of this species was prolonged (
Figure 6D). Similarly, although Rab4a and Rab11a depletion did not affect peak levels of VEGF-A-stimulated intracellular signalling and activation of endothelial nitric oxide synthase (eNOS), the duration of the activated phospho-eNOS (peNOS) species was also prolonged; an affect more pronounced upon Rab11a depletion (
Figure 6E). The activation of p38 MAPK was unaffected by the depletion of either Rab4a or Rab11a.
Figure 6.
Effects of Rab silencing on VEGF-A-stimulated intracellular signalling in endothelial cells. (A) HUVECs were subjected to RNAi (see Materials and Methods) and the control; Rab4a- or Rab11a-depleted HUVECs were then stimulated with VEGF-A followed by quantitative immunoblotting for (B) VEGFR2-pY1175, (C) phosphorylated Akt (p-Akt S473), (D) phosphorylated MAPK (p42/44 MAPK or p-ERK1/2 T202/Y204) and (E) phosphorylated eNOS (p-eNOS S1179). The data shown are derived from the analysis of three or more independent experiments. Error bars denote ±SEM, with asterisks indicating significance (*, p < 0.05).
Figure 6.
Effects of Rab silencing on VEGF-A-stimulated intracellular signalling in endothelial cells. (A) HUVECs were subjected to RNAi (see Materials and Methods) and the control; Rab4a- or Rab11a-depleted HUVECs were then stimulated with VEGF-A followed by quantitative immunoblotting for (B) VEGFR2-pY1175, (C) phosphorylated Akt (p-Akt S473), (D) phosphorylated MAPK (p42/44 MAPK or p-ERK1/2 T202/Y204) and (E) phosphorylated eNOS (p-eNOS S1179). The data shown are derived from the analysis of three or more independent experiments. Error bars denote ±SEM, with asterisks indicating significance (*, p < 0.05).
3.4. Rab GTPase Regulation of Intracellular Signalling Cell Migration, Proliferation and Tubulogenesis
To test if altered Rab-dependent VEGFR2 recycling influences VEGF-A responses, we analysed endothelial cell migration in Rab4a or Rab11a-depleted endothelial cells (
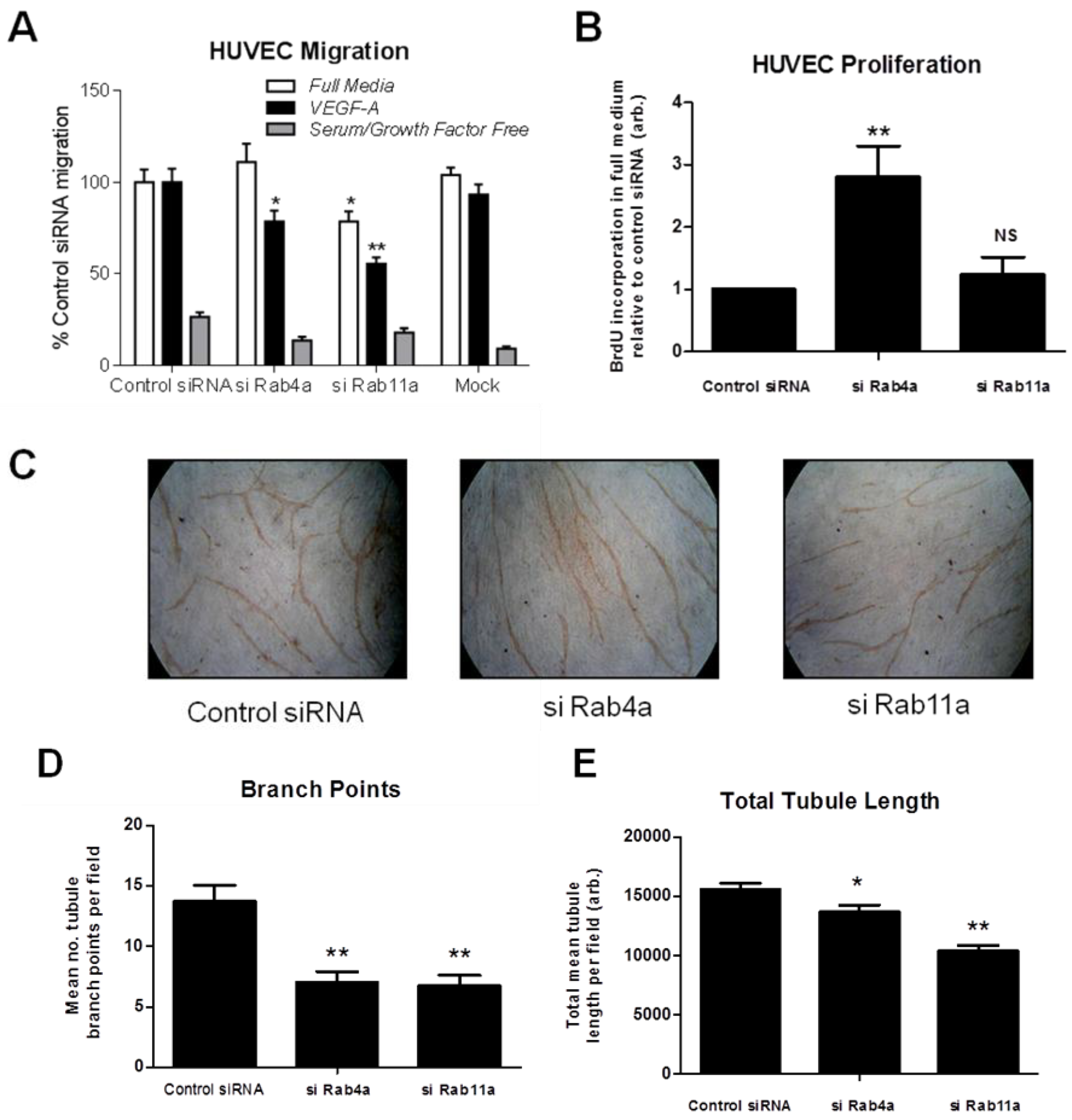
Figure 7A). Endothelial cells subjected to Rab4a knockdown showed ~20% reduction in cell migration in VEGF-A-supplemented minimal medium compared to complete medium (
Figure 7A). In cells subjected to Rab11a knockdown, cell migration was reduced ~20% in complete media and reduced ~40% in VEGF-A-supplemented minimal media (
Figure 7A). To assess such effects for another physiological response, such as VEGF-A-stimulated cell proliferation, endothelial cells subjected to Rab4a knockdown showed nearly a three-fold increase in cell proliferation relative to controls (
Figure 7B). In contrast, Rab11a-depleted endothelial cells did not show altered cell proliferation (
Figure 7B). In the absence of VEGF-A or adequate medium for >6–12 h, there is pronounced endothelial necrosis/apoptosis that is further accentuated by siRNA treatments. To control for this aspect, we used comparisons with scrambled siRNA controls (control siRNA) and other Rab-specific siRNAs as described here.
Next, we used an organotypic endothelial-fibroblast co-culture assay to recapitulate the endothelial tube formation characteristic of VEGF-A-stimulated angiogenesis. We analysed endothelial tubulogenesis in Rab4a or Rab11a-depleted endothelial cells subjected to sustained VEGF-A treatment for seven days (
Figure 7C). Endothelial cells showed ~50% reduction in the incidence of branch points in tubular networks upon depletion of either Rab4a or Rab11a GTPase (
Figure 7C and D). Interestingly, VEGF-A-stimulated endothelial tubule length was only reduced by ~10% in Rab4a-depleted cells; whilst there was a ~30% decrease in tubule length in Rab11a-depleted cells (
Figure 7E). These findings suggested key differences in the mechanism of Rab GTPase action on VEGFR2 recycling linked to long-term cellular responses.
Figure 7.
Rab-mediated regulation of endothelial cell migration, proliferation and in vitro angiogenesis. HUVECs were subjected to RNAi (see Materials and Methods) and the control; Rab4a- or Rab11a-depleted HUVECs were then analysed for (A) cell migration using a Transwell assay in complete media, VEGF-A-supplemented minimal media or in minimal media alone, (B) endothelial cell proliferation using a 5-bromo-2-deoxyuridine (BrdU)-modified nucleotide DNA incorporation ELISA or (C) endothelial tubule formation (tubulogenesis) using an organotypic endothelial-fibroblast co-culture assay. RNAi-activated HUVECs were trypsinized and plated on a bed of confluent human fibroblasts and allowed to grow in the presence of excess VEGF-A for seven days before endothelial tubules became visible. Co-cultures were fixed, stained with anti-PECAM-1 (CD31) antibody and HRP-conjugated secondary antibody followed by processing for light microscopy. Endothelial tubules were then analysed for (D) the number of branch points and (E) the total tubule length after tubulogenesis using Image J software. Error bars denote ±SEM; *, p < 0.05; **, p < 0.01.
Figure 7.
Rab-mediated regulation of endothelial cell migration, proliferation and in vitro angiogenesis. HUVECs were subjected to RNAi (see Materials and Methods) and the control; Rab4a- or Rab11a-depleted HUVECs were then analysed for (A) cell migration using a Transwell assay in complete media, VEGF-A-supplemented minimal media or in minimal media alone, (B) endothelial cell proliferation using a 5-bromo-2-deoxyuridine (BrdU)-modified nucleotide DNA incorporation ELISA or (C) endothelial tubule formation (tubulogenesis) using an organotypic endothelial-fibroblast co-culture assay. RNAi-activated HUVECs were trypsinized and plated on a bed of confluent human fibroblasts and allowed to grow in the presence of excess VEGF-A for seven days before endothelial tubules became visible. Co-cultures were fixed, stained with anti-PECAM-1 (CD31) antibody and HRP-conjugated secondary antibody followed by processing for light microscopy. Endothelial tubules were then analysed for (D) the number of branch points and (E) the total tubule length after tubulogenesis using Image J software. Error bars denote ±SEM; *, p < 0.05; **, p < 0.01.
![Cells 03 00363 g007]()
To address whether Rab4a and Rab11a activity is important for blood vessel growth and angiogenesis
in vivo, morpholino-mediated knockdown of these Rab GTPases was carried out in transgenic
Fli1-GFP zebrafish embryos, where GFP expression is restricted to vascular endothelium in the developing embryo [
20,
35]. Control scrambled, Rab4a- or Rab11a-specific morpholino antisense oligonucleotides were used to target Rab4a or Rab11a gene expression, and vascular development was analysed (
Figure 8).
Figure 8.
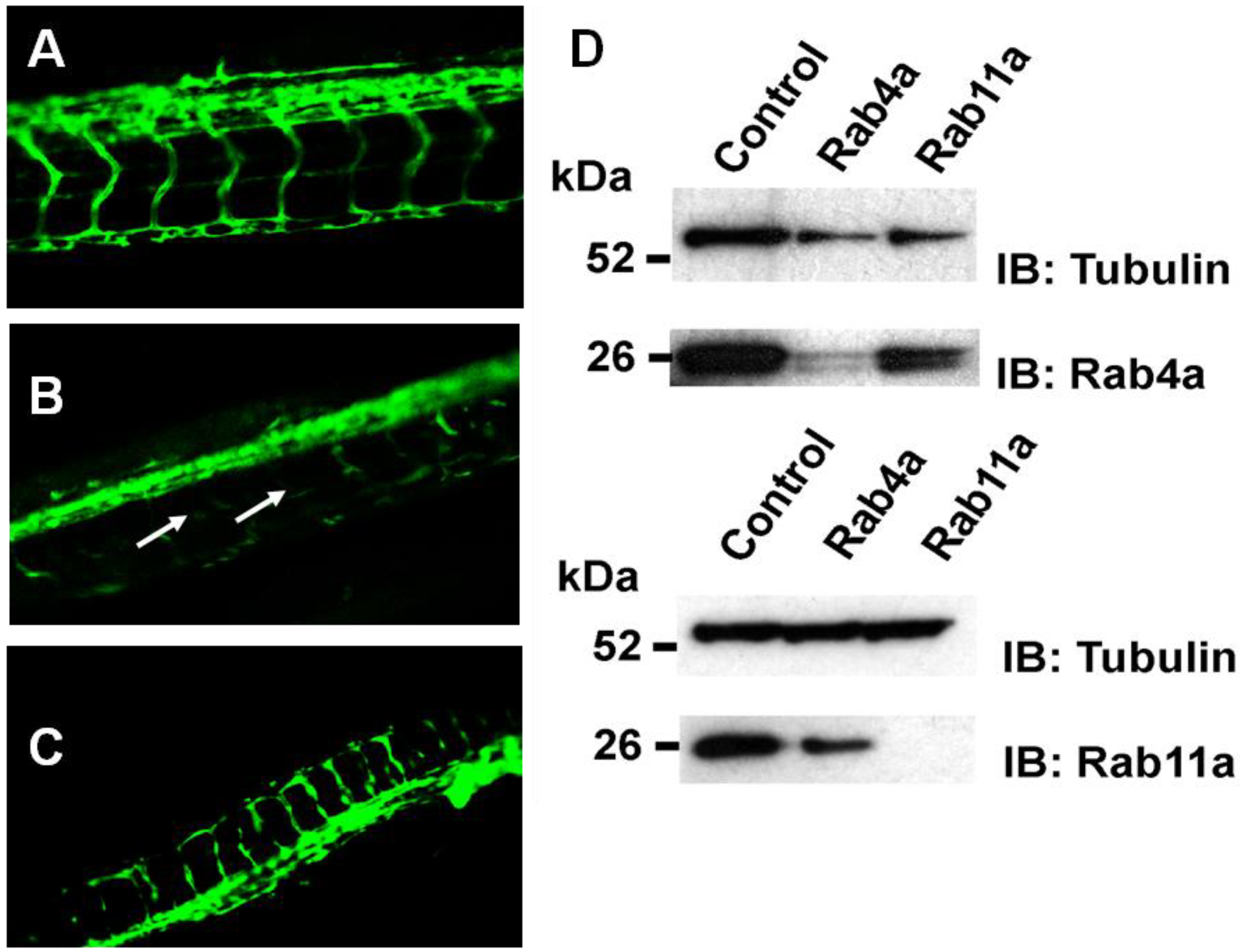
Rab4a regulates vascular development in transgenic zebrafish. Fluorescence microscopy on representative (A) control, (B) Rab4a-depleted or (C) Rab11a-depleted zebrafish transgenic lines (morphants) expressing Fli1:EGFP. Arrows in panel B denote defects in blood vessel formation in Rab4a-depleted animals. (D) Immunoblotting of control, Rab4a- or Rab11a-depleted transgenic zebrafish extracts.
Figure 8.
Rab4a regulates vascular development in transgenic zebrafish. Fluorescence microscopy on representative (A) control, (B) Rab4a-depleted or (C) Rab11a-depleted zebrafish transgenic lines (morphants) expressing Fli1:EGFP. Arrows in panel B denote defects in blood vessel formation in Rab4a-depleted animals. (D) Immunoblotting of control, Rab4a- or Rab11a-depleted transgenic zebrafish extracts.
In controls (
Figure 8A), a well-defined vascular system was evident with intersegmental vessels (ISV) connecting the major dorsal aorta and dorsal longitudinal anastomotic vessel (DLAV). Specific morpholino oligonucleotide-mediated Rab4a depletion (Rab4a morphants) caused severe impairment of both ISV and DLAV formation, with ISVs often missing or prematurely terminated (
Figure 8B). In contrast, although Rab11a morphants displayed marked developmental defects (
Figure 8C), including some disruption to the embryonic vasculature, both ISV and DLAV appeared to be better preserved than in Rab4a morphants (
Figure 8C). Immunoblotting revealed substantial knockdown in protein levels of either Rab4a or Rab11a using specific morpholino antisense oligonucleotides (
Figure 8D). Zebrafish morphology upon depletion of either Rab4a or Rab11a also revealed pleiotropic morphological effects, higher developmental abnormalities and mortalities in comparison to controls (S.P.
et al., unpublished findings).
3.5. Discussion
Here, we provide evidence that a Rab GTPase-dependent endosome-to-plasma membrane recycling step regulates VEGF-A-dependent intracellular signalling, cell migration, proliferation and angiogenesis. VEGFR2 recycles in both the presence and absence of VEGF-A stimulation. The Rab4a and Rab11a GTPases exhibited differential effects on VEGF-A-stimulated cell proliferation and migration. Rab4a depletion caused a dramatic three-fold increase in the VEGF-A-dependent proliferative response. However, Rab11a depletion did not affect endothelial cell proliferation. Rab4a displayed a modest reduction in VEGF-A-stimulated cell migration, whilst Rab11a depletion affected cell migration mediated by both serum and VEGF-A. A VEGF-A-dependent model of endothelial tubulogenesis showed a requirement for both Rab4a and Rab11a, but regulation by both GTPases appeared more crucial for tubule branching rather than extension of tubule length. This observation could in part explain the discrepancy between increased cell proliferation and decreased cell migration in Rab4a-depleted endothelial cells. Rab4a or Rab11a depletion in a transgenic zebrafish model produced different effects on vascular development, indicating the involvement in VEGF-A-regulated processes. However, it is also possible that such effects on animal development are not directly related to VEGF-VEGFR2 function, but rather, are a consequence of the essential roles of such GTPases in trafficking and the function of other membrane receptors.
Overexpression of Rab4a and Rab11a wild-type, or mutant GTPases, indicated a role for Rab4a in VEGFR2 trafficking from early endosomes to the plasma membrane. Comparison of VEGFR2 trafficking upon overexpression of a constitutively active GTP-bound Rab4a-Q67L mutant (S.P. et al., unpublished findings) showed that different Rab4a mutants can cause a block in activated VEGFR2 trafficking through the endosome-lysosome system. Depletion of Rab4a did not significantly affect VEGFR2 levels or phosphorylation status, although downstream activation of Akt, a serine/threonine protein kinase and master regulator, was prolonged. In contrast, Rab11a depletion did not affect Akt activation. Depletion of Rab4a also perturbed activated VEGFR2 proteolysis, consistent with the requirement for VEGFR2 recycling from early endosome to the plasma membrane following ligand stimulation. Thus, a link exists between endosomal Rab4a and the VEGF-A-dependent response in the endothelium. However, it cannot be discounted that other components of signalling pathways (e.g., MAPK enzymes, PDK1, Akt) that localise to the endocytic pathway may also be disrupted by Rab depletion, thus also modulating VEGF-A-stimulated responses.
It has been previously shown that VEGFR2 undergoes recycling from endosomes to the plasma membrane [
6,
15,
21]. However, Mellor and colleagues postulated that although VEGFR2 showed co-distribution with Rab4a-positive endosomes, they argued that VEGFR2 recycles from peripheral endosomes via a Src-dependent pathway [
6]. Ballmer-Hofer and colleagues showed that VEGFR2 recycling via Rab4a- or Rab11a-regulated pathways is dependent on VEGFR2 association with NRP1 [
21]. We had previously employed both biochemical and microscopy-based assays to show that VEGFR2 does indeed recycle between the cell surface and endosomes, and this was dependent on VEGFR2 tyrosine kinase activity [
15]. This study suggests that Rab4a, but not Rab11a, is required for VEGFR2 recycling. This is at variance with previous findings [
21]. However, we do observe significant differences between TfR and VEGFR2 recycling, suggesting that the two receptors share common regulatory features (e.g., Rab4a), but also differences in trafficking from endosomes to the plasma membrane.
The localisation of VEGFR2 along the endocytic pathways is regulated by different GTPases [
2,
3]. Our previous work showed that Rab5a regulated VEGFR2 trafficking through early endosomes [
36]. VEGFR2 early endosome localization was associated with pro-angiogenic signalling from early endosomes, including ERK1/2 activation and cell migration. However, although Rab7a regulated VEGFR2 trafficking out of late endosomes, retention in this compartment decreased pro-angiogenic signal transduction [
36]. These findings suggest that quiescent VEGFR2 is efficiently recycled back to the cell surface via Rab4a-positive endosomes; however, this recycling may also occur through Rab11a-positive endosomes, depending on the association with NRP1 [
21]. However, if VEGFR2 undergoes activation, phosphorylation and ubiquitination and these post-translational modifications are not removed in the early endosome, activated VEGFR2 progresses towards late endosomes and eventual degradation in lysosomes; compartments that are positioned close to the nucleus [
37].
The different effects noted for unchanged VEGFR2 phosphorylation status
versus prolonged Akt activation following Rab4a depletion represents a possible differential spatio-temporal regulation of these signalling molecules. A mutant Rab4a does not prevent internalisation of VEGFR2 from the plasma membrane (where phosphorylation is initiated), suggesting that such downstream signalling events may be initiated in internal membrane compartments, a phenomenon which has been previously suggested [
13,
14]. A VEGFR1- and Rab4a-dependent step is also implicated in regulating endothelial αVβ3 integrin recycling and the pro-angiogenic response [
38]. Inhibition or depletion of endocytic regulators, including clathrin, ESCRT or Rab proteins, can also affect VEGFR2 trafficking, processing and downstream signalling [
12,
14,
36]. Expression of dominant-negative Rab4a in epithelial cells also inhibits ligand-stimulated degradation of another receptor tyrosine kinase, EGFR/ErbB1 [
39].
VEGFR2 undergoes constitutive endocytosis, but displays a lower internalisation rate than the constitutively endocytosed transferrin receptor (~8% per min); this is still a relatively high level of constitutive endocytosis (S.P.
et al., unpublished observations). VEGFR2 can localise to both the plasma membrane and intracellular vesicles in endothelial cells. A large fraction of VEGFR2 resides within early endosomes, whilst the remaining fraction is present at the plasma membrane, concordant with previous studies. VEGFR2-mediated cellular outputs have been observed to be linked to lipid rafts and associated factors, which can also modulate signal transduction, trafficking and recycling in other pathways [
40,
41,
42,
43,
44]. Increased VEGFR2 endocytosis triggered by VEGF-A could be linked to VEGFR2 exit from plasma membrane caveolae or lipid rafts to facilitate receptor-mediated endocytosis. Although plasma membrane VEGFR2 internalisation increased ~1.5-fold upon VEGF-A stimulation (S.P.
et al., unpublished observations), this does not account for increased endosome-to-plasma membrane recycling. Inhibition of endosome-to-plasma membrane recycling using the ionophore, monensin, in quiescent cells resulted in the accumulation of VEGFR2 in a perinuclear compartment and a decrease in plasma membrane VEGFR2, when new protein synthesis was also inhibited. This suggests that VEGFR2 is continually recycling through the endosome-plasma membrane system, even in the absence of the VEGF-A ligand.
In contrast to VEGFR2, other transmembrane receptor kinases, such as TGF-β receptor Type I and Type II transmembrane proteins, undergo endocytosis, but recycle via a Rab11a-dependent pathway [
45,
46]. Rab11a GTPase activity is also implicated in regulating β
2-adrenergic receptor recycling and degradation [
32]. However, other G-protein coupled receptors appear to differentially utilize Rab4a and Rab11a recycling pathways for recycling [
25,
47]. A comparison of two different prostaglandin G-protein-coupled receptors also suggests differential use of Rab4a
versus Rab11a recycling pathways [
47]. There is also likely to be some overlap between these two routes, as transferrin receptor endosome-plasma membrane recycling is dependent on both Rab4a and Rab11a. One possibility is that membrane proteins are segregated into ‘fast’ or ‘slow’ recycling routes from endosomes depending on the physiological context.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}