PKC-dependent Phosphorylation of the H1 Histamine Receptor Modulates TRPC6 Activity


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Cell Culture and Transfection
2.2. Fluorescence Imaging
2.3. Molecular Biology
2.4. Materials
2.5. Statistics
3. Results and Discussion
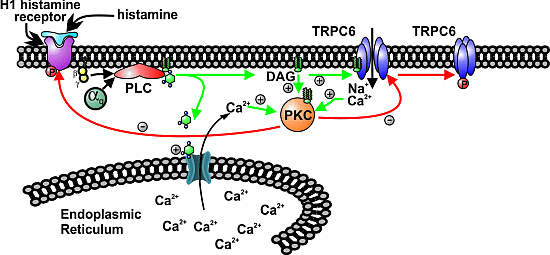
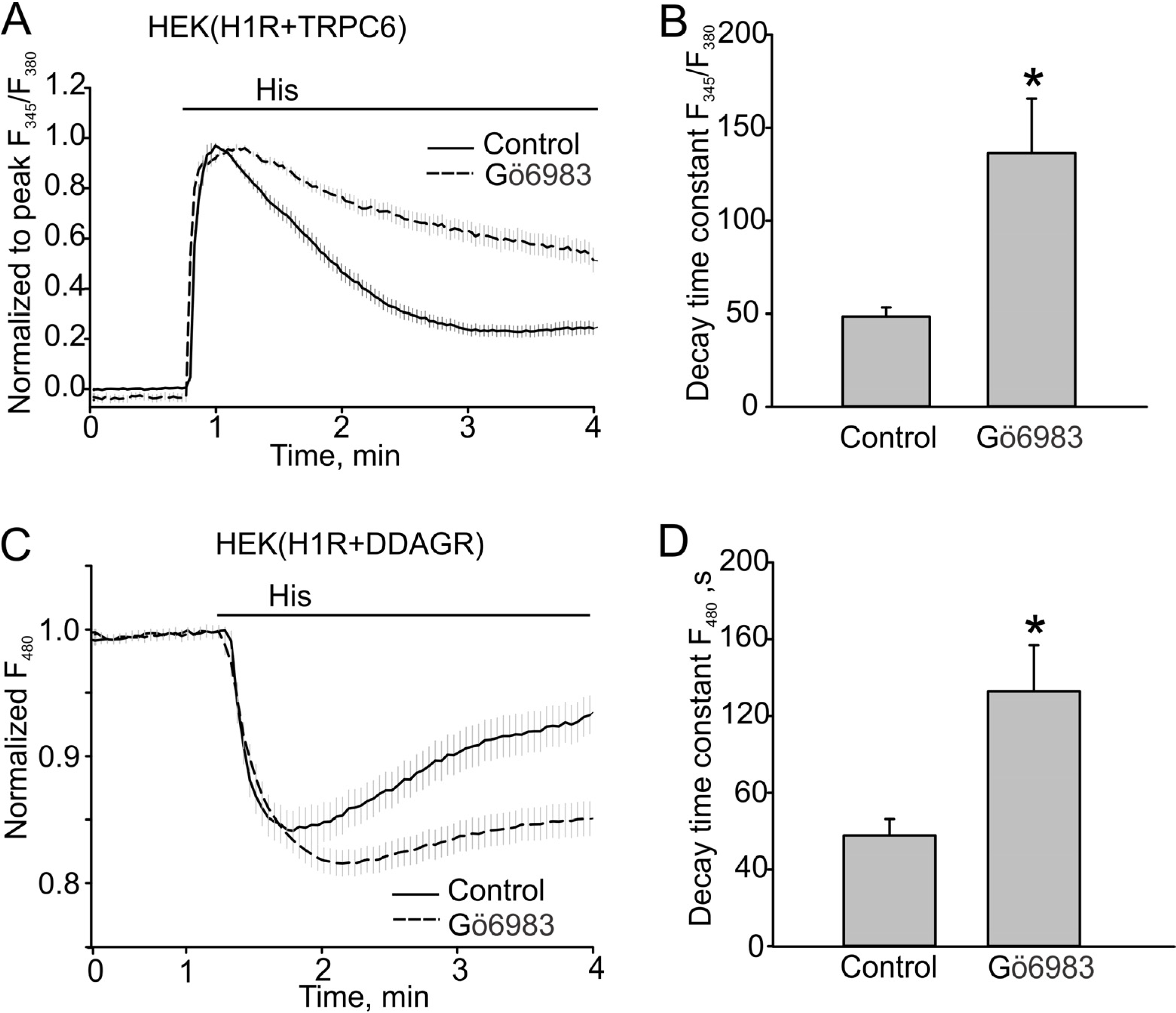
3.1. Histamine-induced TRPC6 Activity Decays in the Present of Histamine
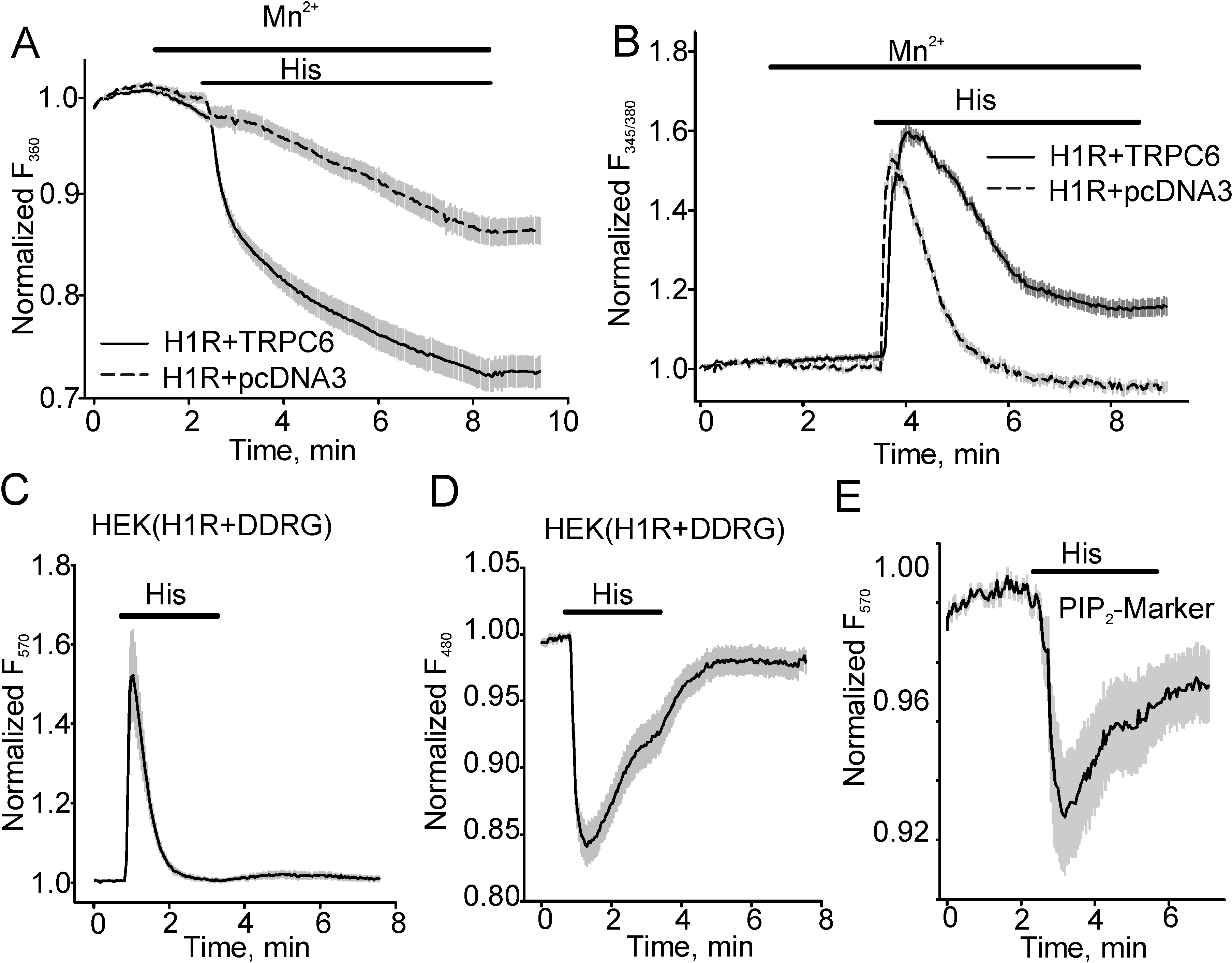
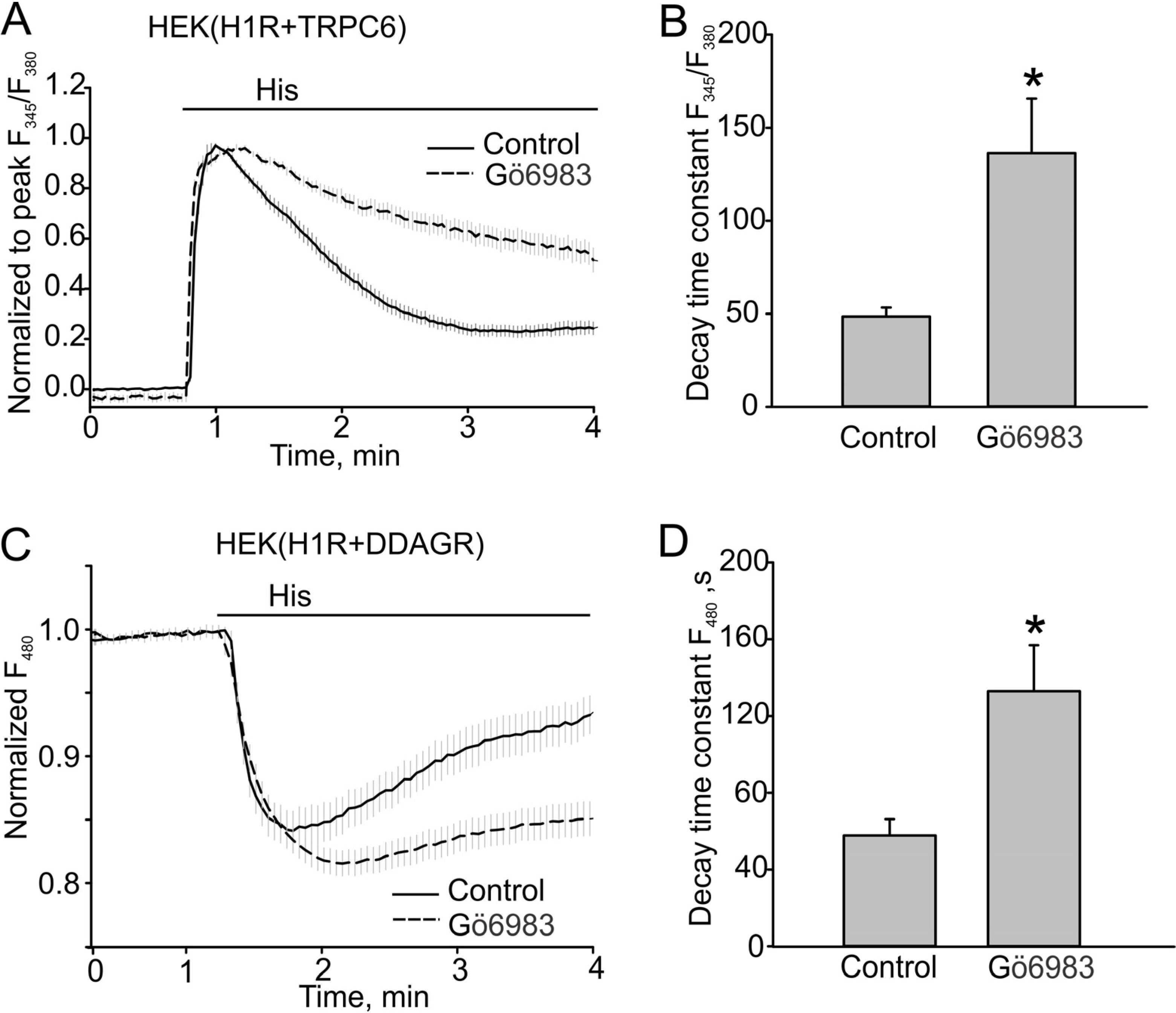
3.2. The Effect of PKC Inhibition on TRPC6 Inactivation Rate and Decay of DAG
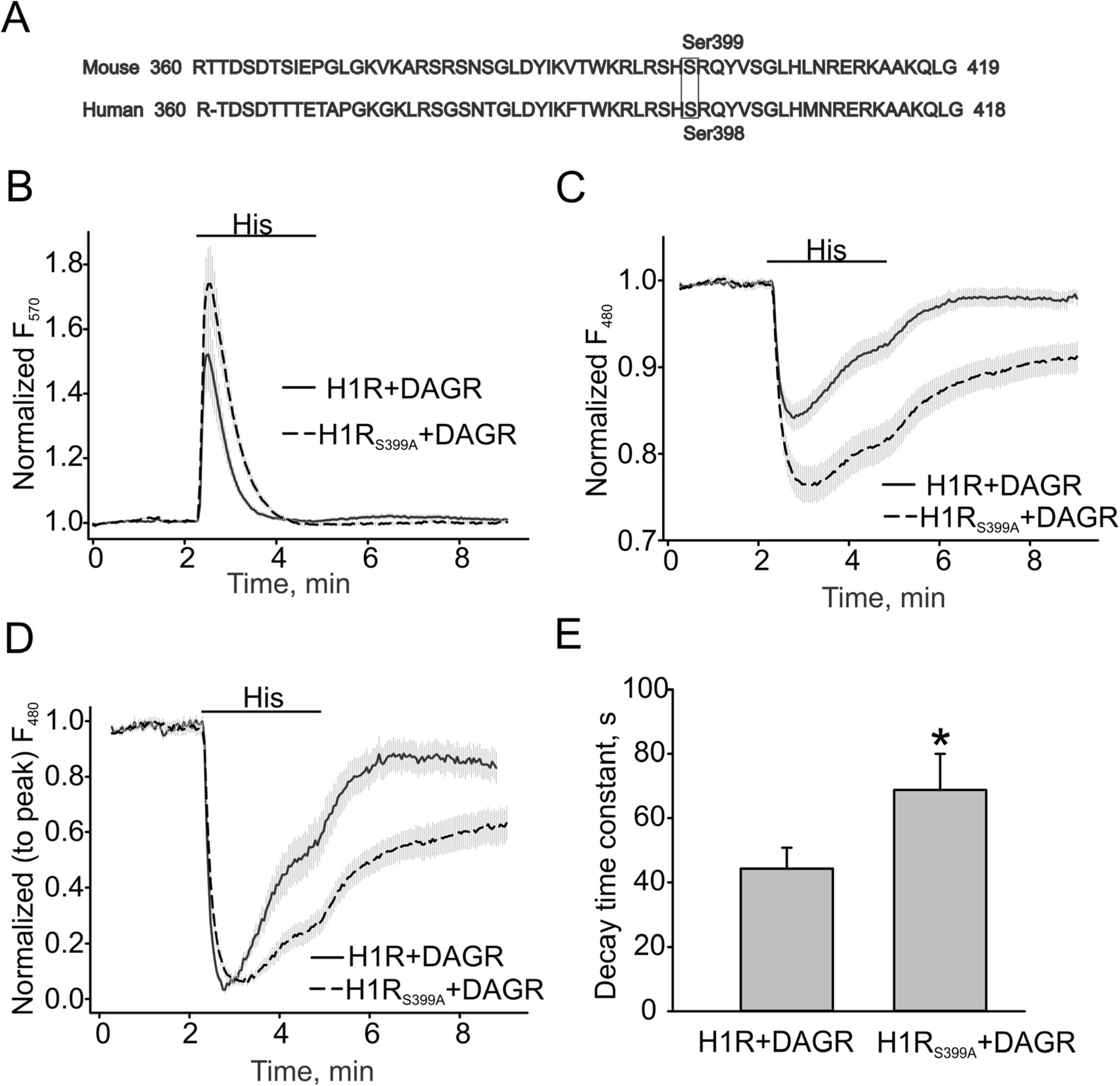
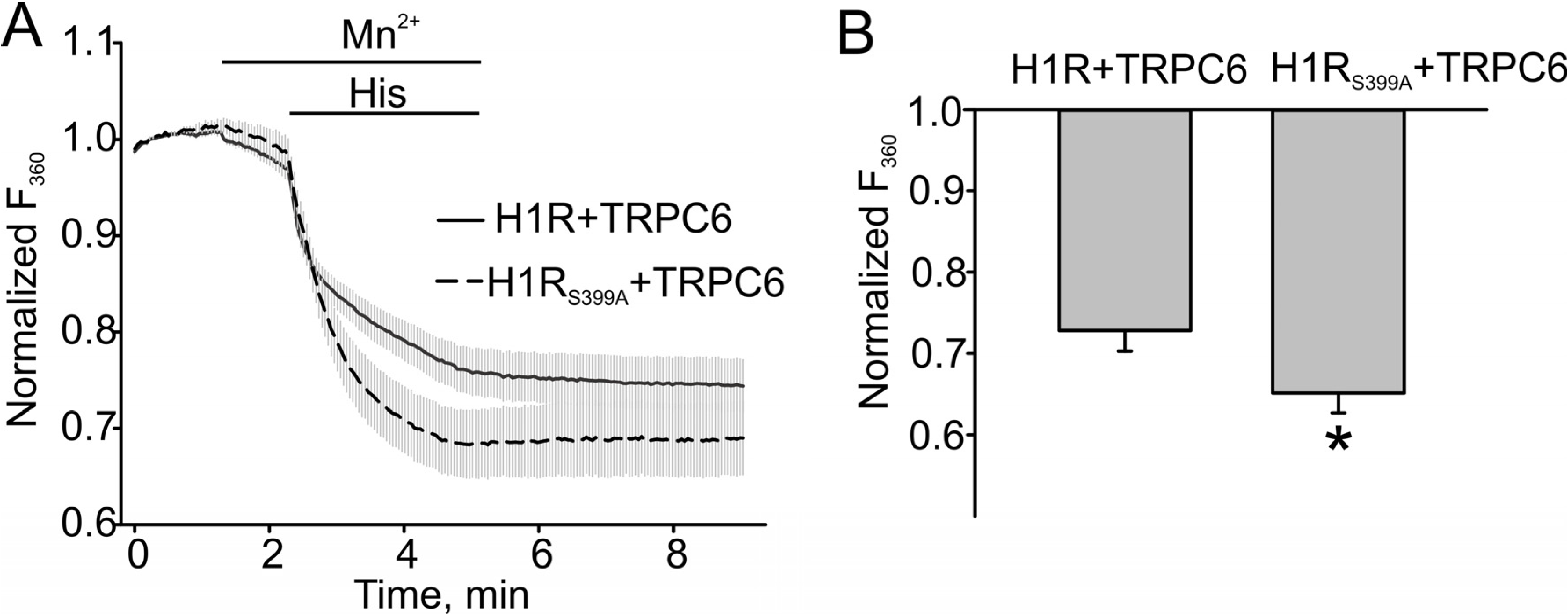
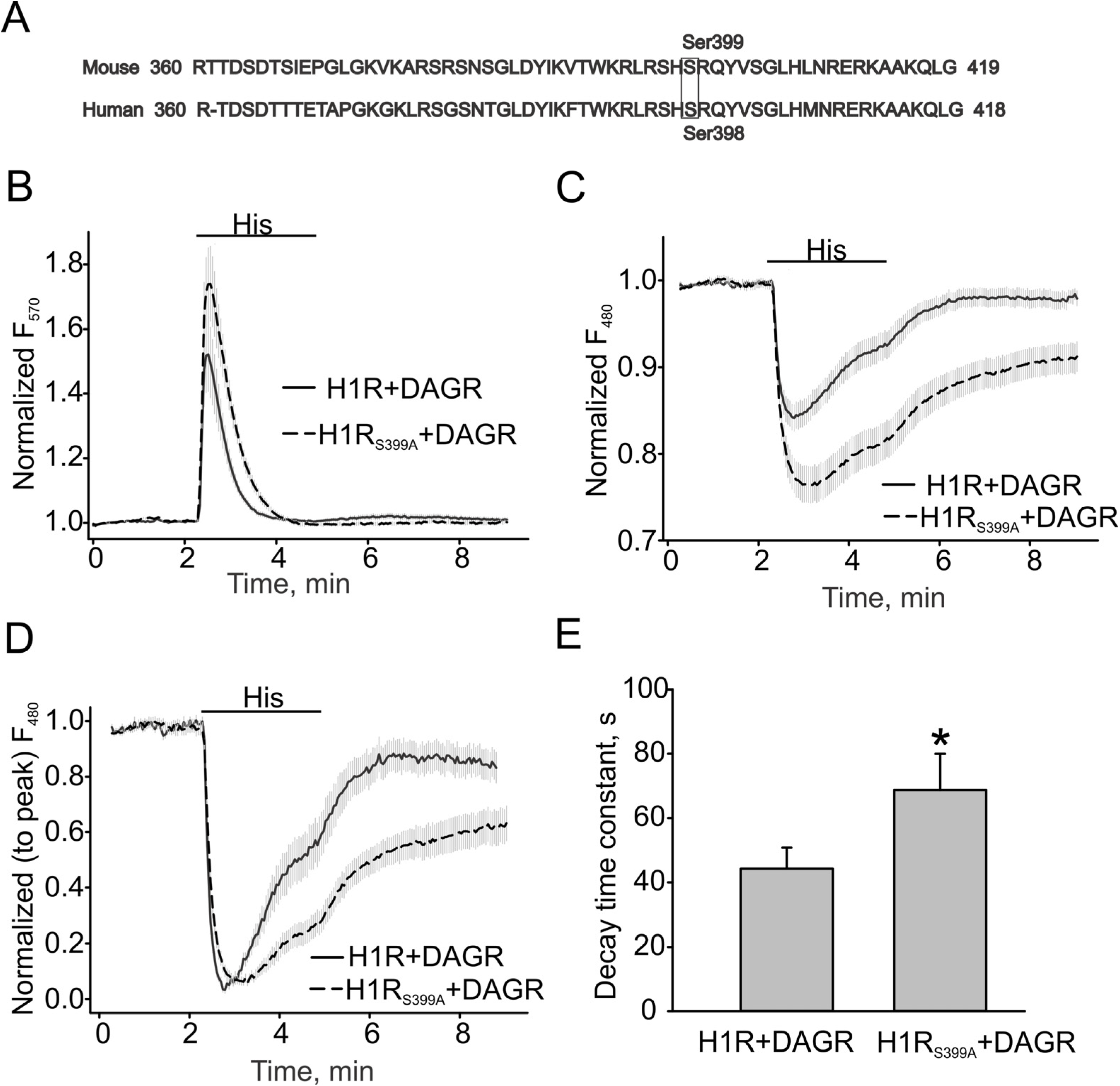
3.3. H1 Receptor Mutant (H1RS399A) Maintained A Prolonged DAG Production
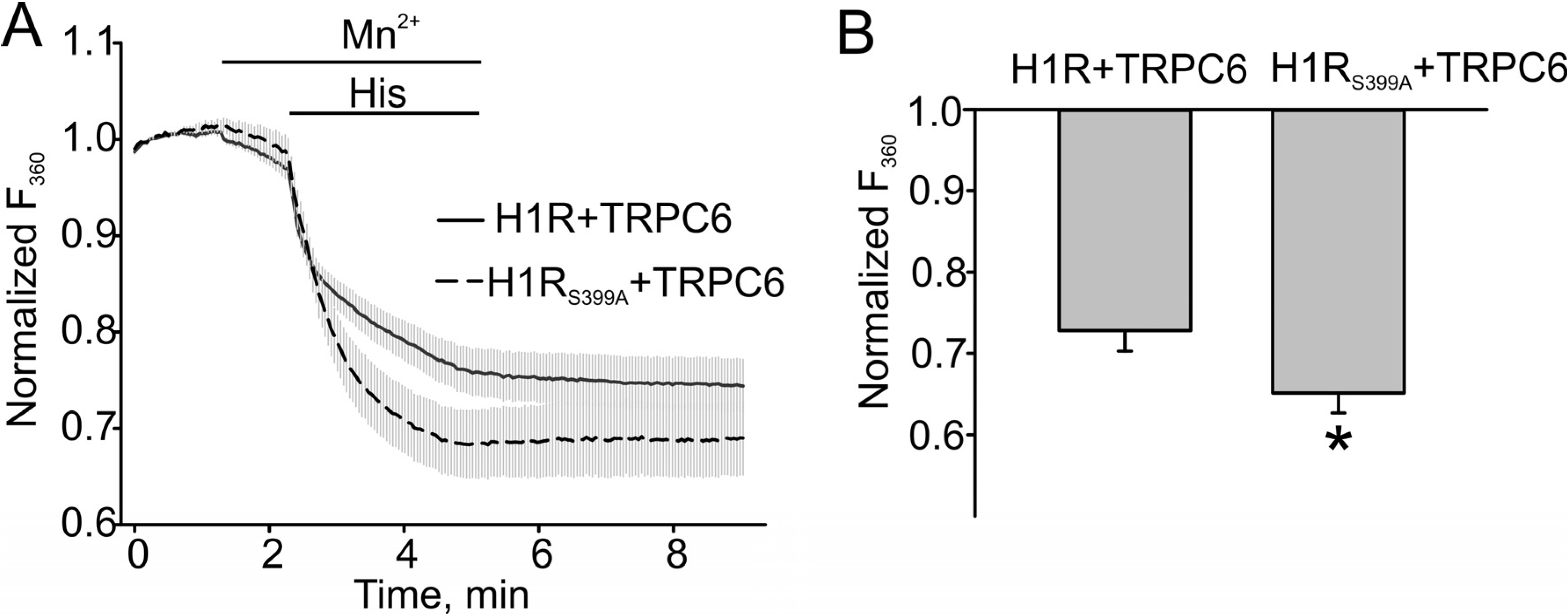
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Loga, F.; Domes, K.; Freichel, M.; Flockerzi, V.; Dietrich, A.; Birnbaumer, L.; Hofmann, F.; Wegener, J.W. The role of cGMP/cGKI signalling and trpc channels in regulation of vascular tone. Cardiovasc. Res. 2013, 100, 280–287. [Google Scholar] [CrossRef]
- Kistler, A.D.; Singh, G.; Altintas, M.M.; Yu, H.; Fernandez, I.C.; Gu, C.; Wilson, C.; Srivastava, S.K.; Dietrich, A.; Walz, K.; et al. Transient receptor potential channel 6 (TRPC6) protects podocytes during complement-mediated glomerular disease. J. Biol. Chem. 2013, 288, 36598–36609. [Google Scholar] [CrossRef]
- Ding, Y.; Winters, A.; Ding, M.; Graham, S.; Akopova, I.; Muallem, S.; Wang, Y.; Hong, J.H.; Gryczynski, Z.; Yang, S.H.; et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J. Biol. Chem. 2011, 286, 31799–31809. [Google Scholar] [CrossRef]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef]
- Krall, P.; Canales, C.P.; Kairath, P.; Carmona-Mora, P.; Molina, J.; Carpio, J.D.; Ruiz, P.; Mezzano, S.A.; Li, J.; Wei, C.; et al. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS One 2010, 5, e12859. [Google Scholar] [CrossRef]
- El, B.C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Bousquet, S.M.; Monet, M.; Boulay, G. Protein kinase C-dependent phosphorylation of transient receptor potential canonical 6 (TRPC6) on serine 448 causes channel inhibition. J. Biol. Chem. 2010, 285, 40534–40543. [Google Scholar] [CrossRef]
- Kim, J.Y.; Saffen, D. Activation of M1 muscarinic acetylcholine receptors stimulates the formation of a multiprotein complex centered on TRPC6 channels. J. Biol. Chem. 2005, 280, 32035–32047. [Google Scholar] [CrossRef]
- Miyoshi, K.; Das, A.K.; Fujimoto, K.; Horio, S.; Fukui, H. Recent advances in molecular pharmacology of the histamine systems: Regulation of histamine H1 receptor signaling by changing its expression level. J. Pharmacol. Sci. 2006, 101, 3–6. [Google Scholar] [CrossRef]
- Horio, S.; Kato, T.; Ogawa, M.; Fujimoto, K.; Fukui, H. Two threonine residues and two serine residues in the second and third intracellular loops are both involved in histamine H1 receptor downregulation. FEBS Lett. 2004, 573, 226–230. [Google Scholar] [CrossRef]
- Horio, S.; Ogawa, M.; Kawakami, N.; Fujimoto, K.; Fukui, H. Identification of amino acid residues responsible for agonist-induced down-regulation of histamine H(1) receptors. J. Pharmacol. Sci. 2004, 94, 410–419. [Google Scholar] [CrossRef]
- Kawakami, N.; Miyoshi, K.; Horio, S.; Yoshimura, Y.; Yamauchi, T.; Fukui, H. Direct phosphorylation of histamine H1 receptor by various protein kinases in vitro. Methods Find. Exp. Clin. Pharmacol. 2003, 25, 685–693. [Google Scholar] [CrossRef]
- Liggett, S.B. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci. Signal. 2011, 4, e36. [Google Scholar] [CrossRef]
- Tewson, P.; Westenberg, M.; Zhao, Y.; Campbell, R.E.; Quinn, A.M.; Hughes, T.E. Simultaneous detection of Ca2+ and diacylglycerol signaling in living cells. PLoS One 2012, 7, e42791. [Google Scholar]
- Hu, G.; Oboukhova, E.A.; Kumar, S.; Sturek, M.; Obukhov, A.G. Canonical transient receptor potential channels expression is elevated in a porcine model of metabolic syndrome. Mol. Endocrinol. 2009, 23, 689–699. [Google Scholar] [CrossRef]
- Tewson, P.H.; Quinn, A.M.; Hughes, T.E. A multiplexed fluorescent assay for independent second-messenger systems: Decoding GPCR activation in living cells. J. Biomol. Screen. 2013, 18, 797–806. [Google Scholar] [CrossRef]
- Merritt, J.E.; Jacob, R.; Hallam, T.J. Use of manganese to discriminate between calcium influx and mobilization from internal stores in stimulated human neutrophils. J. Biol. Chem. 1989, 264, 1522–1527. [Google Scholar]
- Fujimoto, K.; Ohta, K.; Kangawa, K.; Kikkawa, U.; Ogino, S.; Fukui, H. Identification of protein kinase C phosphorylation sites involved in phorbol ester-induced desensitization of the histamine H1 receptor. Mol. Pharmacol. 1999, 55, 735–742. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, X.; Egly, C.; Riley, A.M.; Li, W.; Tewson, P.; Hughes, T.E.; Quinn, A.M.; Obukhov, A.G. PKC-dependent Phosphorylation of the H1 Histamine Receptor Modulates TRPC6 Activity. Cells 2014, 3, 247-257. https://doi.org/10.3390/cells3020247
Chen X, Egly C, Riley AM, Li W, Tewson P, Hughes TE, Quinn AM, Obukhov AG. PKC-dependent Phosphorylation of the H1 Histamine Receptor Modulates TRPC6 Activity. Cells. 2014; 3(2):247-257. https://doi.org/10.3390/cells3020247
Chicago/Turabian StyleChen, Xingjuan, Christian Egly, Ashley M. Riley, Wennan Li, Paul Tewson, Thomas E. Hughes, Anne Marie Quinn, and Alexander G. Obukhov. 2014. "PKC-dependent Phosphorylation of the H1 Histamine Receptor Modulates TRPC6 Activity" Cells 3, no. 2: 247-257. https://doi.org/10.3390/cells3020247