1. Introduction
Hyaluronic acid [hyaloid (vitreous) + uronic acid] (HA) was isolated for the first time in 1934 from the vitreous humor of bovine eyes by Meyer and Palmer [
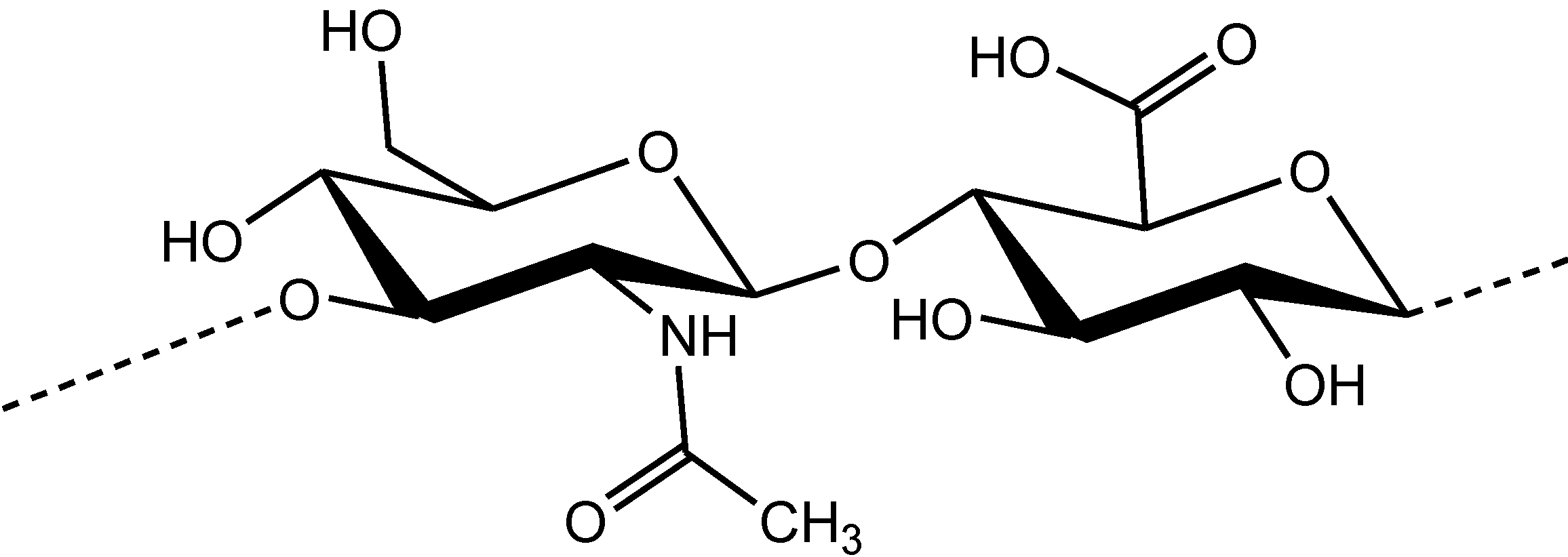
1]. It is a naturally occurring linear polysaccharide with repeating units of
d-glucuronic acid and
N-acetyl-
d-glucosamine disaccharide (
Figure 1).
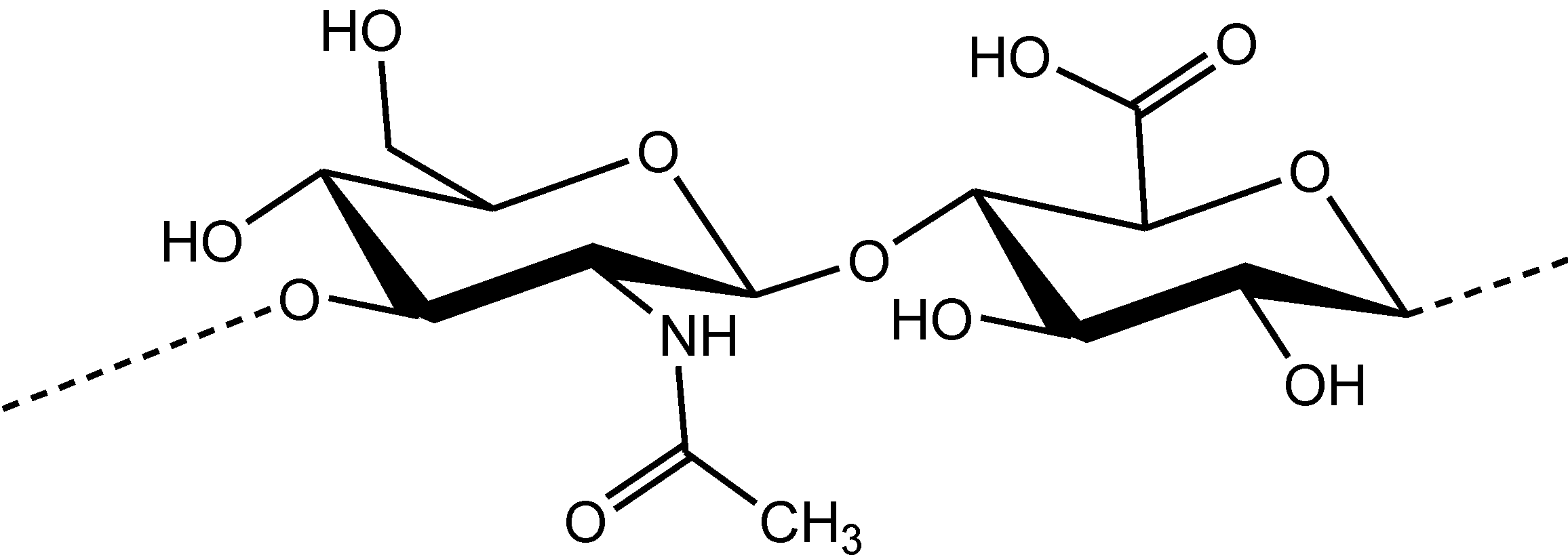
Figure 1.
Chemical structure of the disaccharide unit of hyaluronic acid.
Figure 1.
Chemical structure of the disaccharide unit of hyaluronic acid.
The pK
a of HA carboxyl groups is 3–4; at pH = 7, these groups being ionized, the hyaluronan molecule is a polyanion associated with cations (the counterions). HA is a highly hydrophilic polymer that can absorb a large amount of water and expand up to 1000 times its solid volume, forming a loose hydrated network [
2]. The molar mass of HA can be as high as 10 MDa, and such a molar mass accounts for the important physiological roles of HA in living organism, including maintenance of the viscoelasticity of liquid connective tissues (such as synovial fluid in the joints or eye vitreous humor), control of tissue hydration, water transport, proteoglycan organization in the extracellular matrix (ECM), tissue repair and various receptor-mediated functions in cell detachment, tumor development and inflammation [
3].
Hyaluronan is the major constituent of the skin. In the epidermis, it is metabolized and participates actively in many regulatory processes, such as cell proliferation, migration and differentiation [
4]. In the dermis, it plays the role of a space-filling material in the extracellular spaces.
HA in the ECM shows the important capacity to interact with some cell receptors, “hyaluronan binding proteins”, such as the CD-44, RHAMM, LYVE-1, IVd4 and LEC receptors [
5]. Among these, CD44, or the cluster of common leukocyte antigens (clusters of differentiation, CD), is the most studied HA receptor [
6].
Due to its versatile biological properties, its important characteristics of biocompatibility and biodegradability and viscoelastic properties, HA of high, moderate or low molar mass has found numerous applications in medicine and in cosmetics preparations [
7,
8,
9]. The success relies also on its non-antigenicity and the commercial availability in a wide range of forms, ranging from gels, to tubes, sheets of solid material and lightly woven meshes. Interestingly HA can be easily modified to tune its physical and biological properties, such as solubility, rate of degradation, viscosity and amphiphilicity. So far, many HA derivatives have been synthesized and employed in tissue and wound healing [
10,
11], viscosupplementation in joint diseases, dermal filling in anesthetic surgery, drug delivery and tissue engineering.
In the field of drug delivery, HA has become a carrier of great interest owing to its advantages, like: (i) biodegradability; (ii) biocompatibility; (iii) ease of chemical modification; (iv) high potential drug loading; and (v) its intrinsic targeting properties, due to the selective interactions with receptors, such as CD44 or hyaluronan receptors for endocytosis (HARE). CD44–hyaluronan interaction is known to participate in a wide variety of cellular functions, including cell–cell aggregation, pericellular matrix retention, matrix-cell and cell-matrix signaling, receptor-mediated internalization/degradation of hyaluronan and cell migration. Interestingly, it has been noted that CD44 receptors are overexpressed in a number of solid cancers. This last opportunity is at the basis of the use of hyaluronic acid as a carrier, as well as an active targeting moiety for the delivery of anticancer drugs [
12,
13,
14]. Many papers are already available in the literature on the conjugation of anti-inflammatory or anti-cancer drugs to HA [
15,
16,
17,
18].
Low molecular weight drugs, especially anticancer drugs, suffer from several limitations that limit their potentials in the clinic. They have often low water solubility, a lack of specificity and several side effects, due to their indiscriminate body distribution and short
in vivo half-life. Drug delivery systems (DDSs) can overcome the above-mentioned issues by increasing drug solubility and by selectively accumulating the drugs in solid tumors, thanks to the known enhanced permeability and retention (EPR) effect [
19]. In this context, HA can be exploited as a polymeric drug carrier through chemical conjugation of the drug or as a targeting agent of other DDSs, like nanoparticles and liposomes. HA, when used as main DDS, can play the role of carrier and targeting agent at the same time, offering the opportunity to reach a high drug loading. HA can overcome the limits of other polymeric carriers, like polyethylene glycol (PEG), which is not biodegradable and has a low payload.
Beyond its application in the delivery of anticancer drugs, HA has also been proposed as a polymer for proteins and peptide conjugation. Biologics represent a relevant and fast increase in pharmaceuticals. To date, there are over 200 protein-based products approved for therapeutic applications and more than 600 under development [
20]. However, this growing success is not devoid of issues and shortcomings associated with rapid blood clearance, physical and chemical instability, enzymatic degradation, immunogenicity, low bioavailability via non-invasive routes and the need to do invasive and inconvenient administrations [
21]. For these reasons, it is often necessary to administer high amounts of biologics, thus increasing the risk of side effects and the cost of the therapy.
The above issues of proteins have been addressed by different kinds of protein delivery systems to reduce the frequency of injections, the amount of costly biologics and the onset of adverse side effects, this maximizing the dose-response. Fusion proteins, hyper-glycosylation, incorporation in liposomes, hydrogels or nano-particles or a chemical modification using polymers have been designed for this scope [
22,
23]. HAylation, the covalent conjugation of HA, is another tool for researchers aiming to increase both the hydrodynamic volume of proteins and their half-life and to decrease the proteolytic degradation. The polymer biodegradability accounts for at least a partial recovery of the protein activity, which is usually reduced after protein conjugation [
24].
Here, we want to focus on the use of HA as a carrier for the covalent conjugation of drugs, peptides and proteins. Several studies will be reviewed discussing the advantages of HA in this field. The use of HA as a device for the sustained release of proteins is also reported by illustrating the advantages and limits of this technology.
3. HA Anticancer Drug
The most successful and thoroughly studied conjugations of drugs to HA involve anticancer agents, because HA receptors (CD44 and RHAMM) are overexpressed in a large number of cancers [
13]. Furthermore, beside this targeting advantage, HA–drug conjugates provide advantages in terms of drug solubilization, stabilization, localization and controlled release. Platt and Szoka reviewed a number of HA–drug conjugates for selective CD44 receptor-mediated cell internalization by cells [
27]. Once internalized, intracellular enzymes hydrolyze the hydrolytically labile HA–drug bond, thus releasing the drug inside the target cell [
28,
29,
30].
Numerous antineoplastic drugs have been conjugated to hyaluronic acid, generating new compounds with promising antitumor effects [
29,
30,
31,
32]. Paclitaxel was one of the lead compound studied. The free drug has shown tremendous potential as an anticancer compound [
33], but its use is compromised by its poor aqueous solubility. Moreover, its commercial formulation contains a castor oil (Cremophor®), which is responsible for most of the side effects and adverse drug reactions [
34,
35]. The selective targeting of paclitaxel to tumor cells might be achieved by covalent conjugation to HA. In the literature, paclitaxel has been linked to HA by several chemical strategies, differing mainly in the spacer between HA and paclitaxel, which yielded derivatives with different chemical linkages and, consequently, with diverse performances.
Luo and Prestwich [
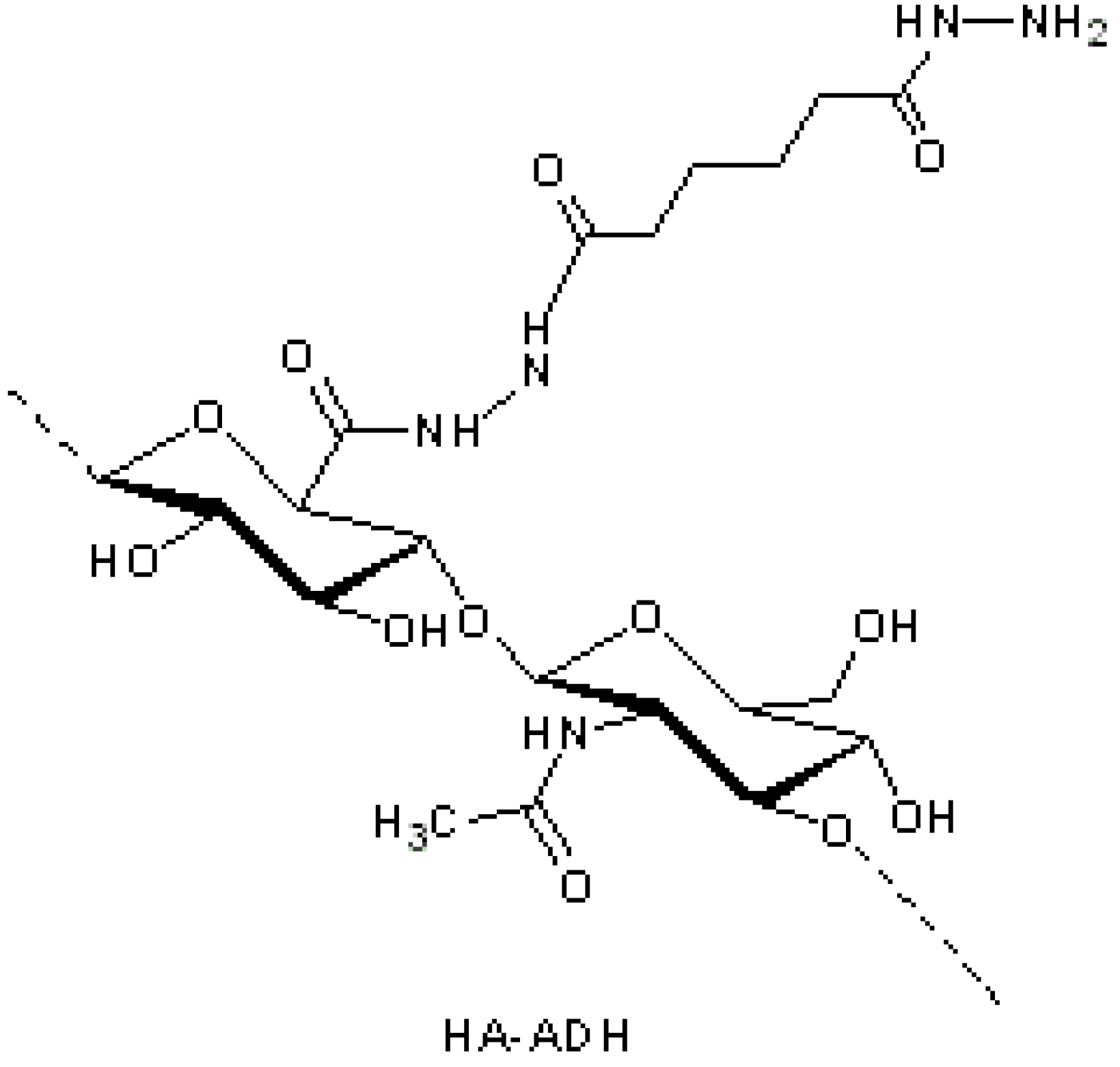
29] suggested the modification of HA with adipic dihydrazide (ADH) (
Figure 2), followed by the conjugation with a hemisuccinate
N-hydroxysuccinimide (NHS) activated ester of paclitaxel. The hydrolytically unstable ester bond allowed drug release inside cells by intracellular enzymatic hydrolysis. The HA–paclitaxel conjugate (
Figure 3a) is internalized by receptor-mediated endocytosis followed by esterase-catalyzed drug release in the lysosomal compartment. Consequently, only in CD44 overexpressing cells did HA–paclitaxel show a relevant cytotoxicity [
30].
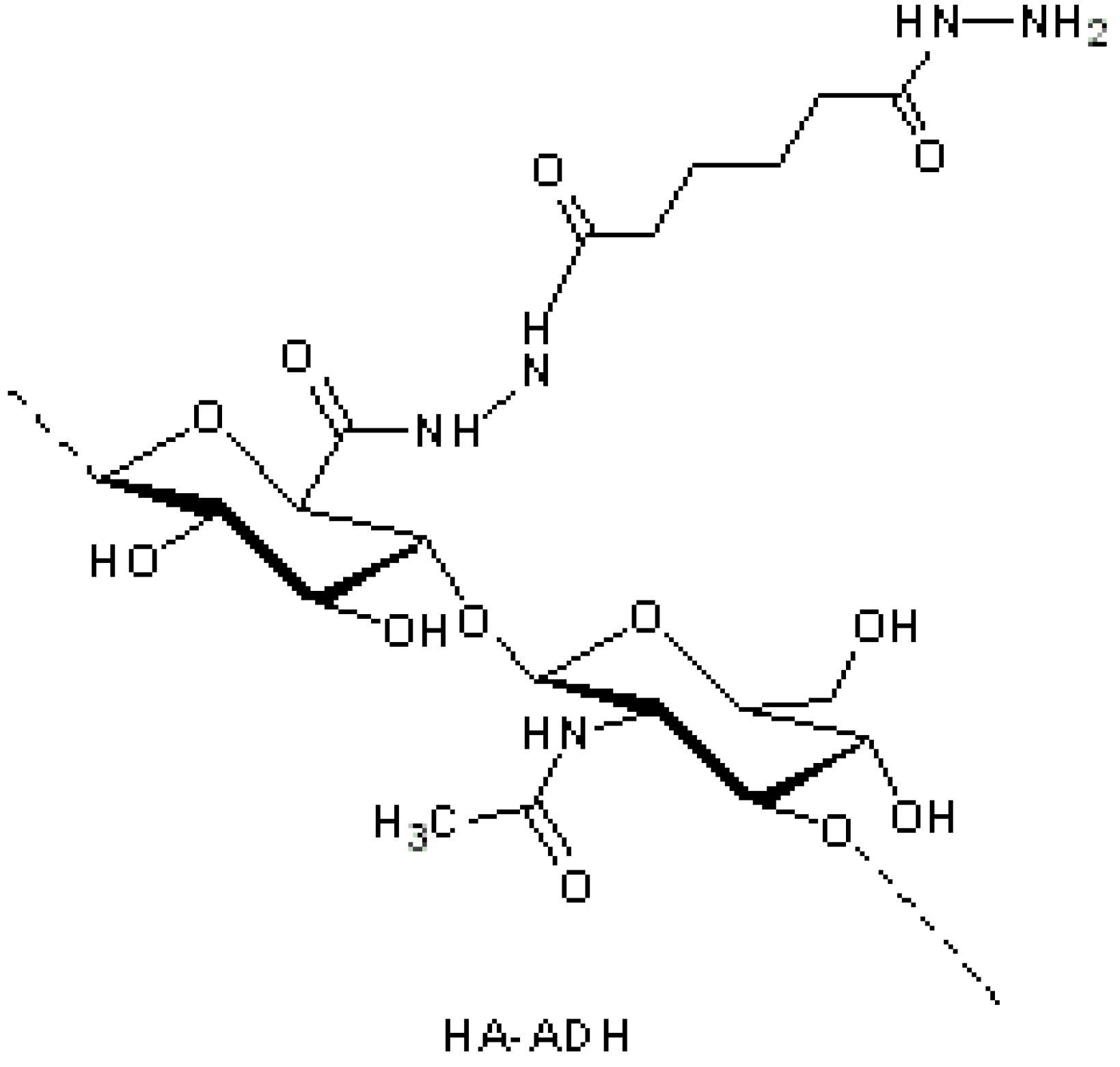
Figure 2.
Hyaluronic acid (HA)-adipic dihydrazide (ADH) intermediate for further derivatization with paclitaxel [
29].
Figure 2.
Hyaluronic acid (HA)-adipic dihydrazide (ADH) intermediate for further derivatization with paclitaxel [
29].
Tabrizian and co-workers coupled the hemisuccinate NHS activated ester of paclitaxel to a HA previously partially derivatized at some of the carboxylic groups by ethylenediamine (EDA) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), as coupling carbodiimide, in a buffered medium (pH 5) [
36]. The new conjugate (
Figure 3b) was assembled in a polyelectrolyte multilayer (PEM) construct with chitosan by using the layer-by-layer technique, which allowed for a precise control of the degradation and drug release. The presence of amide linkages between the HA and the spacer prejudiced the water solubility, differently to the HA–paclitaxel conjugate described by Prestwich [
29]; thus only the derivative with 3% (mol) paclitaxel loading exhibited the required hydrosolubility for the PEM preparation.
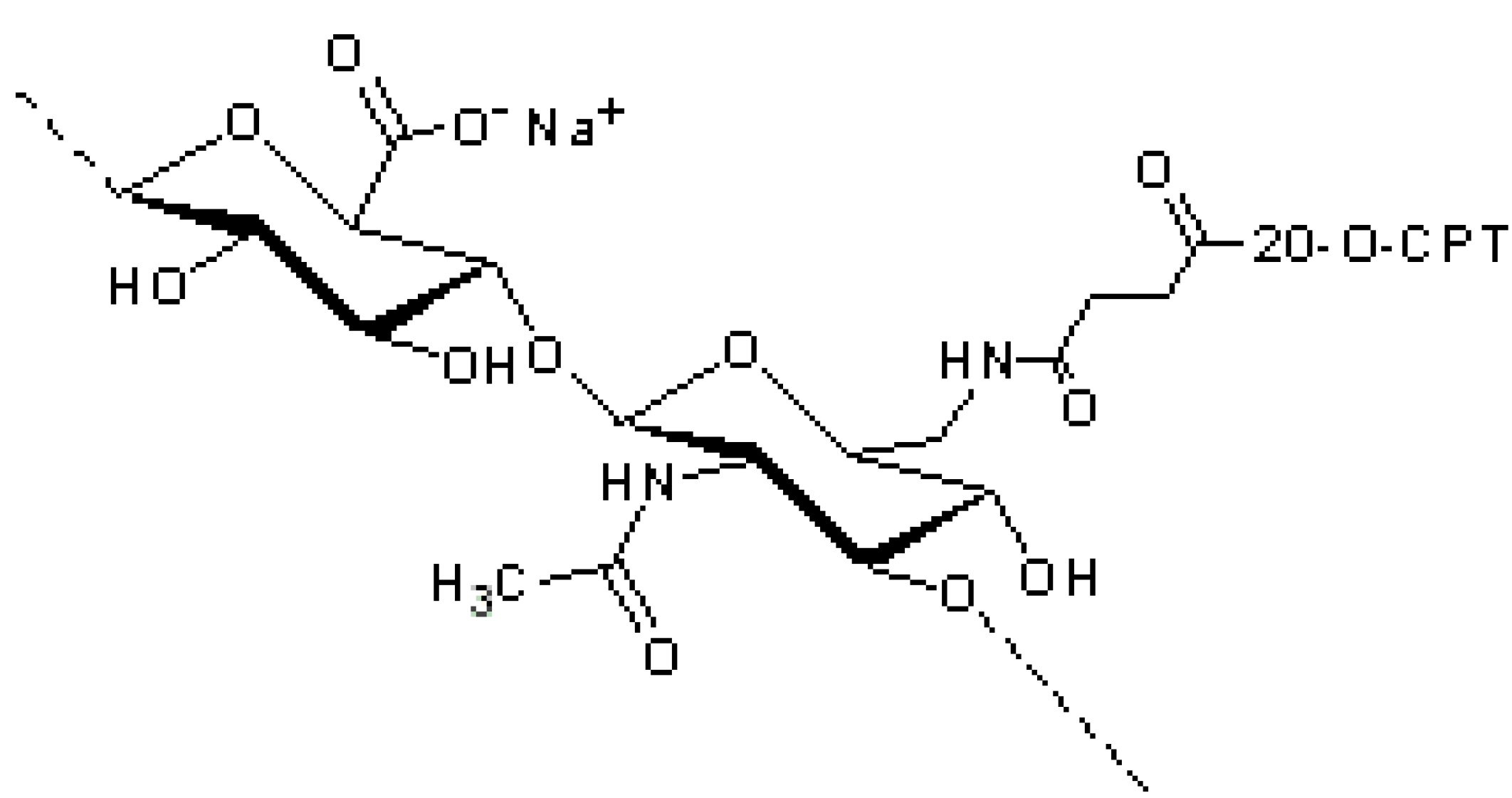
Crescenzi and co-workers in collaboration with Fidia Farmaceutici suggested another approach to conjugate paclitaxel to HA (200 KDa): firstly, the hydroxyl group of paclitaxel was esterified with 4-bromobutyric acid, previously activated via a carbodiimide, and then, the 4-bromobutyric-paclitaxel was linked to HA by forming an ester bond with the carboxylic group of HA in an organic solvent [
31,
37]. Therefore, the spacer was linked to both paclitaxel and HA via ester bonds (
Figure 3c). The conjugate (ONCOFID™-P) achieved a remarkably high drug loading (up to 12 mol %) and solubility (up to 15 mg/mL conjugate equivalent). Rosato
et al. reported that ONCOFID™-P easily released paclitaxel after cell internalization and presented a stronger antitumor activity than the free drug [
38]. De Stefano
et al. reported the safety and tolerability of ONCOFID™-P after loco-regional treatment of ovarian cancer xenograft in mice, it being capable of providing higher dosages without increasing adverse effects [
39]. Currently, ONCOFID™-P is undergoing phase II clinical studies in six European countries for the treatment of refractory bladder cancer [
38,
40]. The same conjugation strategy of ONCOFID™-P was successfully applied to other antineoplastic agents, presently under development by Fidia Farmaceutici [
41], such as docetaxel, doxorubicin and the active metabolite of irinotecan (7-ethyl-10-hydroxycamptothecin), leading to the ONCOFID™ platform. ONCOFID™-S (
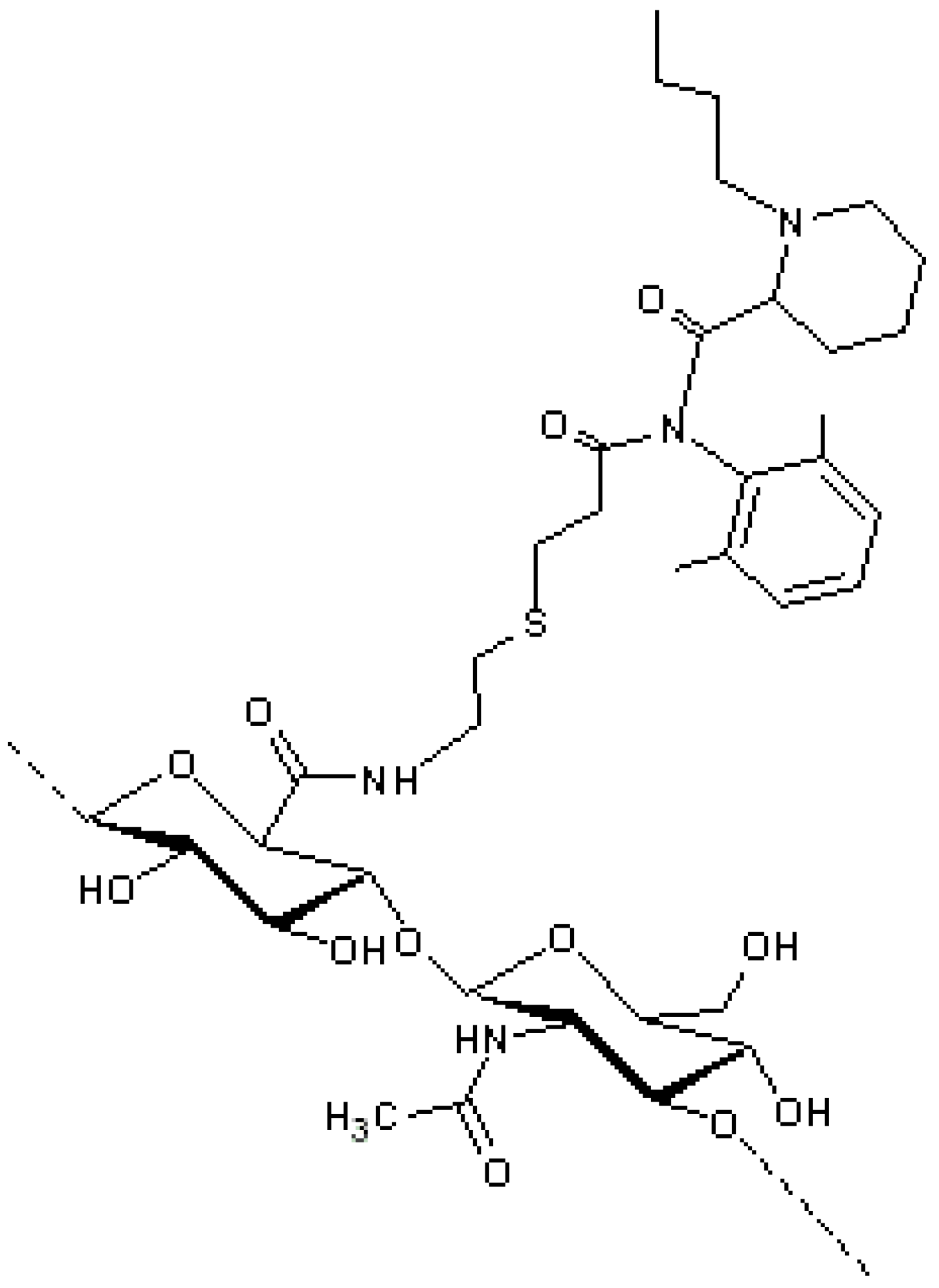
Figure 4), for example, was synthesized by coupling 7-ethyl-10-hydroxycamptothecin (SN38) to HA. The conjugate demonstrated the ability to block the cellular proliferation by a mechanism different from the apoptotic action of the reference drug, this making the HA derivative a drug with a new therapeutic activity and a higher efficacy [
40,
42]. ONCOFID™-S has been developed for the loco-regional intraperitoneal treatment of peritoneal carcinomatosis; the
in vivo study demonstrated that once administered intraperitoneally in rats, it was poorly absorbed into the systemic circulation, even after the administration of a high dose [
43].
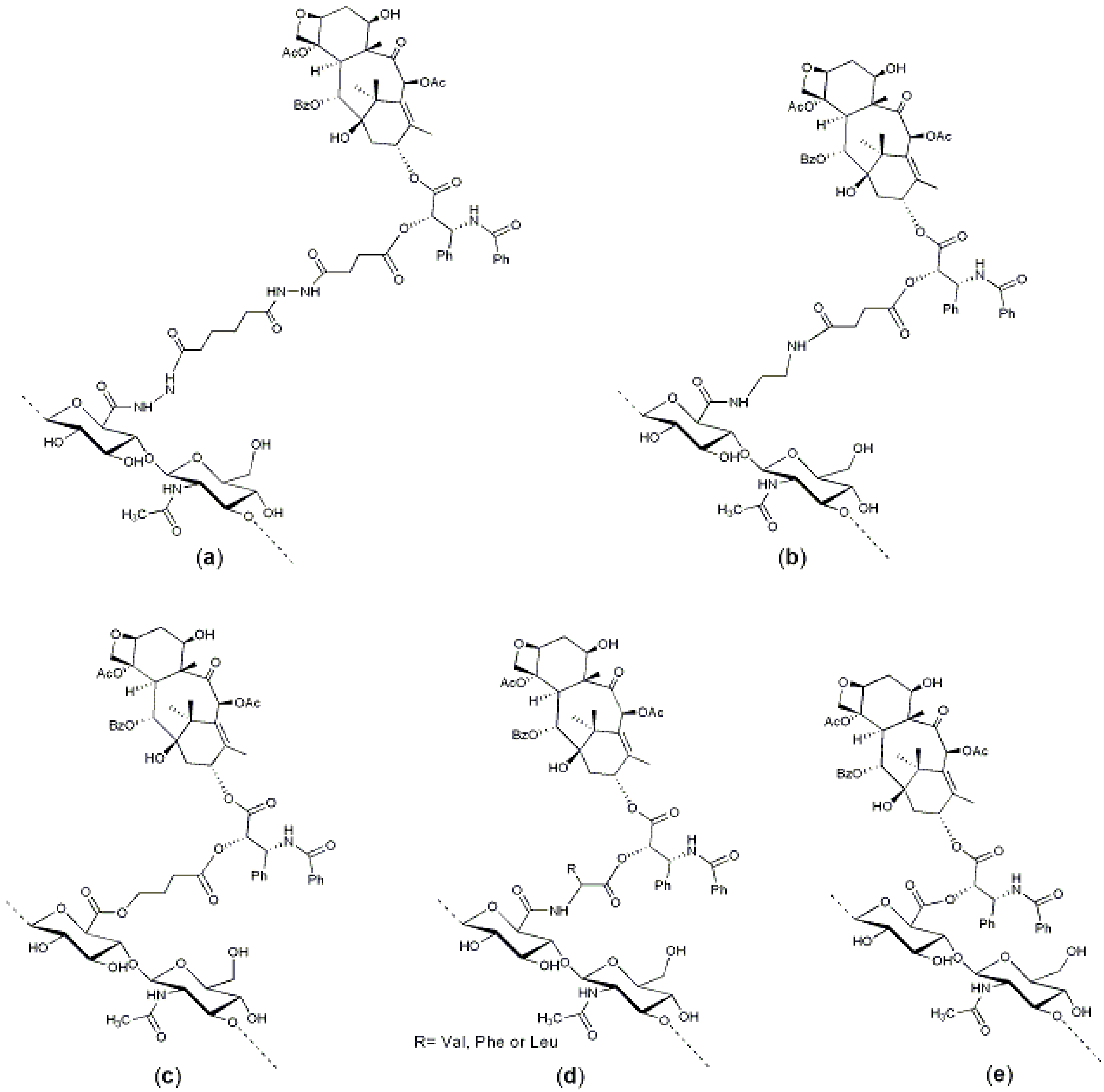
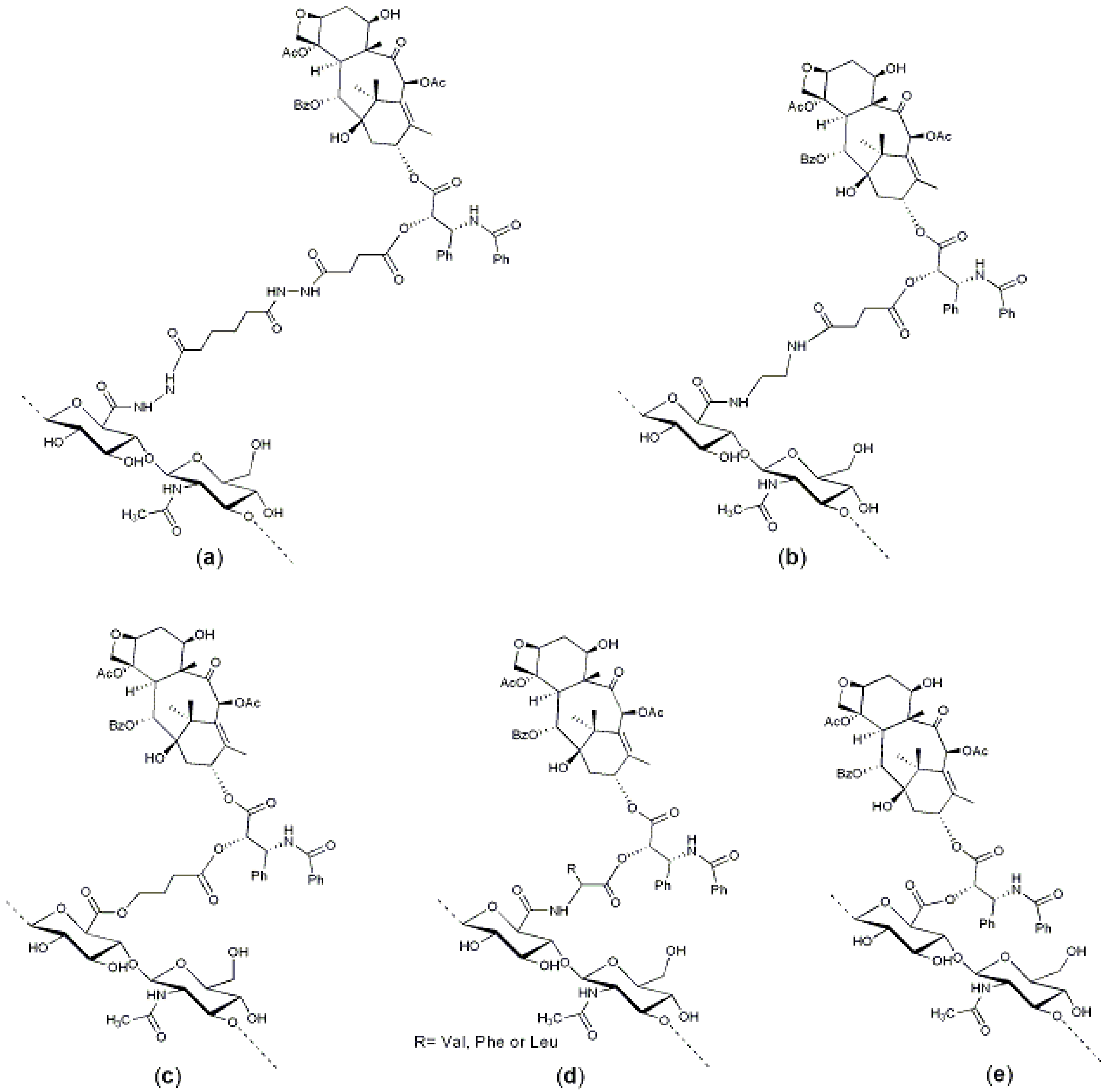
Figure 3.
HA–paclitaxel conjugates: (
a) A hemisuccinate NHS activated ester of paclitaxel was linked to an HA-ADH derivative [
29]; (
b) A hemisuccinate NHS activated ester of paclitaxel was linked to an HA–EDA derivative [
36]; (
c) A 2′-paclitaxel-4-bromobutyrate was conjugate to HA [
37]; (
d) An amino acid-paclitaxel derivative was linked to HA activated by carbodiimide [
44]; and (
e) Paclitaxel was directly conjugated to carboxylic groups of HA by means of dicyclohexylcarbodiimide (DCC)/dimethylaminopyridine (DMAP), leading to polymeric micelles [
45].
Figure 3.
HA–paclitaxel conjugates: (
a) A hemisuccinate NHS activated ester of paclitaxel was linked to an HA-ADH derivative [
29]; (
b) A hemisuccinate NHS activated ester of paclitaxel was linked to an HA–EDA derivative [
36]; (
c) A 2′-paclitaxel-4-bromobutyrate was conjugate to HA [
37]; (
d) An amino acid-paclitaxel derivative was linked to HA activated by carbodiimide [
44]; and (
e) Paclitaxel was directly conjugated to carboxylic groups of HA by means of dicyclohexylcarbodiimide (DCC)/dimethylaminopyridine (DMAP), leading to polymeric micelles [
45].
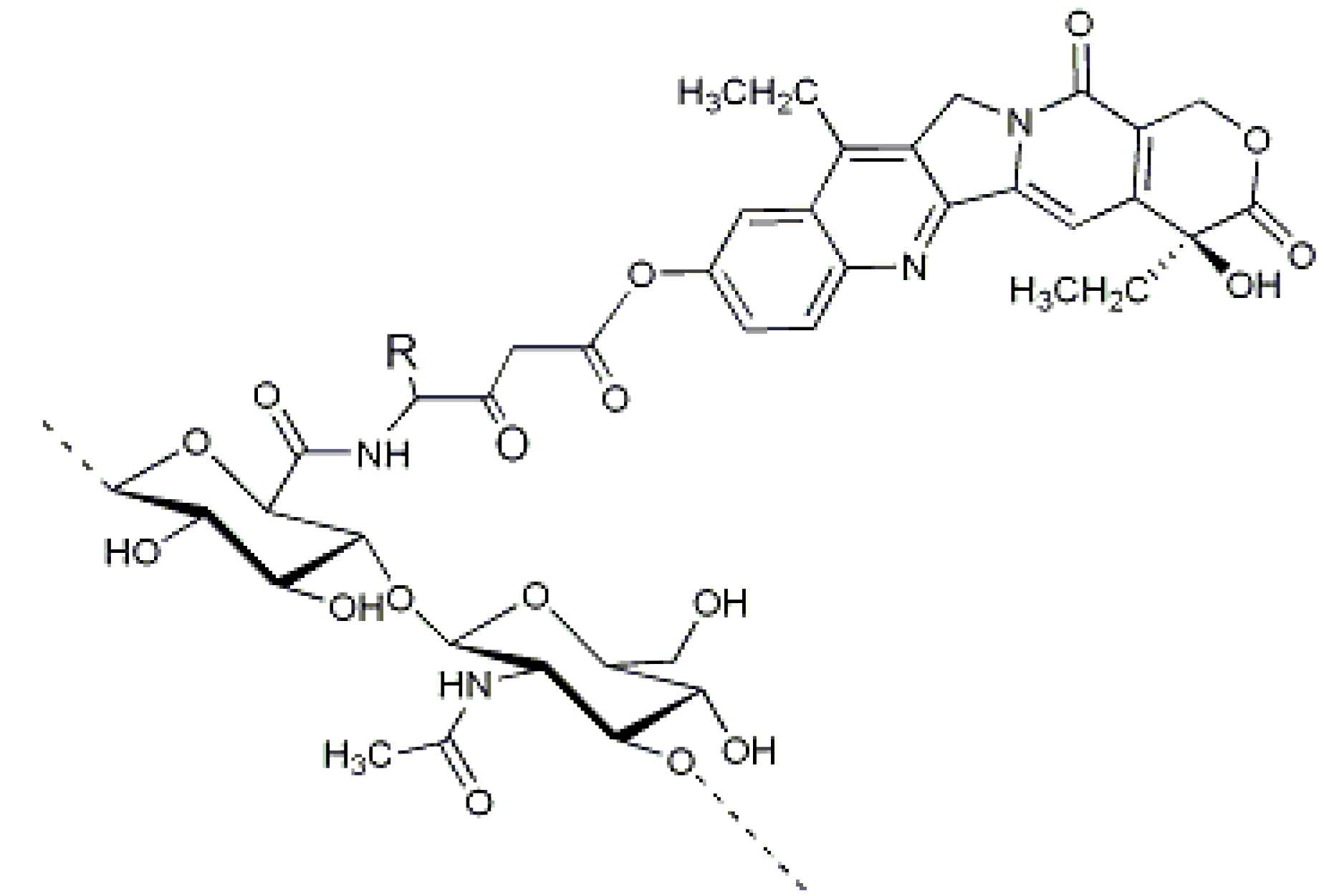
Figure 4.
ONCOFID™-S, the HA-7-ethyl-10-hydroxycamptothecin (SN38) derivative obtained by conjugation of SN38-4-bromobutyrate to HA [
41].
Figure 4.
ONCOFID™-S, the HA-7-ethyl-10-hydroxycamptothecin (SN38) derivative obtained by conjugation of SN38-4-bromobutyrate to HA [
41].
Xin, Wang and Xiang described the conjugation of paclitaxel onto HA using amino acid linkers (
Figure 3d) [
44]. Firstly, the carboxylic group of the chosen amino acid was linked to the hydroxyl group of paclitaxel, and then, the amino group of the amino acid-paclitaxel derivative was grafted and conjugated to the carboxylic group of HA via EDC and NHS activation in DMF. Furthermore, this strategy led to a high drug loading (10%–15 mol %), even if the authors highlighted the formation of nanoparticles in aqueous solution due to the amphiphilic nature of the HA–amino acid–paclitaxel conjugate. The presence of an amino acid spacer allowed for a higher hydrolysis rate and release of paclitaxel compared to that observed in the HA–ADH–paclitaxel conjugate described by Luo and Prestwich, owing to a better recognition by the esterase enzymes [
28]. The ability of HA-hydrophobic drugs to self-assemble in aqueous milieu is sometimes exploited to obtain polymeric micelles, composed of a hydrophobic inner core, containing the hydrophobic drug, and a hydrophilic HA shell layer.
Among the extensive literature, here, we report the approach used by Lee and co-workers that used dimethoxy-PEG (dmPEG) to solubilize HA in DMSO (with a dmPEG/HA weight ratio of five) for the following reaction with paclitaxel [
45]. Differently from the above described works that used spacers, here, hydroxyl groups of paclitaxel were directly conjugated to carboxylic groups of HA in the organic phase, using DCC/DMAP as the coupling agent and leading to an acid-labile ester linkage (
Figure 3e). It should be noted that, however, direct conjugation did not avoid partial crosslinking of HA during the DCC/DMAP coupling process. Nevertheless, the HA–paclitaxel micelles obtained present likely a core/shell structure composed of a hydrophobic core containing aggregated paclitaxel molecules and a hydrophilic HA shell layer. These micelles exhibited a greater cytotoxicity in CD44 overexpressing cells with respect to the reference paclitaxel formulation [
45].
Beyond paclitaxel, other anticancer agents were successfully linked to HA, aiming to overcome toxicity and to confer new physico-chemical characteristics [
27,
28,
46,
47,
48]. Adipic acid dihydrazide and succinic anhydride were used as spacers to connect 5-fluro uracil (5-FU) to HA, reaching a loading of 8 mol %. The polymeric prodrug (
Figure 5) presented intrinsic active tumor targeting of HA, strengthening the anti-proliferative effect of HA-5-FU, evidenced by low IC
50 values in different cancer cell lines [
49].
Figure 5.
HA–5-flurouracil (5-FU) conjugate [
49].
Figure 5.
HA–5-flurouracil (5-FU) conjugate [
49].
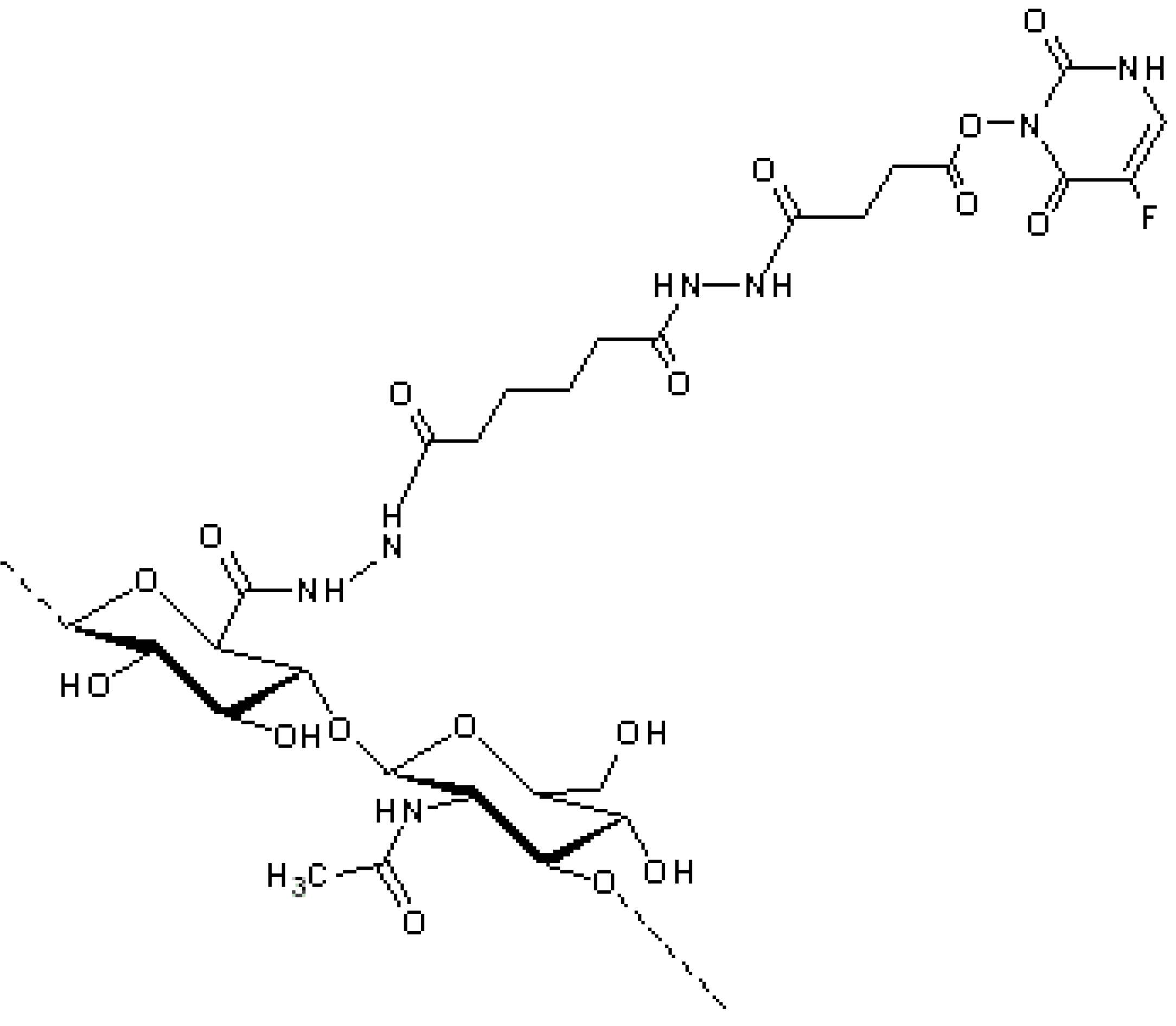
Norbedo
et al., from Eurand Pharmaceuticals (now Aptalis), developed a new strategy of conjugation based on the regio-selective conversion of C-6 hydroxyl groups into amino groups, which are suitable for conjugation with drugs. In particular, camptothecin (CPT), converted into the corresponding 20-
O-hemisuccinate, was conjugated on 6-amino-6-deoxyhyaluronan derivative by a carbodiimide-mediated coupling (
Figure 6) [
50].
Figure 6.
HA-camptothecin (HA-CPT) derivative, obtained by the conjugation of 20-
O-hemisuccinate-CPT to 6-amino-6-deoxyhyaluronan derivative by a carbodiimide-mediated coupling [
50].
Figure 6.
HA-camptothecin (HA-CPT) derivative, obtained by the conjugation of 20-
O-hemisuccinate-CPT to 6-amino-6-deoxyhyaluronan derivative by a carbodiimide-mediated coupling [
50].
This novel approach allowed for a high drug loading (up to 25 mol %) without affecting either the solubility in physiological solution (the high hydrophilic carboxylate groups are free) or CD44 recognition, because C-6 hydroxyl groups are basically not involved in the receptor recognition [
51].
Eurand Pharmaceuticals applied this strategy also to methotrexate (MTX), an antimetabolite and an analogue of folic acid, used as an antineoplastic drug. In the first step, a chloride was introduced at the C-6 position, as the leaving group, which was then substituted by MTX by means of an ester linkage. Acetyl or butyryl groups were introduced at the C-6 position to substitute the remaining chloride, thus producing mixed esters. The conjugates can be obtained with 10 to 18% of drug loading. The HA–MTX conjugate showed significant activity on a liver metastasis tumor model and activity on a mammary carcinoma model. Toxicology and pharmacokinetics investigations in rodent and non-rodent species have shown a prolonged half-life and increased area under the curve (AUC) values with respect to free MTX, with a toxicity profile that is similar to that of the unconjugated drug [
52].
4. HA Anti-Inflammatory Drug
Due to the short half-life of hyaluronic acid in the blood [
53], the majority of HA–drug conjugates developed and described here were designed for local (
i.e., intratumoral, subcutaneous, intravesical, intraperitoneal, intraarticular,
etc.) rather than for intravenously administrations. For example, drugs comprising a hydroxyl group, such as hydrocortisone, prednisone, prednisolone, fluorocortisone, dexamethasone, betamethasone and corticosterone, were conjugated directly to HA by esterification and were studied for intraarticular administration in the treatment of arthritis [
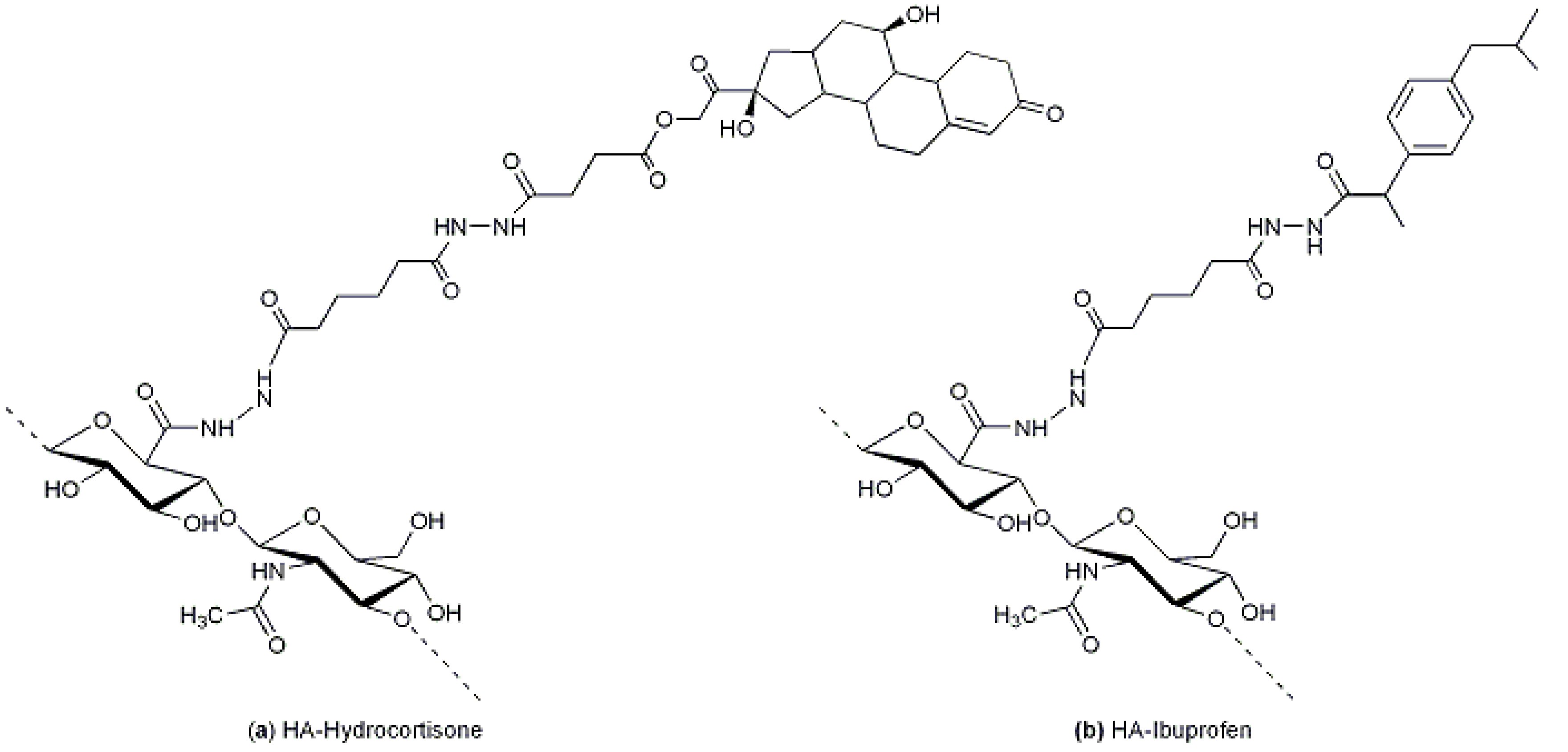
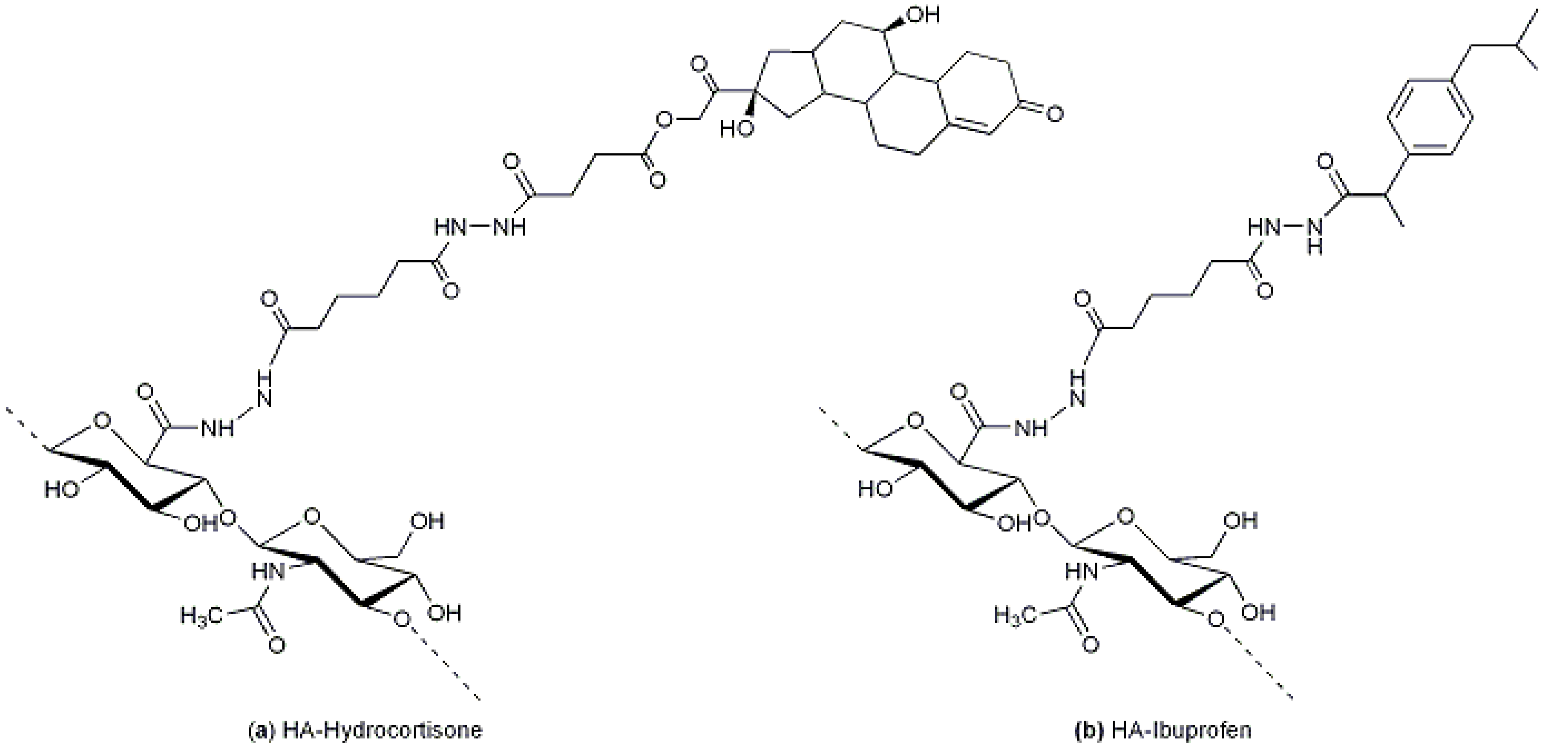
54]. Pouyani and Prestwich described the grafting of anti-inflammatory drugs (hydrocortisone or ibuprofen,
Figure 7a,b, respectively) by means of a spacer for local injections into arthritic joints [
55]. After modification of HA with adipic dihydrazide (ADH), the conjugation involved an activated ester of ibuprofen, using a carbodiimide and NHS, or a hemisuccinate derivative of hydrocortisone activated with NHS in the same way as ibuprofen.
Figure 7.
(
a) HA-hydrocortisone: the HA-ADH derivative was conjugated to a hemisuccinate derivative of hydrocortisone previously activated with NHS [
55]; and (
b) HA-ibuprofen: the HA-ADH derivative was conjugated to an activated ester of ibuprofen, using a carbodiimide and NHS [
55].
Figure 7.
(
a) HA-hydrocortisone: the HA-ADH derivative was conjugated to a hemisuccinate derivative of hydrocortisone previously activated with NHS [
55]; and (
b) HA-ibuprofen: the HA-ADH derivative was conjugated to an activated ester of ibuprofen, using a carbodiimide and NHS [
55].
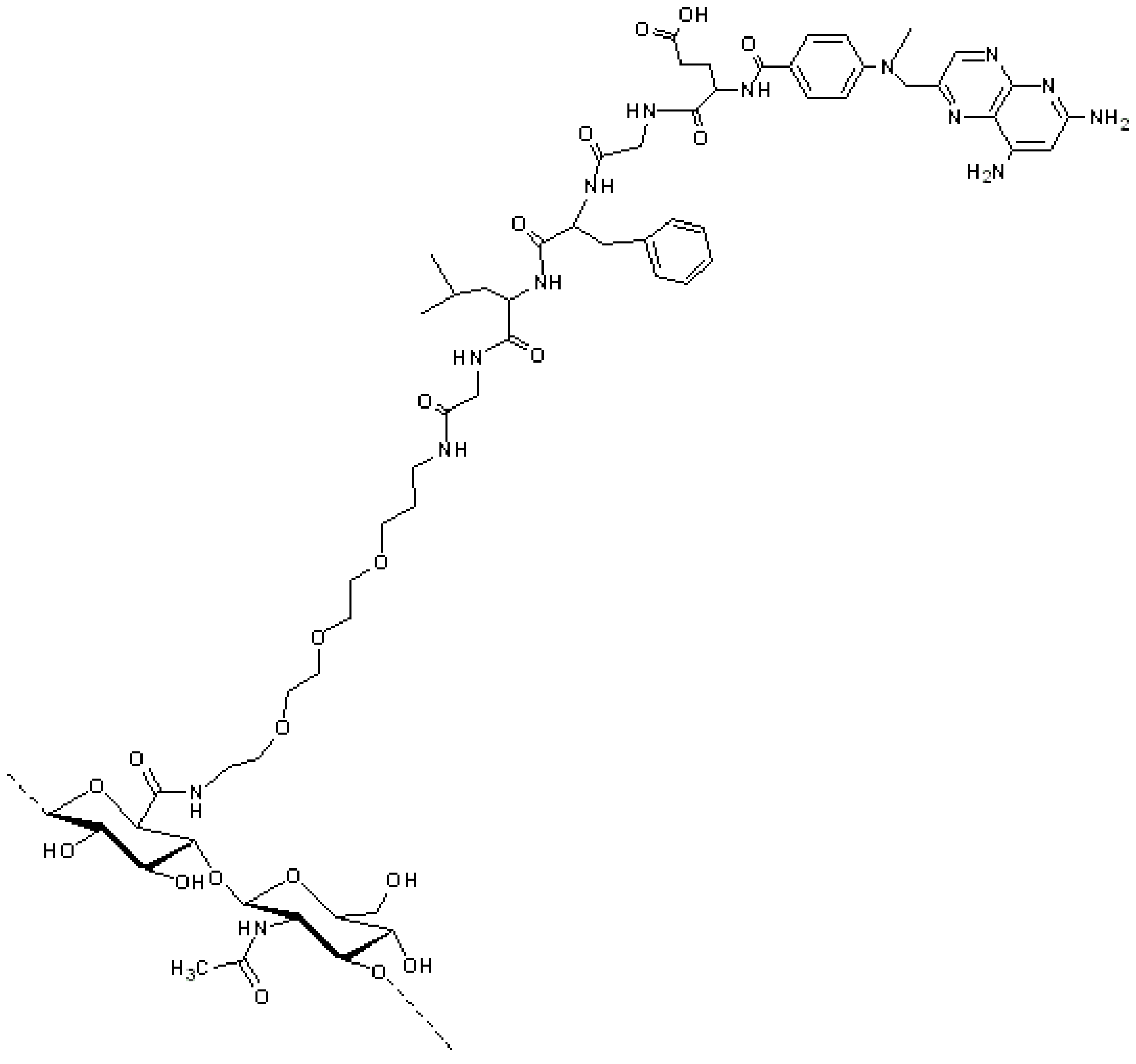
In addition to its antitumor activity, MTX is also an immunosuppressant, very effective in the treatment of autoimmune diseases, such as rheumatoid arthritis [
56]. HA–MTX conjugate was also synthesized by Homma and co-workers (Chugai Pharmaceuticals Co., Tokio, Japan) for the delivery of MTX into the synovium, aiming to reach low systemic side effects after intra-articular administration [
18]. In this attempt, MTX was firstly linked to a lysosomal enzyme-sensitive peptide linker. Then, coupling to HA was achieved exploiting a spacer, inserted between the peptide linker and the HA backbone, this to avoiding the potential steric entanglement of the polymer towards the proteases that cleave the peptide. During these coupling steps, the
MW of the conjugates tended to decrease; therefore, for a better analgesic effect, a high
MW HA (2,000,000 Da) was preferred. In antigen-induced arthritis rats, the intra-articular injection of the HA–MTX conjugate containing the Asn-Phe-Phe linker showed significant reduction of knee swelling. In contrast, intra-articularly injected free MTX and the mixture of HA and MTX showed little or no effect. Of note, the conjugate with the Gly-Phe-Leu-Gly peptide linker (
Figure 8), with a similar MTX loading, showed only a slight tendency to inhibit knee swelling, this suggesting that cleavage of the conjugate
in vivo depends on the peptide sequence [
18].
Figure 8.
HA–methotrexate (MTX) conjugate: MTX was firstly linked to a lysosomal enzyme-sensitive peptide linker (Gly-Phe-Leu-Gly) that was further conjugated to HA by means of a polyethylene glycol spacer [
18].
Figure 8.
HA–methotrexate (MTX) conjugate: MTX was firstly linked to a lysosomal enzyme-sensitive peptide linker (Gly-Phe-Leu-Gly) that was further conjugated to HA by means of a polyethylene glycol spacer [
18].
A further study determined that the optimal peptide sequence is Phe-Phe together with the ethylenediamine spacer and that the MTX loading should be higher than 1.3%; ideally, 3.8% [
57].
Being that HA is a viscosupplement widely used for the local treatment of osteoarthritis, different combinations with drugs were explored in order to improve its beneficial effects in the joints. In this approach, HA is not only a carrier, but also an active ingredient (viscosupplement). Miyamoto and co-workers from Seikagaku Co. developed a HA derivative in which non-steroidal anti-inflammatory drugs were covalently bound to the polysaccharide through a biodegradable spacer, for the treatment of arthritic joints and related pain and inflammation through a local delivery approach [
58]. The studied anti-inflammatory drugs had a carboxyl group suitable for the coupling with the hydroxyl group of a bifunctional linker containing at the other site an amino group for HA conjugation at the level of carboxylic group.
In vivo investigations demonstrated that the anti-inflammatory drug (e.g., ketoprofen, ibuprofen, naproxen, diclofenac,
etc.) once conjugated to HA suppressed pain better than the free form and HA alone. In addition, pharmacokinetics residence studies revealed that the HA conjugated anti-inflammatory drug remained in the synovial fluid several days after the intra-articular injection. Differently, the free or HA mixed drug disappeared from the synovial fluid within 6 or 12 h. This suggests that HA conjugation could mitigate the side effects of Non-steroidal-anti-inflammatory-drugs (NSAIDs) compared with oral or topical NSAID administration. Actually, after the completion of a Phase I clinical trial in September 2012, Phase II of clinical trials is ongoing in Japan for intraarticular (i.a.) administration of an HA–diclofenac conjugate.
6. HA–Peptide Conjugates
The chemical modification and bioconjugation of HA to biologics have been carried out mostly in aqueous solution through two main approaches of HA derivatization: the direct activation of HA carboxyl groups and the oxidation with periodate of the HA backbone to form a limited amount of aldehyde groups. The main limitation of these approaches, especially for the first one, is the random coupling to the several protein amino groups with a potential crosslinking and, consequently, a low homogeneity of final conjugates and a difficult batch-to-batch reproducibility [
65].
HA–Anti-Flt1 peptide was synthetized for the treatment of retinal and choroidal vascular disease and diabetic retinopathy [
66]. In these pathologies, there is an overexpression of vascular endothelial growth factor (VEGF) in the retina, this leading to angiogenesis and hyperpermeability [
67]. Anti-FLT1 peptide is an antagonist peptide of seven amino acids against VEGF receptor 1 (VEGFR1) [
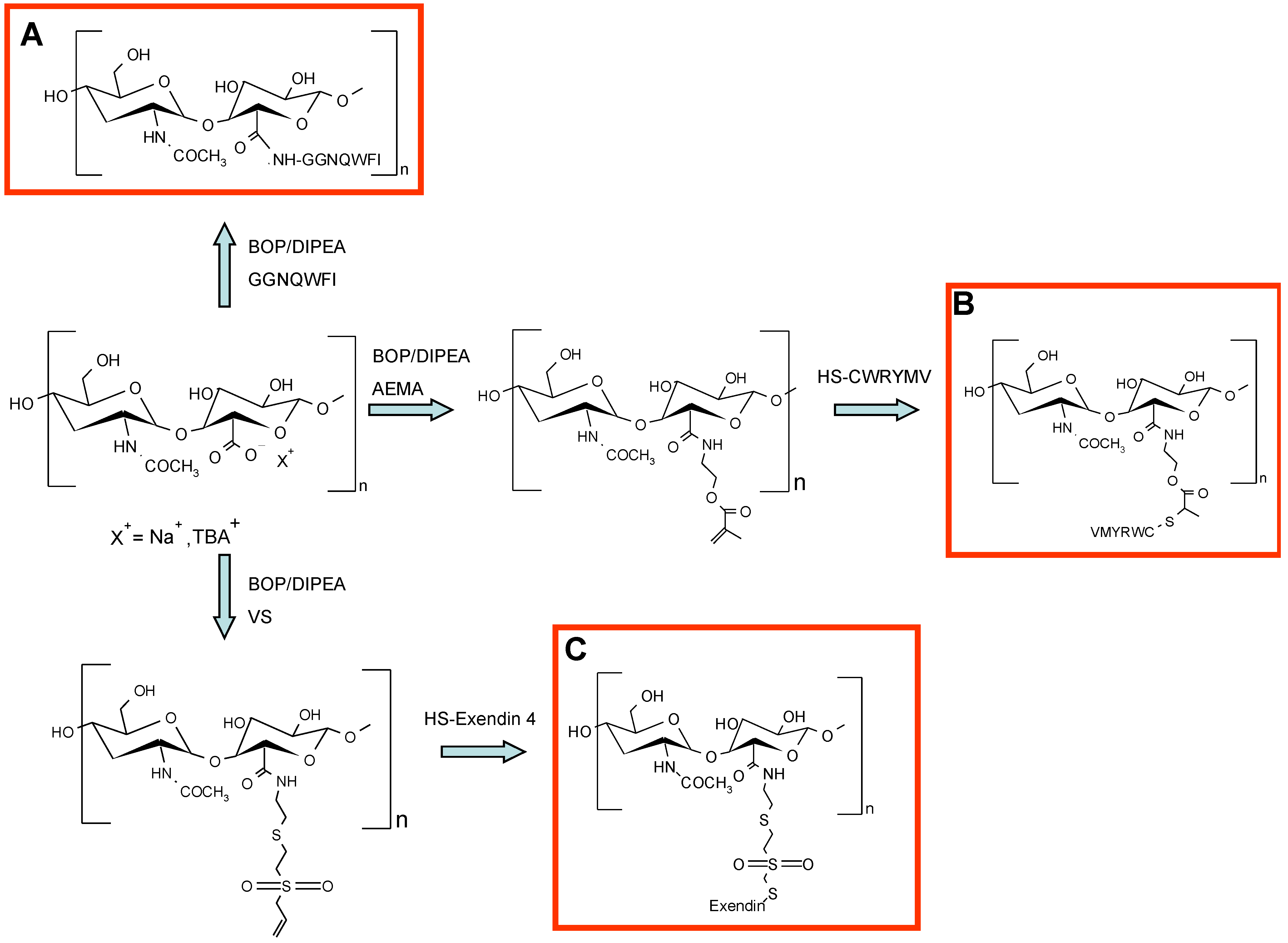
68]. The peptide was chemically conjugated to HA in anhydrous DMSO using benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP) as a coupling reagent (
Figure 10, Panel A). HA was chosen because it allows for a significant increase of peptide solubility in aqueous medium. Furthermore, for this specific application, the viscoelastic and mucoadhesive properties of the polymer can improve the outcome of the therapy.
In vitro study demonstrated that the conjugate preserved the biological activity of the peptide. The conjugate effectively inhibited retinal choroidal neovascularization (CNV) in laser-induced CNV model rats. The retinal vascular permeability and the deformation of retinal vascular structure were also significantly reduced in diabetic retinopathy model rats after treatment with HA–Anti-Flt1 conjugate. PK analysis confirmed the increased residence time of conjugated peptide for more than two weeks.
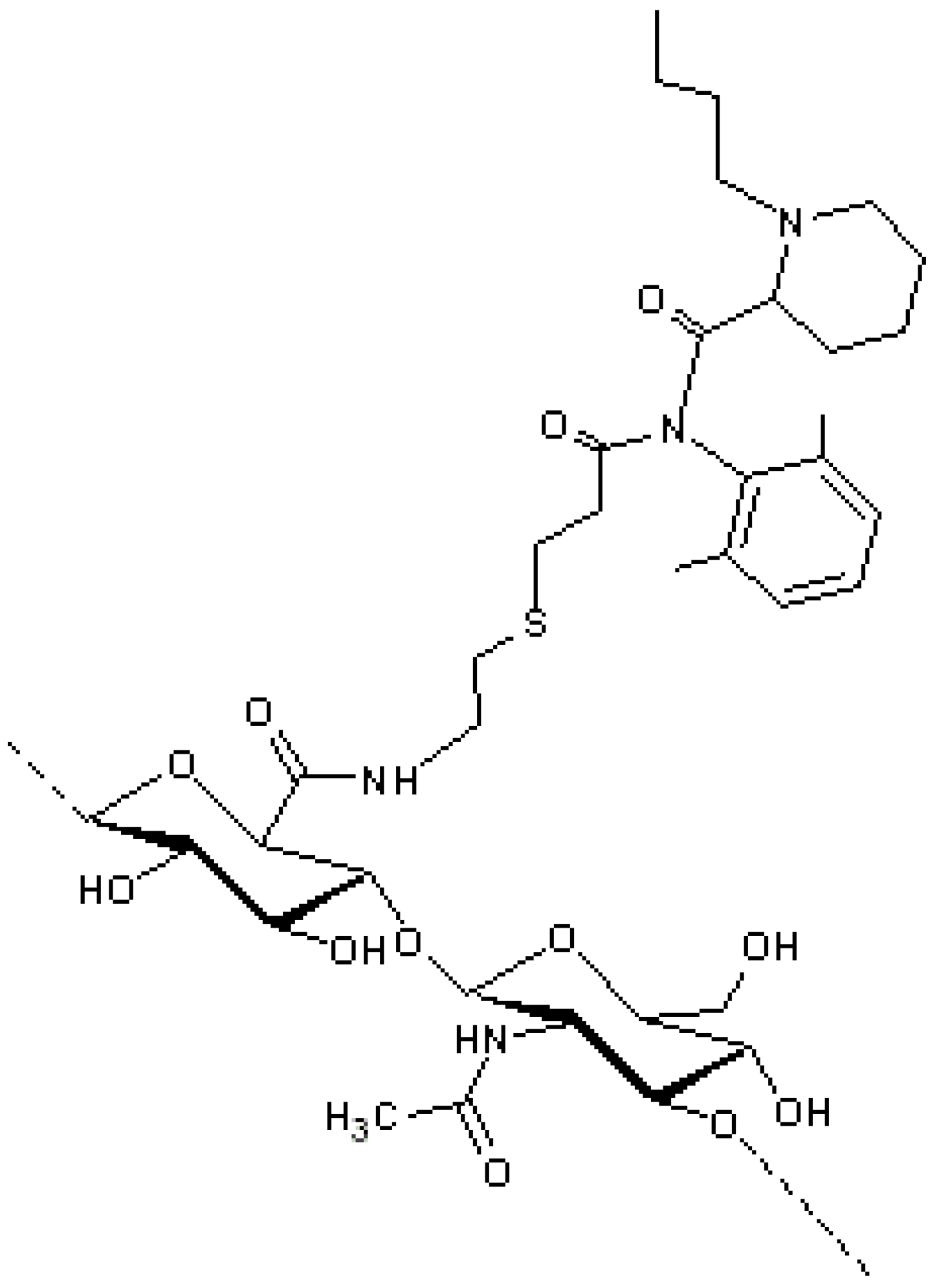
A selective thiol conjugation was also developed for the conjugation of HA to exendin-4 [
69] (
Figure 10, Panel C). The polypeptide is currently on the market for the treatment of type 2 diabetes, and it is injected subcutaneously twice a day, owing to its short circulating half-life, about 60–90 min [
70,
71]. Conjugation with HA has been proposed to increase its residence time in blood and to reduce the frequency of administration. The HAylation was carried out by a Michael addition involving a HA-vinyl sulfone derivative and a thiolated exendin-4. The coupling of vinyl sulfone moieties occurred at the level of HA carboxylic groups in order to reduce the HA recognition by HA receptors and hyaluronidase, thus elongating the conjugate half-life. HA–exendin-4 conjugates showed an increased stability in human serum with respect free exendin-4, further demonstrating the protective and anti-fouling capacity of the polymer. An
in vivo study on db/db mice showed that the glucose-lowering effect of HA–exendin-4 conjugates was maintained up to three days, while free exendin-4 exhausted its effect within a day.
Figure 10.
Schematic representation of different HA activation and peptide conjugation. (
A) Chemical conjugation of anti-Flt1 peptide to HA in anhydrous dimethylsulfoxide (DMSO) using BOP and diisopropylethylamine (DIPEA) as a coupling reagents [
66]; (
B) Conjugation of aminoethyl methacrylated HA (HA–AEMA) with CWRYMV peptide via Michael addition between the methacryloyl group of AEMA and the thiol group of cysteine in the peptide [
72]; and (
C) Conjugation of HA to exendin-4 by a Michael addition involving a HA–vinyl sulfone (VS) derivative and a thiolated peptide [
69].
Figure 10.
Schematic representation of different HA activation and peptide conjugation. (
A) Chemical conjugation of anti-Flt1 peptide to HA in anhydrous dimethylsulfoxide (DMSO) using BOP and diisopropylethylamine (DIPEA) as a coupling reagents [
66]; (
B) Conjugation of aminoethyl methacrylated HA (HA–AEMA) with CWRYMV peptide via Michael addition between the methacryloyl group of AEMA and the thiol group of cysteine in the peptide [
72]; and (
C) Conjugation of HA to exendin-4 by a Michael addition involving a HA–vinyl sulfone (VS) derivative and a thiolated peptide [
69].
Another strategy presented by Oh and co-workers was applied on agonistic peptide of the FPRL1 receptor (formyl peptide receptor-like-1), a promising drug for the treatment of inflammatory diseases, such as asthma, rheumatoid arthritis or sepsis [
72]. Aminoethyl methacrylated HA (HA-AEMA) was synthesized and conjugated with CWRYMV peptide via Michael addition between the methacryloyl group of AEMA and the thiol group of cysteine in the peptide (
Figure 10, Panel B). The formation of three HA–peptide conjugates, prepared with a different drug loading, was confirmed by
1H-NMR and gel permeation chromatography. The serum stability of the conjugates were studied
in vitro and demonstrated that the conjugated peptide was not degraded after five days of incubation, whereas the free peptide was reduced by 30% after three days. The signal transduction activity of HA–peptide conjugate for the FPRL1 receptor was assessed by measuring the elevation level of phospho-extracellular signal-regulated kinase (pERK) in RBL-2H3/FPRL1 cells. Western blots demonstrated that the conjugates showed less activity with respect free peptides, due to the steric hindrance of the polymer, but after hyaluronidase treatment, the activity increased substantially. As expected, the conjugate with the highest loading showed the best activity. This study further confirms the usefulness of HA for peptide modification; in fact, it has preserved the coupled peptides from enzymatic degradation, increased their serum stability and preserved their activity, thanks to the polymer degradation by hyaluronidase that allowed for the recovery of activity during time.
7. HA–Protein Conjugates
Duncan
et al. [
24] proposed the use of HA for protein conjugation in the Polymer Masked-Unmasked Protein Therapy (PUMPT) concept. The hypothesis is that conjugation of a biodegradable polymer to a biologically active protein can mask activity and enhance stability in the bloodstream, while subsequent recovery of the activity can be achieved by degradation of the polymer at the target site.
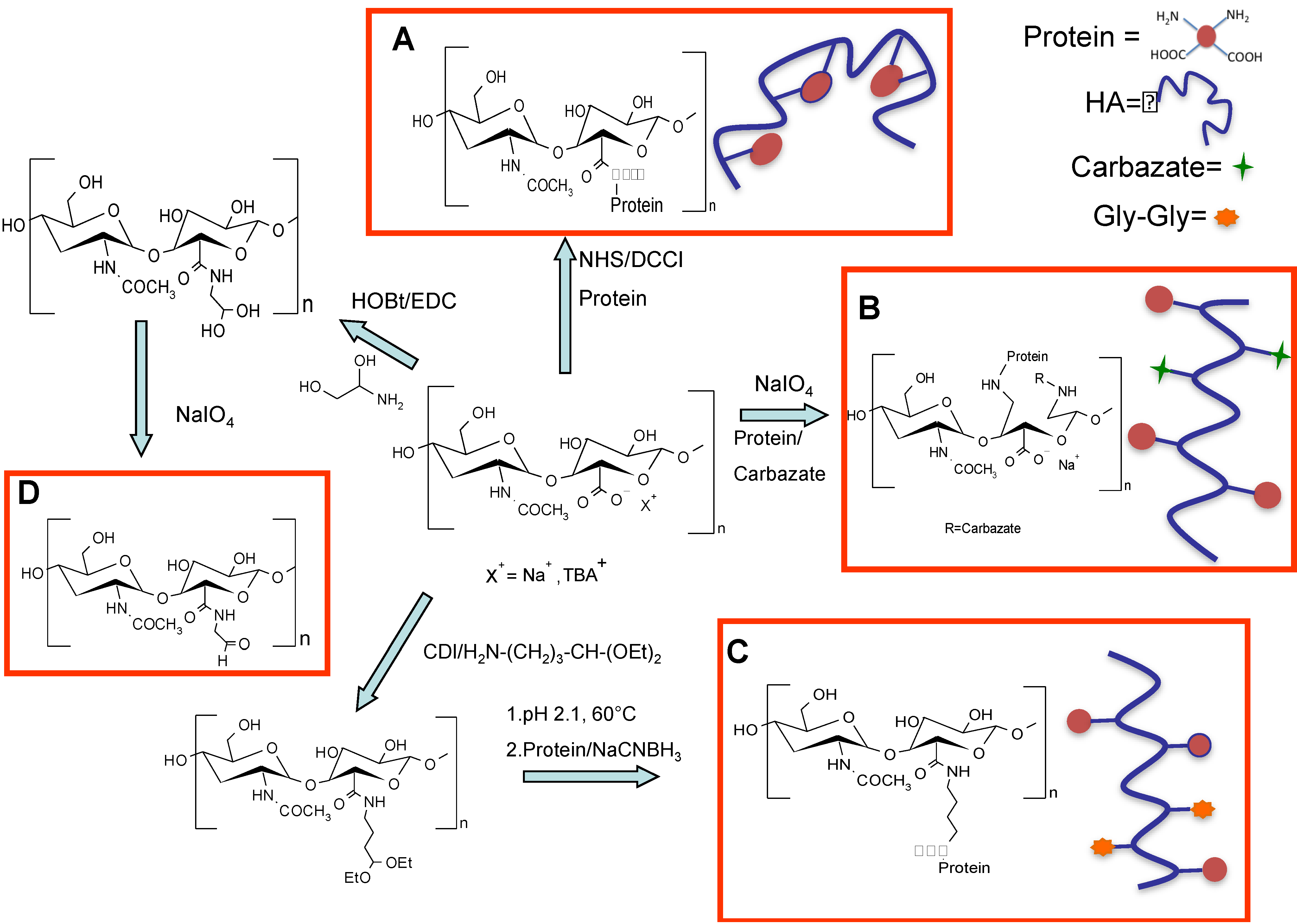
Hyaluronic acid (HA) has been conjugated to trypsin and ribonuclease A (RNase A) as proof of concept of PUMPT. The model proteins were randomly modified exploiting the free amino groups present on the protein surface and activating the carboxyl groups of the polymer via NHS/DCC (figure 11, panel A). This kind of approach was successful with only a slight decrease of enzymatic activity in vitro while a relevant increase of protein stability was achieved in the presence of proteolytic enzymes. The same group also developed HA-EGF for application in the field of wound healing. Unfortunately, the conjugate showed low activity in vitro on HEp2 cells [
73], probably as consequence to the random conjugation that interested the two lysines of EGF, Lys 28 and 48 [
74]. Lys 28 is situated in close proximity to EGF’s receptor binding domain. It is possible that the polymer entangled the recognition of EGF by its receptor. Hence, a random approach is not always reliable because the protein activity can be compromised especially in the case of proteins that interact with receptors. This simple chemistry of lysine coupling unfortunately faces the common problem of low selectivity owing the fact that this amino acid is well-represented on the surface of proteins. Usually, quite complex mixtures of isomers are obtained with this random conjugation, thus affecting the batch-to-batch reproducibility and the purification and characterization steps. Furthermore, the requirements from regulatory agencies to get approval for human use have been increased, and only thoroughly characterized conjugates can meet the specifications. It is obvious that the characterization/identification methods and the batch-to-batch reproducibility would be more straightforward with a conjugation reaction yielding a single isomer rather than an approach generating two or even more isomers [
21]. For these reasons,
N-terminal conjugation is one of the most studied techniques in order to get site-specific protein modification. The different pKa of
N-terminal and epsilon amino groups can be exploited to direct the conjugation at the
N-terminus by performing the reaction in slightly acidic buffers and using an aldehyde polymer, a well-known approach for site-selective protein conjugation [
75,
76].
Considering the polysaccharide nature of HA, the most used approach has been the partial oxidation of some sugar units in the backbone by periodate, yielding aldehyde groups useful for the selective
N-terminal modification. The advantage of this chemistry is that it can be performed in mild conditions, such as aqueous conditions and neutral pH values [
77].
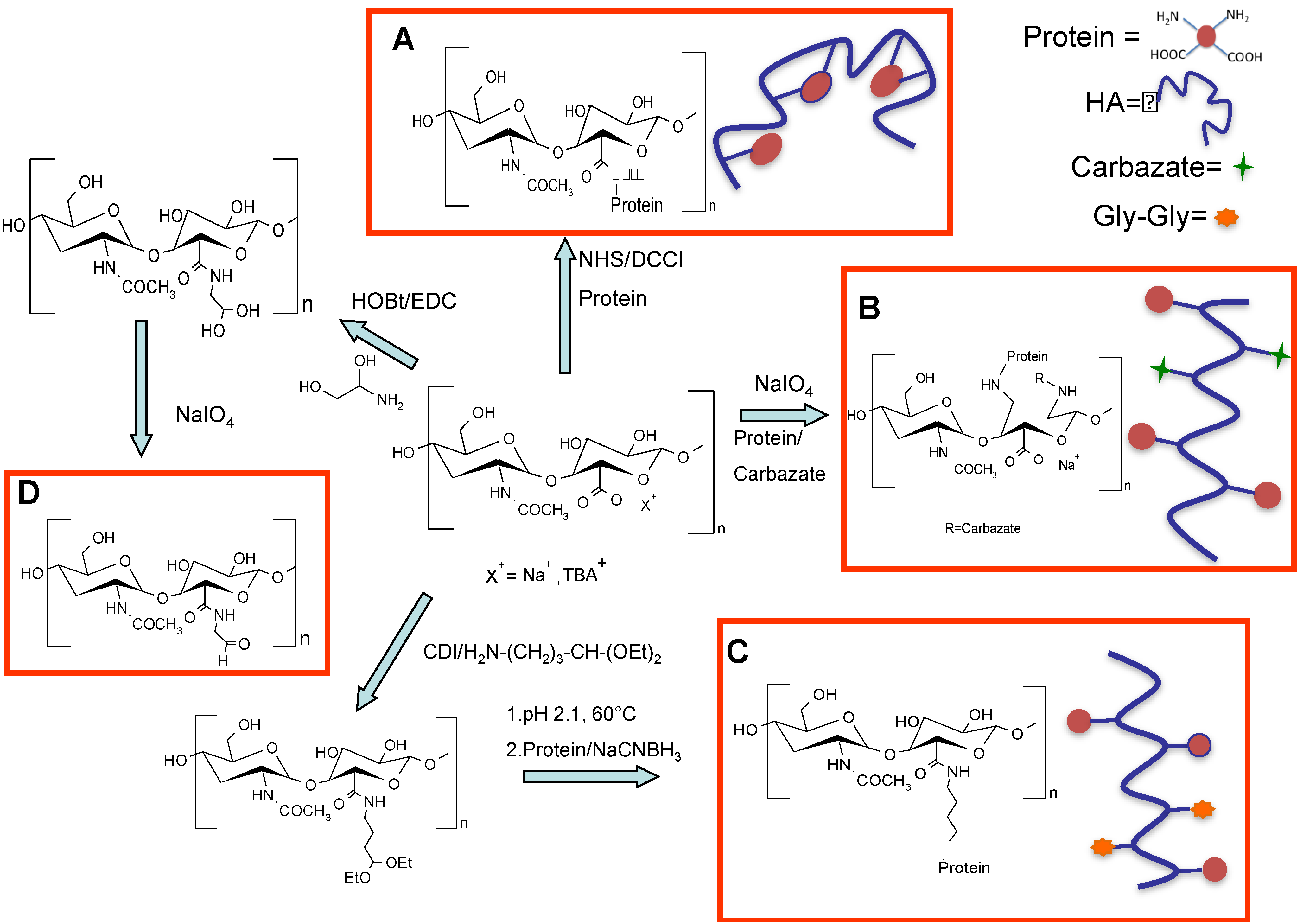
Figure 11.
Schematic representation of different HA activation methods and the following conjugation to proteins: (
A) random conjugation of amino groups of proteins to carboxylic groups of HA [
24]; (
B) Slight HA oxidation in the presence of periodate and conjugation of HA–ALD with
N-terminal primary amine group of proteins [
78,
79]; (
C) Modification of HA with an acetal spacer for selective conjugation to the
N-terminal amino groups after hydrolysis to the active aldehyde of the polymer [
65]; and (
D) Coupling of amino glycerol to HA carboxylic groups and partial oxidation for selective
N-terminal conjugation to proteins [
82].
Figure 11.
Schematic representation of different HA activation methods and the following conjugation to proteins: (
A) random conjugation of amino groups of proteins to carboxylic groups of HA [
24]; (
B) Slight HA oxidation in the presence of periodate and conjugation of HA–ALD with
N-terminal primary amine group of proteins [
78,
79]; (
C) Modification of HA with an acetal spacer for selective conjugation to the
N-terminal amino groups after hydrolysis to the active aldehyde of the polymer [
65]; and (
D) Coupling of amino glycerol to HA carboxylic groups and partial oxidation for selective
N-terminal conjugation to proteins [
82].
Yang and co-workers applied this chemistry for the site-specific modification of human growth hormone (hGH) and interferon-α (IFNα) [
78,
79]. The HA was slightly oxidized in the presence of periodate using a molar ratio of 1:1 for the disaccharide unit. The degree of modification was determined by
1H-NMR after modification of the aldehyde group with carbazate. HA-hGH and HA-IFNα products were successfully synthesized by the coupling reaction of HA–ALD (HA-aldehyde) with the
N-terminal primary amine group of proteins (
Figure 11, Panel B). The number of protein units linked to the polymer was calculated to be in the range of 1–9 molecules, depending on the amount of protein in the starting reaction mixture. The HA–hGH conjugate, for receptor-mediated transdermal delivery, showed
in vivo activity in fibroblasts by elevating the JAK2 levels, the hGH-receptor mediated signaling pathway. The PK study after topical treatment with HA–hGH demonstrated moreover that the conjugate reached the bloodstream, showing an AUC and a bioavailability 10 and 16 fold higher compared to hGH delivered topically.
According to anti-proliferation tests in Daudi cells, in vitro biological activity of HA–IFNα conjugate was lower than that of native IFNα, but comparable to that of PEG-Intron. Dye labeled HA–IFNα conjugate demonstrated an intrinsic liver targeting after administration in mice. Pharmacokinetic analysis revealed that HA–IFNα conjugate has a prolonged residence time, up to five days, depending on the degree of HA modification. Finally, in vivo antiviral activity of the HA–IFNα conjugate was confirmed from the elevated level of OAS 1 (2′,5′-oligoadenylate synthetase 1) in the liver tissues. The higher OAS 1 expression level by the treatment with HA–IFNα conjugate with a low loading than that by high loading HA–IFNα conjugate and PEG-Intron might be attributed to the target-specific delivery of slightly modified HA derivatives to the liver. The two examples have, therefore, demonstrated an increased half-life and a specific liver targeting of HA, which can be exploited to carry the drug to this specific site of action.
However, the approach to slightly oxidize HA presents some difficulties, due to the formation of two close aldehyde groups with a different reactivity, and it would be challenging to determine which is involved in the protein coupling [
65]. Furthermore, it has been shown that the opening of sugar rings by periodate oxidation results in a HA derivative that lacks its cellular recognition properties [
80]. Varghese
et al. proposed a simple and versatile synthetic strategy to graft aldehyde moieties on HA without compromising the structural integrity of the polymer [
81]. Amino glycerol was coupled to some HA carboxylic groups by exploiting the carbodiimide chemistry with hydroxybenzotriazole [
82] (
Figure 11, Panel D). The authors reported that the flexible vicinal diols of glycerol have a faster reaction kinetics of oxidation with respect the C2-C3 vicinal diols in
trans geometry in the HA backbone. The reaction was carried out by using one equivalent of periodate with respect to the disaccharide unit, for a short reaction time (5 min). Under these conditions, the polymer backbone integrity was preserved and only the glycerol unit was oxidized.
Mero and co-workers proposed a different method in order to introduce an aldehyde group in the polymer backbone without altering the polymer integrity [
65]. HA was modified with the 4-amino butyraldehyde diethyl acetal spacer (
Figure 11, Panel C). The acetal spacer was chosen first, because it contains a protected aldehyde functionality that avoids the instability issue of aldehyde groups during long-term storage. In fact, the HA-acetal derivative can be easily deprotected to prepare the aldehyde active form just when required for a protein conjugation. Secondly, the butyraldehyde group is less reactive than other shorter aliphatic aldehydes, namely propyl or ethyl aldehydes [
83], thus allowing for a better selectivity of the coupling reaction towards the protein
N-terminus. Thirdly, the degree of functionalization can be easily determined by
1H-NMR, where the typical signal of the methyl moieties in the acetal group is compared to the acetyl signal of HA. The applicability of the method was firstly verified on model proteins, trypsin and ribonuclease. HA-acetal, hydrolyzed in acidic conditions to aldehyde, was directly added to protein solutions, and then, the conjugates were purified by gel filtration chromatography.
In vitro studies demonstrated that the polymer does not affect both the enzymatic activity of conjugated enzymes and their thermal stabilities. In particular, for trypsin, the polymer reduces the typical autolysis process, and after incubation in physiological conditions at 37 °C for 6 h, the free enzyme has lost more than 90% of activity, whereas for the conjugate, the activity was of 34%. HA-acetal was also conjugated to two important pharmaceutical proteins: insulin (INS) and IFNα. Two HA–INS conjugates were synthetized with different drug payloads. The
in vivo study on diabetic rats demonstrated that the conjugate with a minor degree of INS loading had a glucose lowering effect up to 6 h, while free INS exhausted its action after 1 h. Unexpectedly, the HA conjugate with the higher INS loading showed no significant effect on the glucose titer. Probably the higher insulin loading led to a steric entanglement affecting the receptor/protein recognition.
The rationale that has guided the choice of HA conjugation to IFNα resides in the intrinsic HA targeting to the liver, owing to the recognition by the hyaluronan receptors for endocytosis (HARE) present on liver sinusoidal endothelial cells. This selective delivery could increase the efficacy of IFNα therapy for the treatment of hepatitis. HA–IFNα was synthetized using HA-acetal as reported above. The purified product was tested
in vitro for the capacity to inhibit vesicular stomatitis virus replication and cytolysis of VERO target cells, disclosing that the bioconjugate retained about 25% of the original activity [
84].
All these findings encouraged the use of HAylation technology for the delivery of other proteins of pharmaceutical interest.
9. Conclusions
Thanks to its excellent physico-chemical properties, such as biodegradability, biocompatibility, non-toxicity and non-immunogenicity, HA has been widely used and has gained success in an extraordinarily broad range of biomedical applications, such as the treatment of osteoarthritis, wound healing, ocular surgery, plastic surgery and tissue engineering.
This review highlighted the interest of the research community in HA in the field of drug delivery, because it, beyond the above-mentioned properties, has three qualities rarely present in a single polymer, such as an easy chemistry of activation, a high drug payload and an active targeting. The great potential of HA has fueled thorough and vast investigation from both academia and industry. A variety of commercially available preparations of HA derivatives and crosslinked HA materials have been developed for drug delivery.
As reviewed here, there is a broad spectrum of options for HA chemical modifications to be used for obtaining a specific targeting and a long-lasting delivery of various pharmaceuticals, including proteins, peptides and drugs. Considering the HA peculiar biodistribution and its specific receptors present on the lymphatic vessel, cartilage and liver, HA-based therapeutic bioconjugates could appear suitable for loco-regional treatments or be effectively applied for vaccines, this offering great promise in various fields of medicine.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}