DNA-Promoted Auto-Assembly of Gold Nanoparticles: Effect of the DNA Sequence on the Stability of the Assemblies

Abstract
:1. Introduction

2. Experimental Section
2.1. Synthesis of AuNP
2.2. Functionalization of AuNP

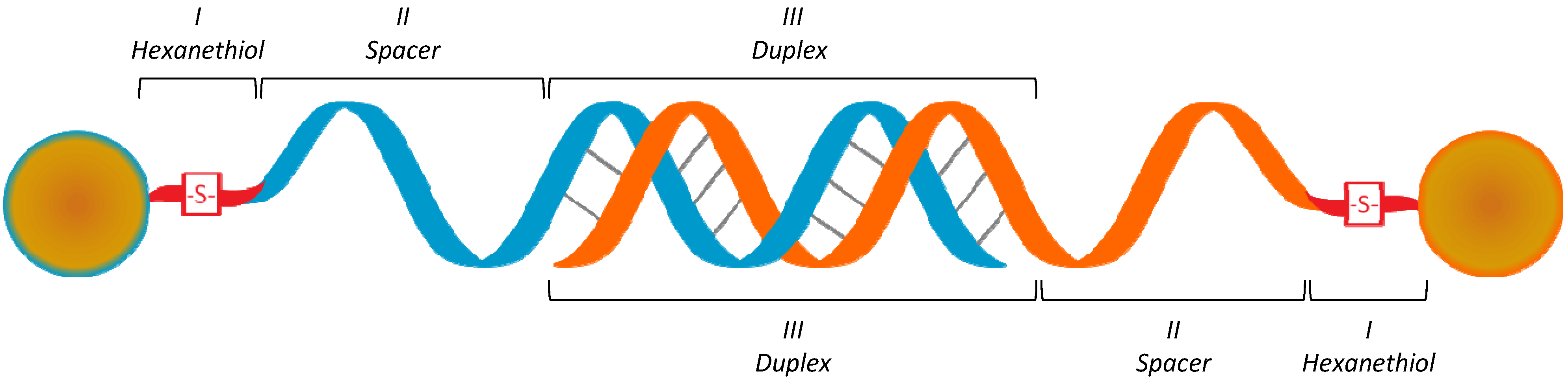{kind=link}
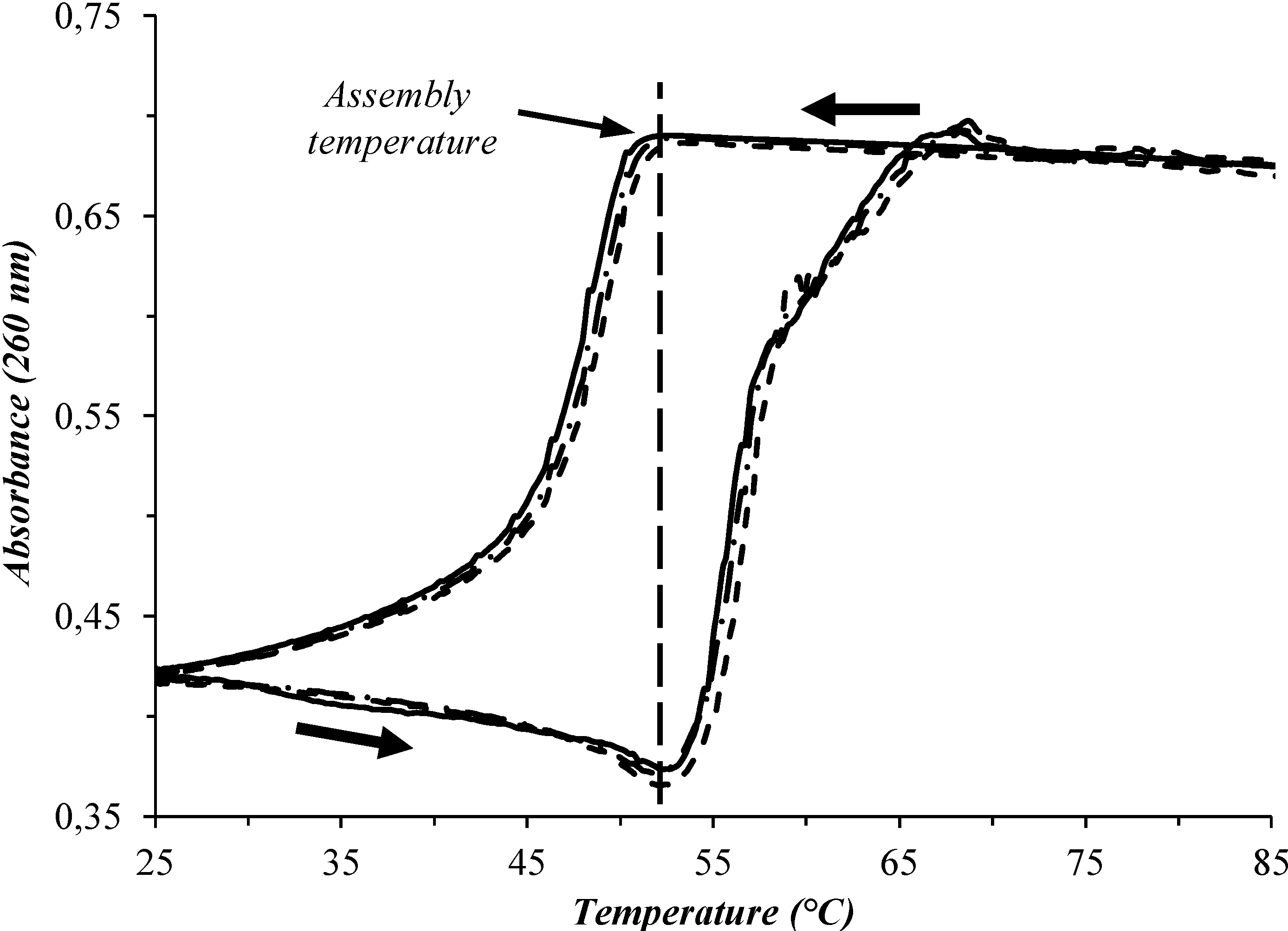{kind=link}
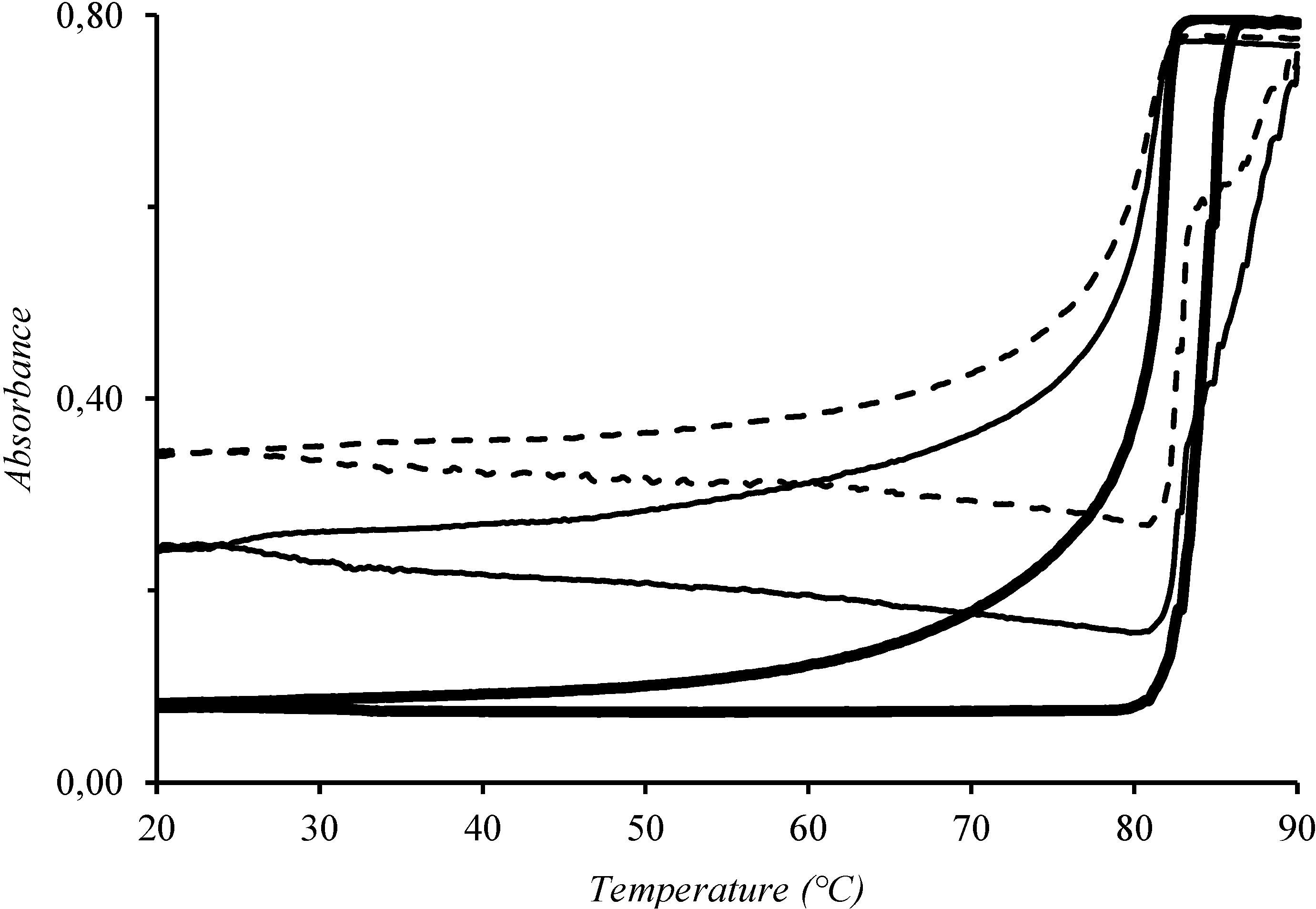{kind=link}
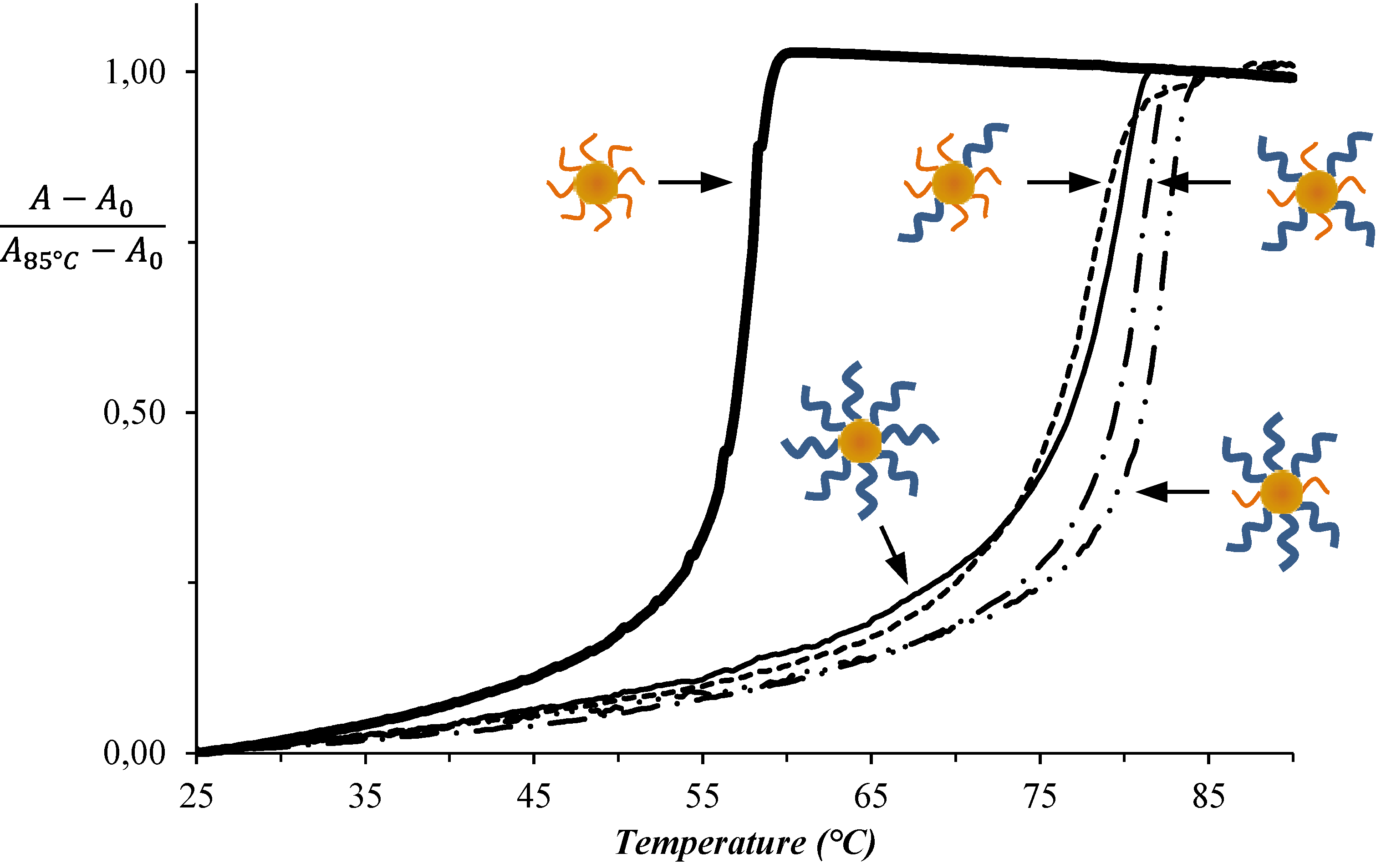{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | Sequence |
|---|---|
| DNAA11 | thiolC6-5′-CGCACACACGC-3′ |
| DNAT11 | thiolC6-5′-GCGTGTGTGCG-3′ |
| T10-DNAA11 | thiolC6-5′-TTTTTTTTTTCGCACACACGC-3′ |
| A10-DNAA11 | thiolC6-5′-AAAAAAAAAACGCACACACGC-3′ |
| T10-DNAT11 | thiolC6-5′-TTTTTTTTTTGCGTGTGTGCG-3′ |
| A10-DNAT11 | thiolC6-5′-AAAAAAAAAAGCGTGTGTGCG-3′ |
| T10-DNAA5 | thiolC6-5′-TTTTTTTTTTCGCAC-3′ |
| T10-DNAT5 | thiolC6-5′-TTTTTTTTTTGCGTG-3′ |
2.3. Assembly of AuNP-DNA
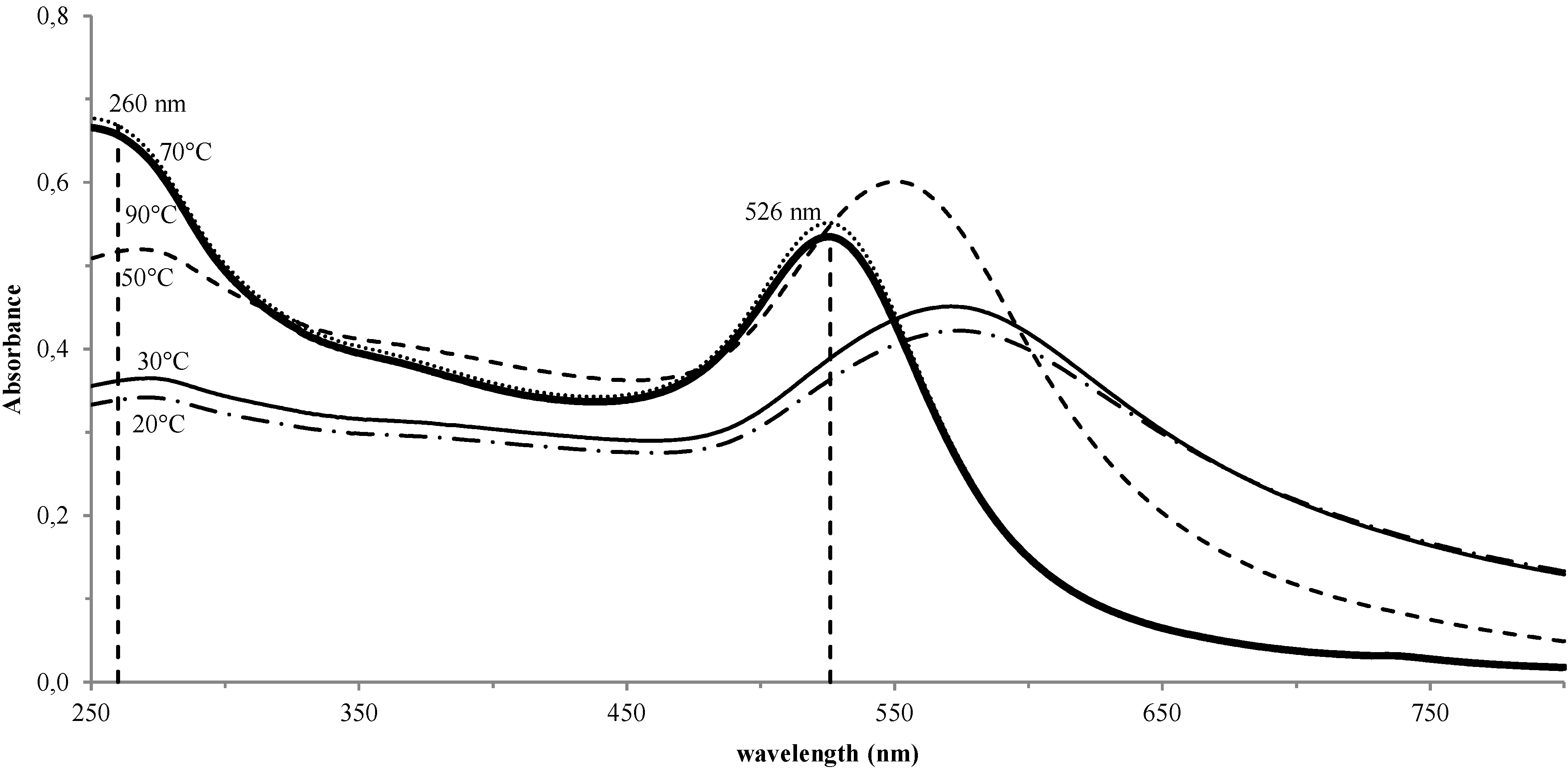
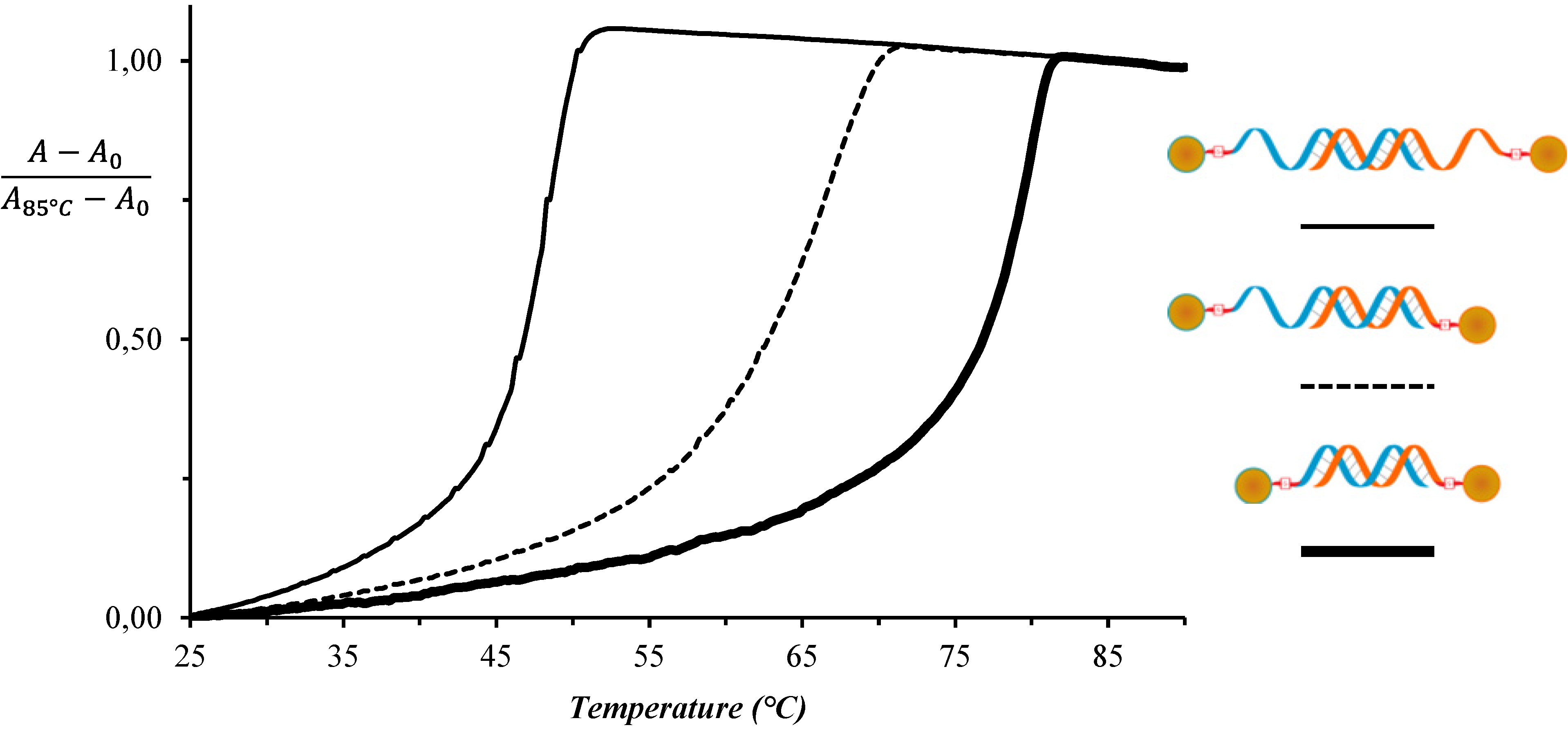
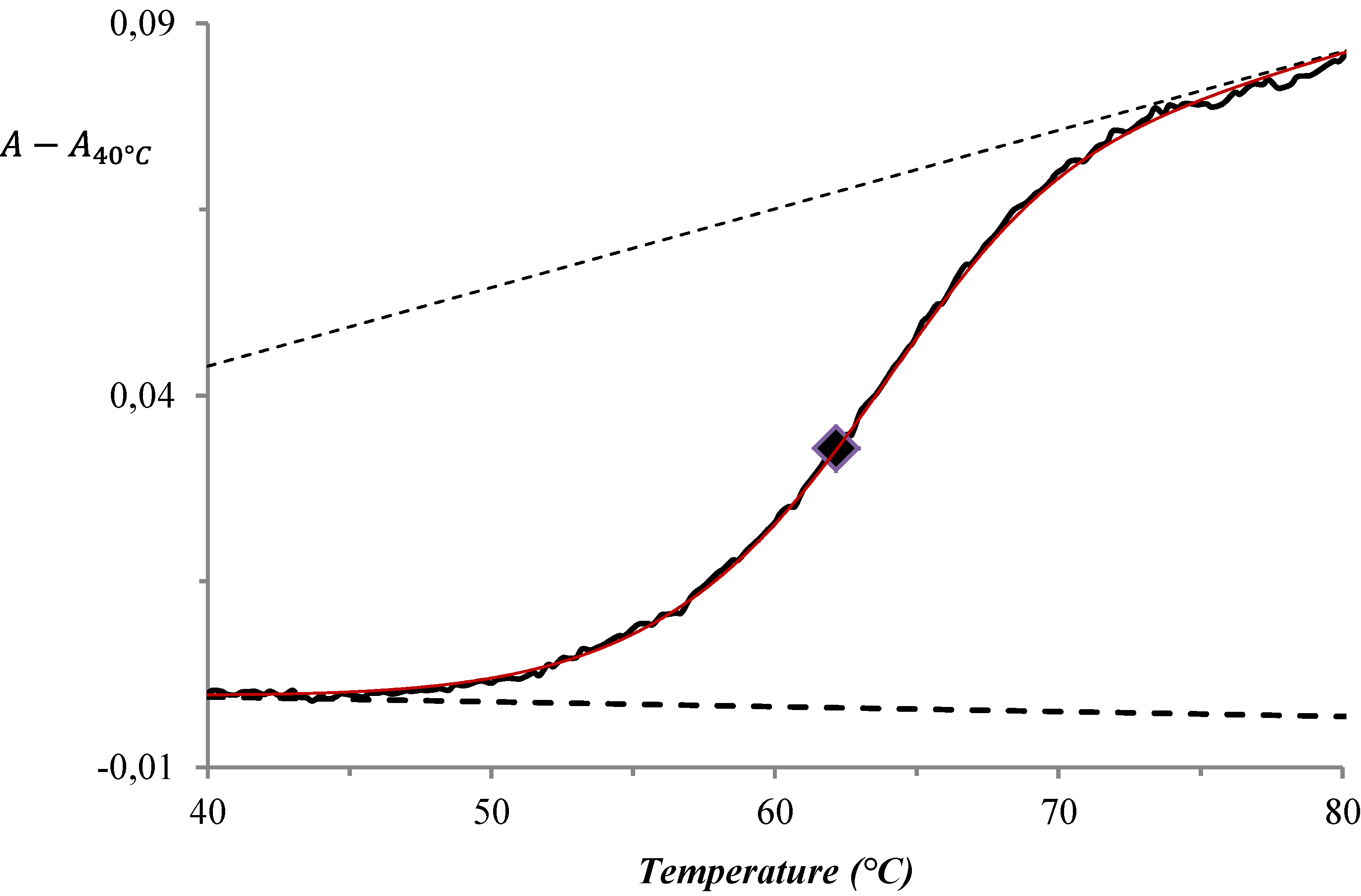
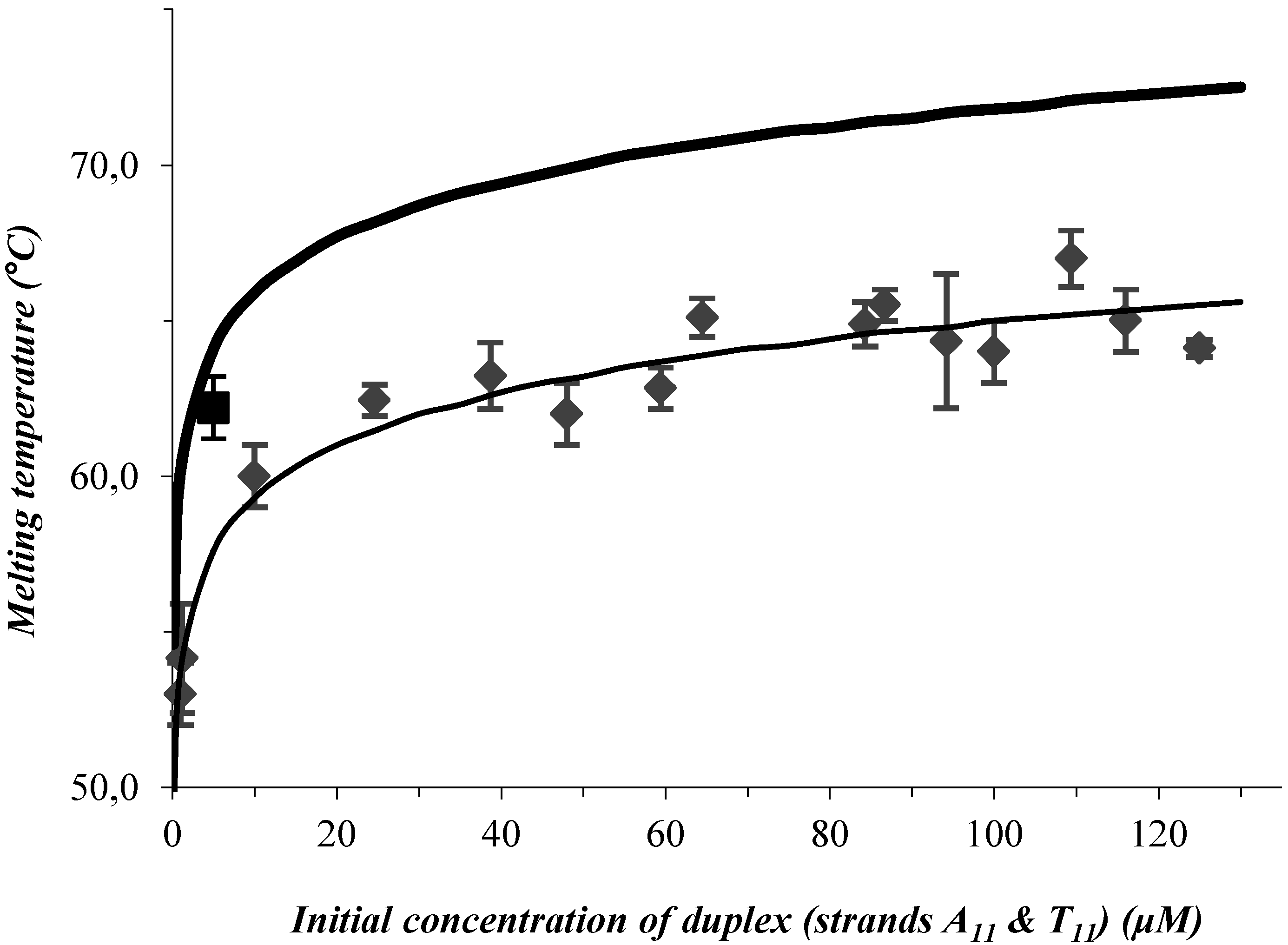
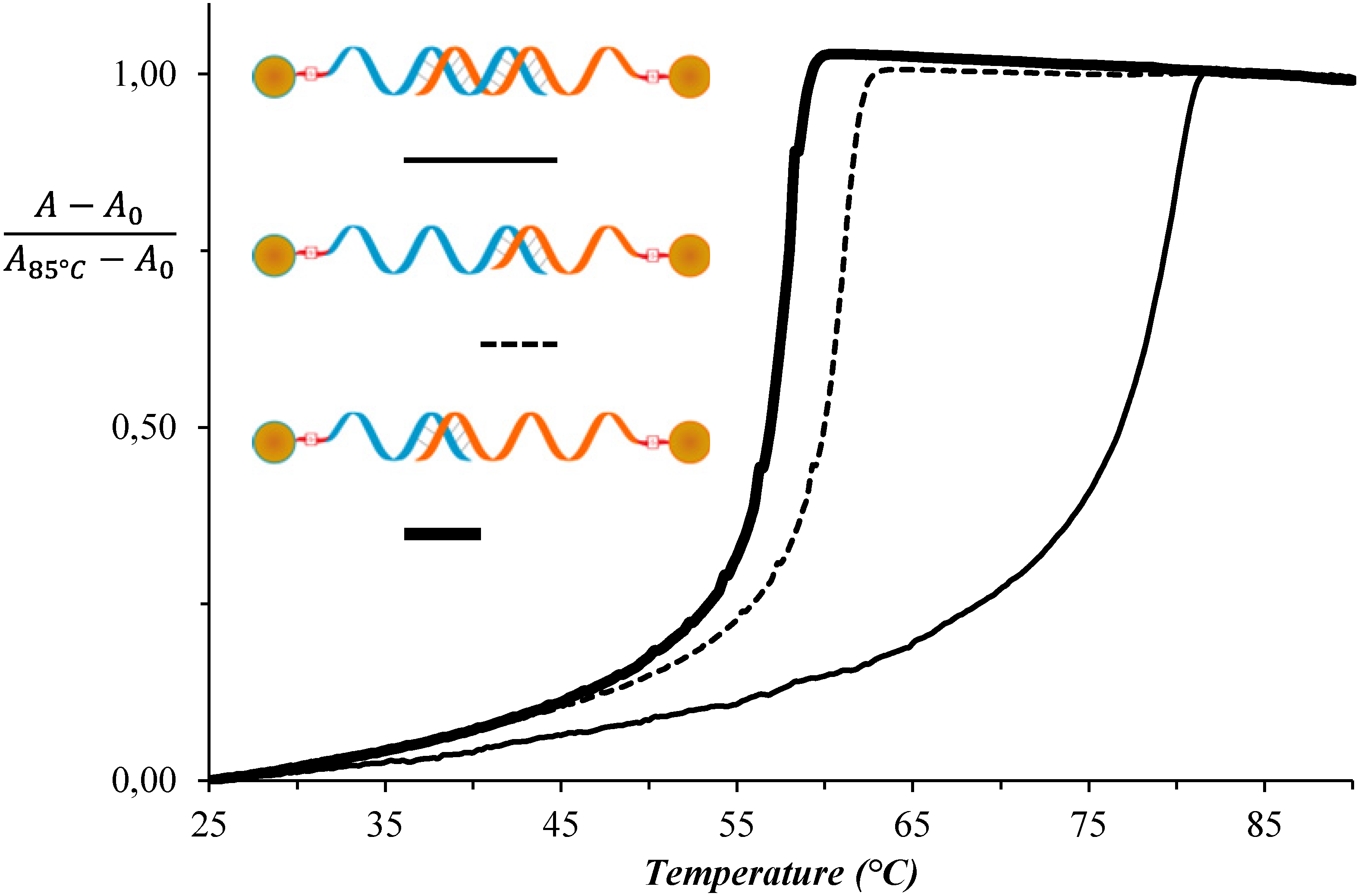
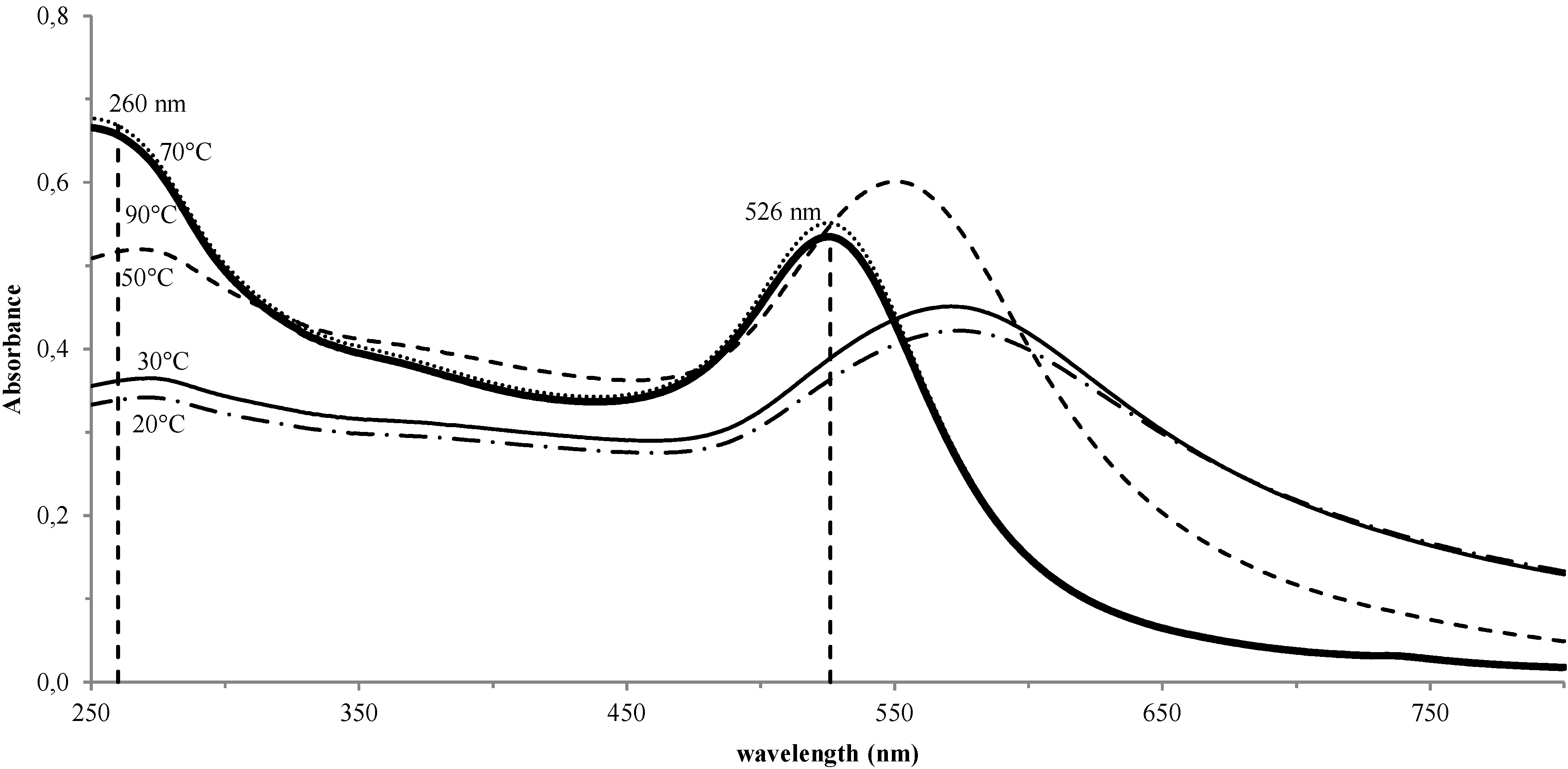
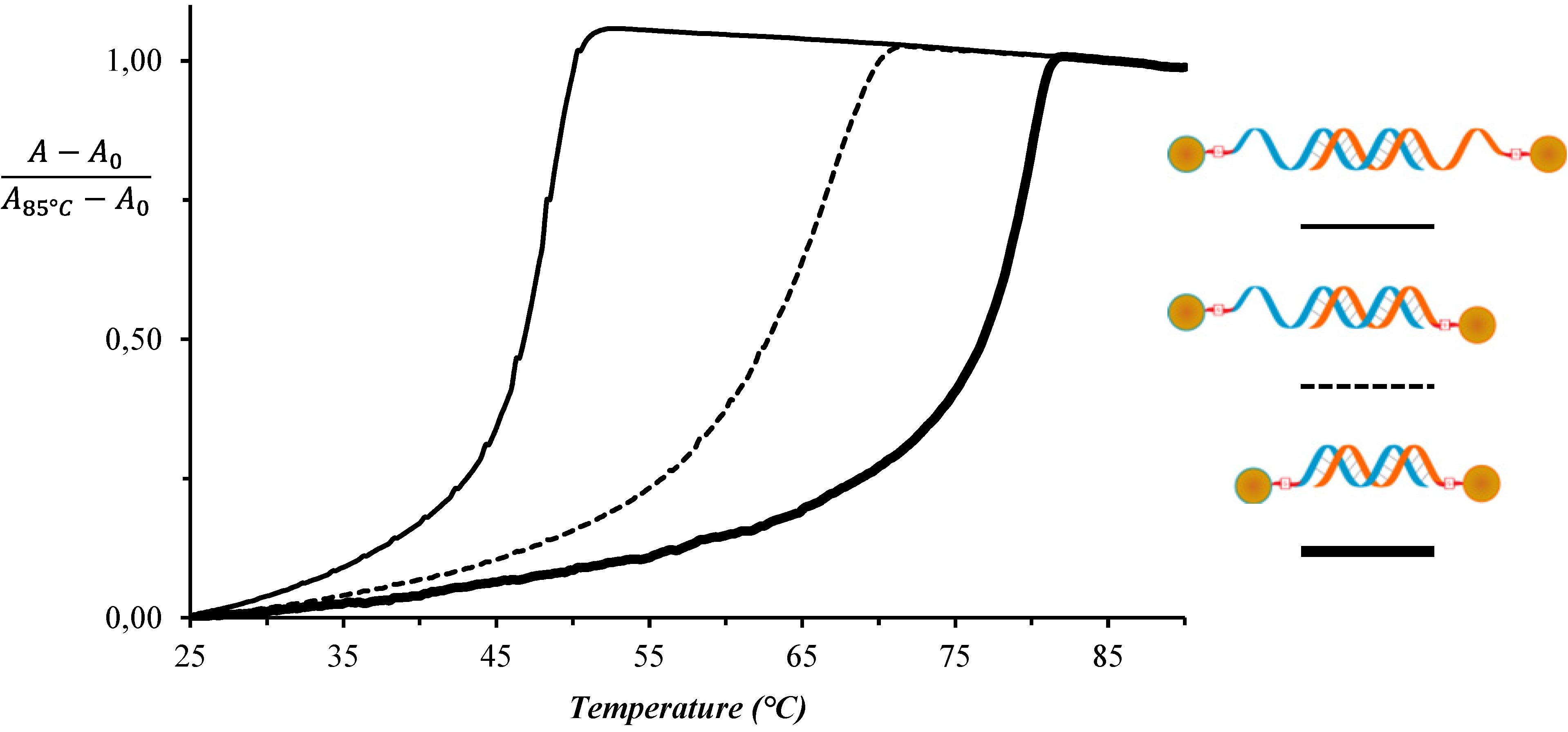
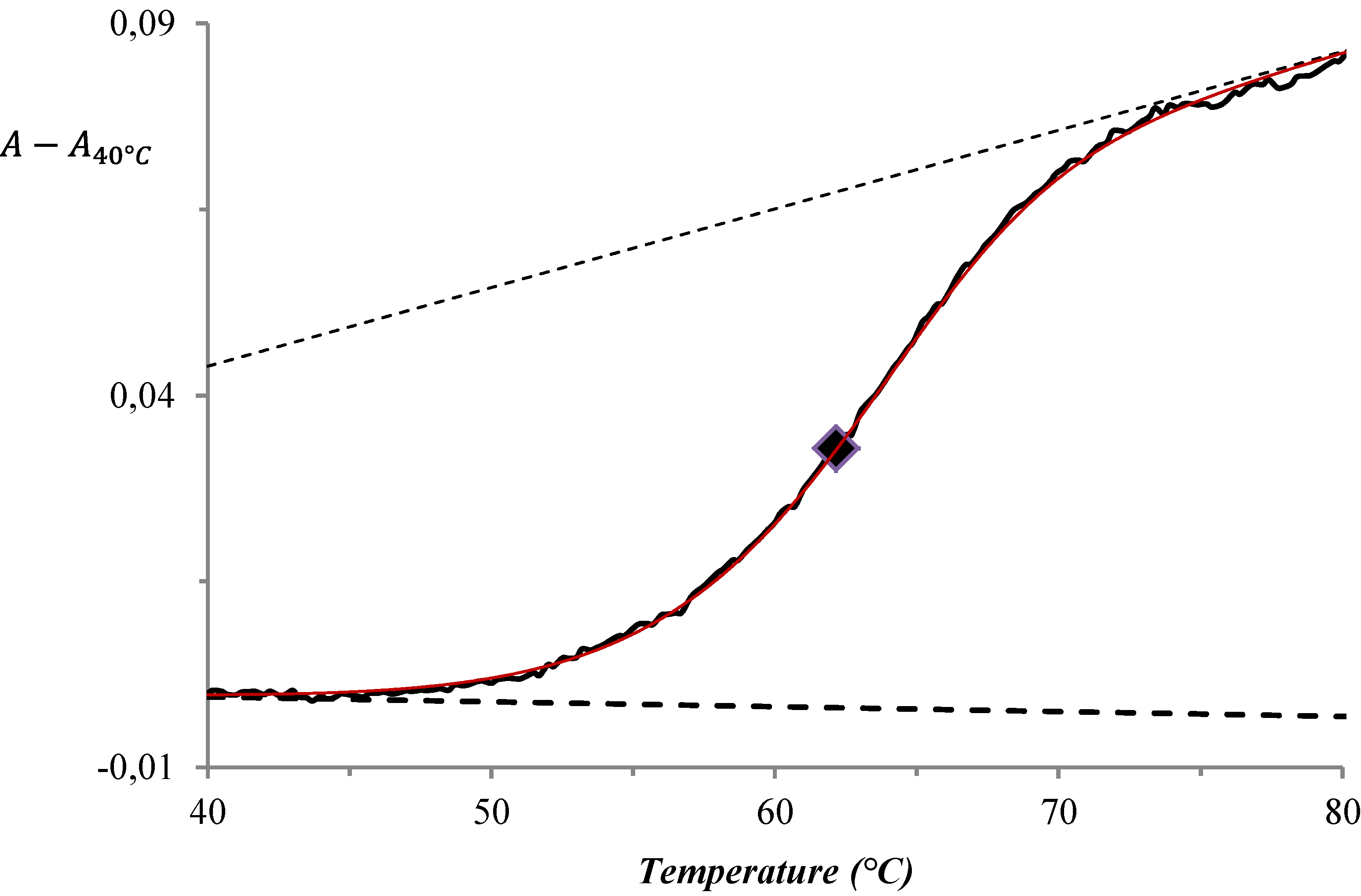
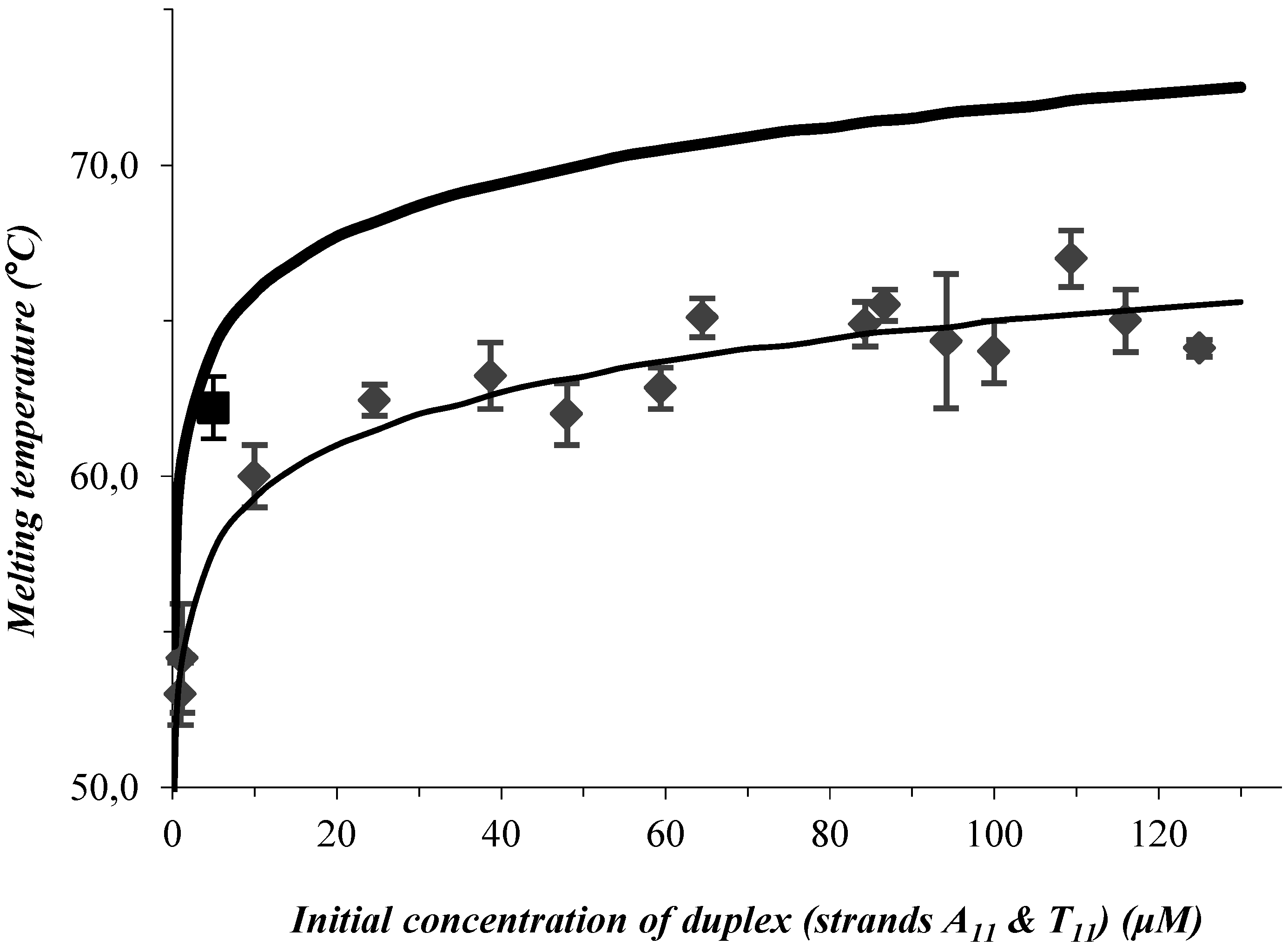
3. Results and Discussion
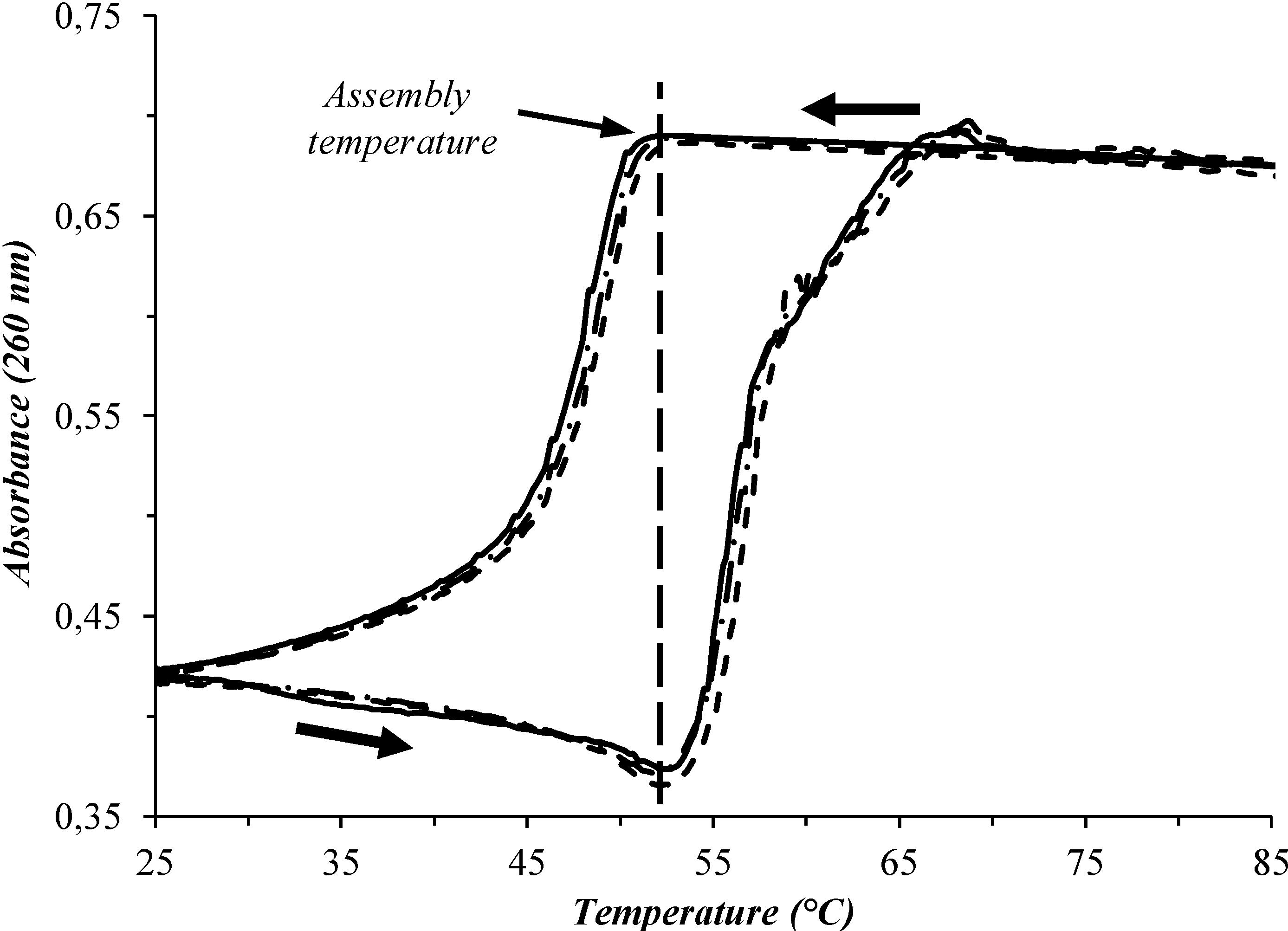

| System No. | AuNP-DNA-1 | AuNP-DNA-2 | TAssembly (°C) |
|---|---|---|---|
| 1 | DNAA11 | DNAT11 | 52 |
| 2 | DNAA11 | T10-DNAT11 | 72 |
| 3 | T10-DNAA11 | T10-DNAT11 | 82 |
| 4 | T10-DNAA11 | T10-DNAT5 | 64 |
| 5 | T10-DNAA5 | T10-DNAT11 | 61 |
| 6 | T10-DNAA11/T10-DNAA5 (25/75) | T10-DNAT11 | 85 |
| 7 | T10-DNAA11/T10-DNAA5 (50/50) | T10-DNAT11 | 84 |
| 8 | T10-DNAA11/T10-DNAA5 (75/25) | T10-DNAT11 | 85 |
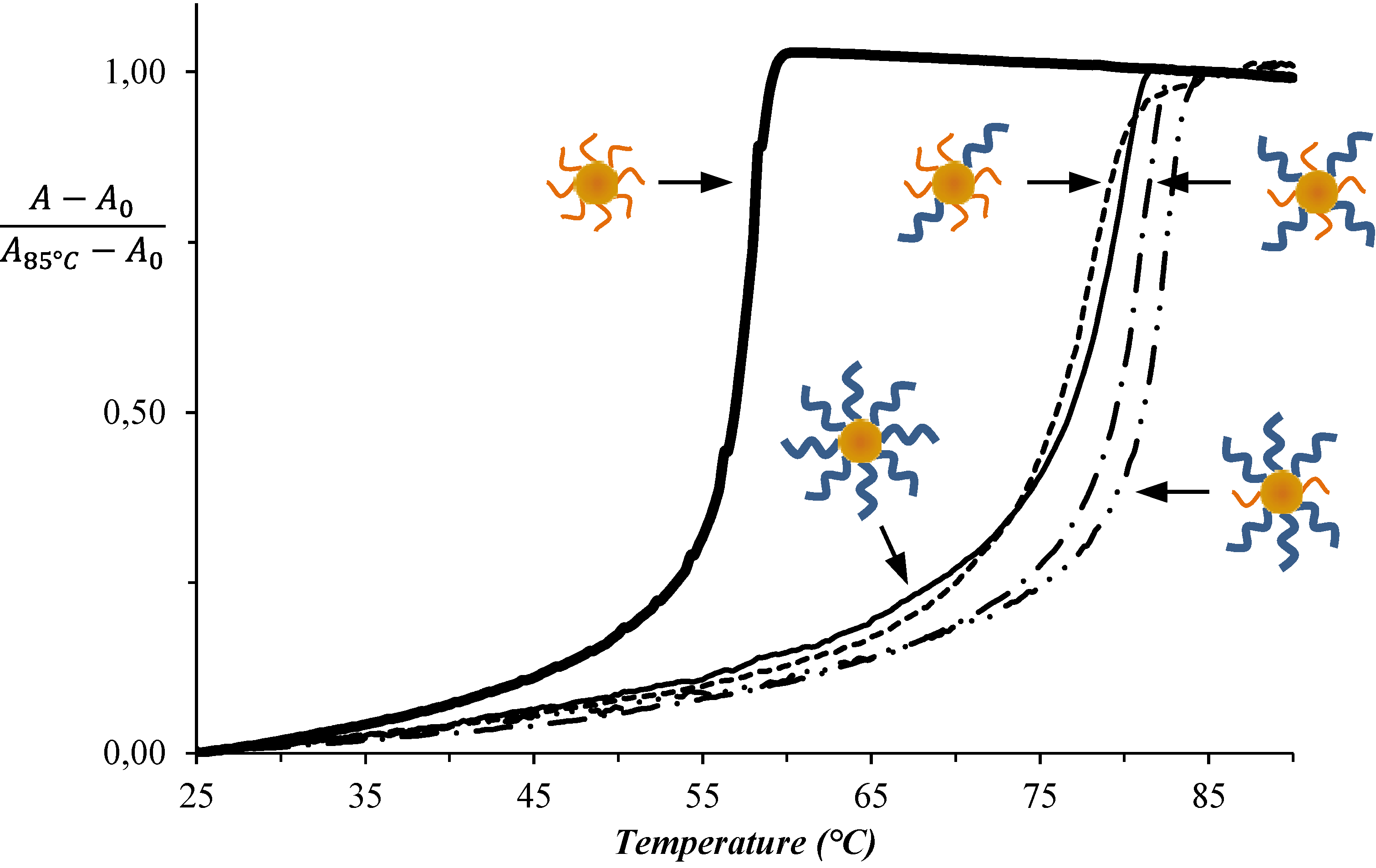
4. Conclusions
Acknowledgments
Conflict of Interest
Appendix
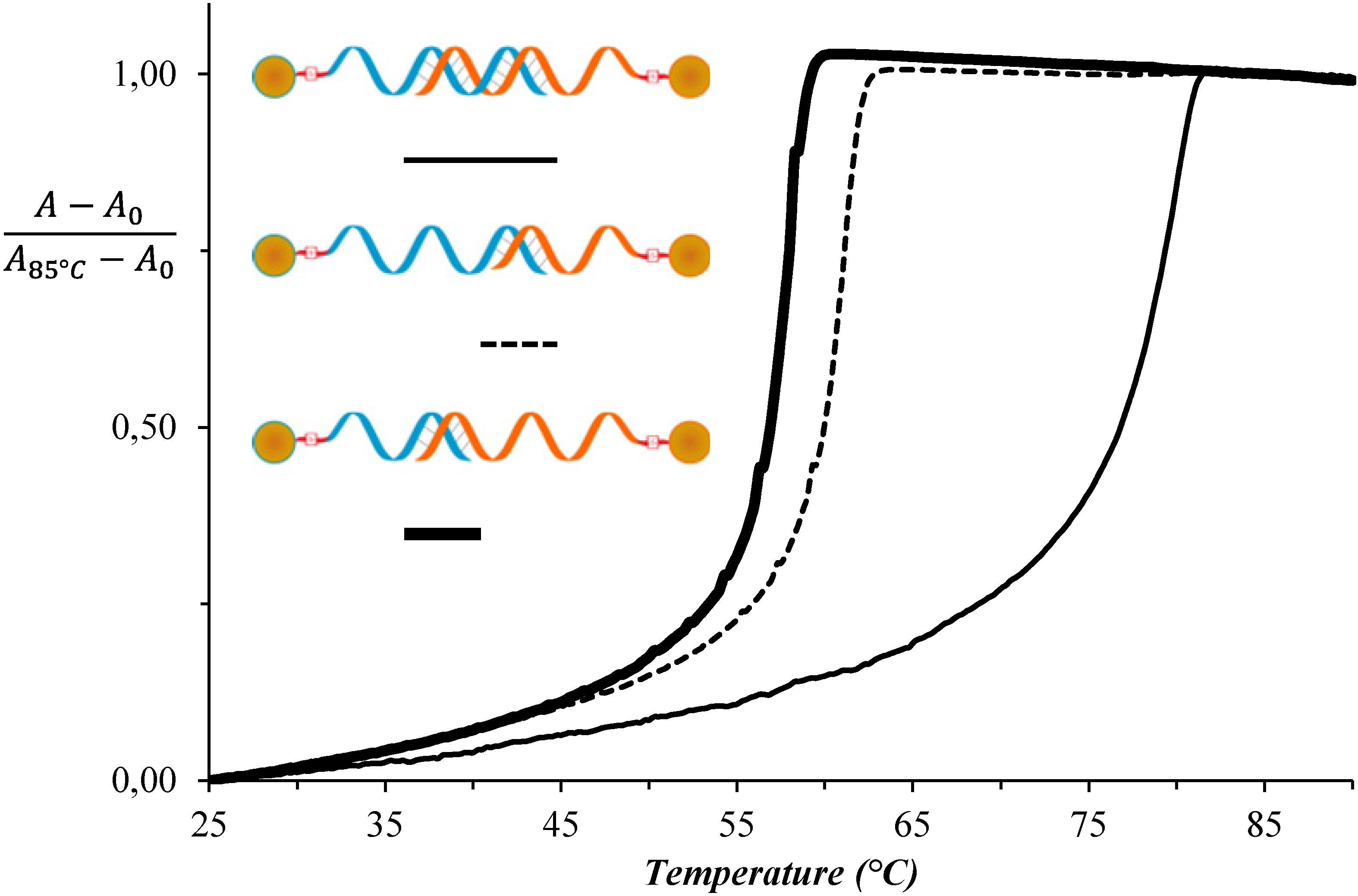
| System No. | DNA-AuNP-1 | DNA-AuNP-2 | TAssembly (°C) |
|---|---|---|---|
| 1 | DNAA11 | DNAT11 | 52 |
| 2 | DNAA11 | T10-DNAT11 | 72 |
| 3 | T10-DNAA11 | T10-DNAT11 | 82 |
| A10-DNAA11 | A10-DNAT11 | 82 | |
| T10-DNAA11 | A10-DNAT11 | 82 | |
| A10-DNAA11 | T10-DNAT11 | 82 | |
| 4 | T10-DNAA11 | T10-DNAT5 | 64 |
| 5 | T10-DNAA5 | T10-DNAT11 | 61 |
| 6 | T10-DNAA11/T10-DNAA5 (25/75) | T10-DNAT11 | 85 |
| 7 | T10-DNAA11/T10-DNAA5 (50/50) | T10-DNAT11 | 84 |
| 8 | T10-DNAA11/T10-DNAA5 (75/25) | T10-DNAT11 | 85 |
References
- Talapin, D.V. LEGO Materials. ACS Nano 2008, 2, 1097–1100. [Google Scholar] [CrossRef]
- Pileni, M.P. Supracrystals of inorganic nanocrystals: An open challenge for new physical properties. Acc. Chem. Res. 2008, 41, 1799–1809. [Google Scholar] [CrossRef]
- Shevchenko, E.V.; Talapin, D.V.; Kotov, N.A.; O’Brien, S.; Murray, C.B. Structural diversity in binary nanoparticle superlattices. Nature 2006, 439, 55–59. [Google Scholar] [CrossRef]
- Urban, J.J.; Talapin, D.V.; Shevchenko, E.V.; Kagan, C.R.; Murray, C.B. Synergism in binary nanocrystal superlattices leads to enhanced p-type conductivity in self-assembled PbTe/Ag2Te thin films. Nat. Mater. 2007, 6, 115–121. [Google Scholar] [CrossRef]
- Bruylants, G.; Bartik, K.; Delplancke-Ogletree, M.-P. Growth Kinetics and Controlled Auto-Assembly of Gold Nanoparticles. In Proceedings of the European Conference on Nano-Films, Liège, Belgium, 24 March 2010; p. 5.
- Hutter, E.; Fendler, J.H. Exploitation of localized surface plasmon resonance. Adv. Mater. 2004, 16, 1685–1706. [Google Scholar] [CrossRef]
- Kalsin, A.M.; Fialkowski, M.; Paszewski, M.; Smoukov, S.K.; Bishop, K.J.M.; Grzybowski, B.A. Electrostatic self-assembly of binary nanoparticle crystals with a diamond-like lattice. Science 2006, 312, 420–424. [Google Scholar] [CrossRef]
- Taleb, A.; Russier, V.; Courty, A.; Pileni, M. Collective optical properties of silver nanoparticles organized in two-dimensional superlattices. Phys. Rev. B 1999, 59, 13350–13358. [Google Scholar] [CrossRef]
- Pinna, N.; Maillard, M.; Courty, A.; Russier, V.; Pileni, M. Optical properties of silver nanocrystals self-organized in a two-dimensional superlattice: Substrate effect. Phys. Rev. B 2002, 66, 045415:1–045415:6. [Google Scholar]
- Pileni, M.P.; Lalatonne, Y.; Ingert, D.; Lisiecki, I.; Courty, A. Self assemblies of nanocrystals: Preparation, collective properties and uses. Faraday Discuss. 2004, 125, 251–264. [Google Scholar]
- Chen, C.-F.; Tzeng, S.-D.; Chen, H.-Y.; Lin, K.-J.; Gwo, S. Tunable plasmonic response from alkanethiolate-stabilized gold nanoparticle superlattices: Evidence of near-field coupling. J. Am. Chem. Soc. 2008, 130, 824–826. [Google Scholar] [CrossRef]
- Sidhaye, D.S.; Prasad, B.L.V. Melting Characteristics of superlattices of alkanethiol-capped gold nanoparticles: The “excluded” story of excess thiol. Chem. Mater. 2010, 22, 1680–1685. [Google Scholar] [CrossRef]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar]
- Alivisatos, A.P.; Johnsson, K.P.; Peng, X.; Wilson, T.E.; Loweth, C.J.; Bruchez, M.P.; Schultz, P.G. Organization of ‘nanocrystal molecules’ using DNA. Nat. Lett. 1996, 382, 609–611. [Google Scholar] [CrossRef]
- Mucic, R.C.; Storhoff, J.J.; Mirkin, C.A.; Letsinger, R.L. DNA-directed synthesis of binary nanoparticle network materials. J. Am. Chem. Soc. 1998, 120, 12674–12675. [Google Scholar] [CrossRef]
- Mitchell, G.P.; Mirkin, C.A.; Letsinger, R.L. Programmed assembly of DNA functionalized quantum dots. J. Am. Chem. Soc. 1999, 121, 8122–8123. [Google Scholar] [CrossRef]
- Sun, D.; Gang, O. Binary heterogeneous superlattices assembled from quantum dots and gold nanoparticles with DNA. J. Am. Chem. Soc. 2011, 133, 5252–5254. [Google Scholar] [CrossRef]
- Storhoff, J.J.; Mirkin, C.A. Programmed materials synthesis with DNA. Chem. Rev. 1999, 99, 1849–1862. [Google Scholar] [CrossRef]
- Mirkin, C.A. Programming the assembly of two- and three-dimensional architectures with DNA and nanoscale inorganic building blocks. Inorg. Chem. 2000, 39, 2258–2272. [Google Scholar] [CrossRef]
- Demers, L.M.; Mirkin, C.A.; Mucic, R.C.; Reynolds, R.A.; Letsinger, R.L.; Elghanian, R.; Viswanadham, G. A fluorescence-based method for determining the surface coverage and hybridization efficiency of thiol-capped oligonucleotides bound to gold thin films and nanoparticles. Anal. Chem. 2000, 72, 5535–5541. [Google Scholar] [CrossRef]
- Jin, R.; Wu, G.; Li, Z.; Mirkin, C. What controls the melting properties of DNA-linked gold nanoparticle assemblies? J. Am. Chem. Soc. 2003, 125, 1643–1654. [Google Scholar] [CrossRef]
- Hurst, S.J.; Lytton-Jean, A.K.R.; Mirkin, C.A. Maximizing DNA loading on a range of gold nanoparticle sizes. Anal. Chem. 2006, 78, 8313–8318. [Google Scholar] [CrossRef]
- Park, S.Y.; Lytton-Jean, A.K.R.; Lee, B.; Weigand, S.; Schatz, G.C.; Mirkin, C.A. DNA-programmable nanoparticle crystallization. Nature 2008, 451, 553–556. [Google Scholar] [CrossRef]
- Hurst, S.J.; Hill, H.D.; Mirkin, C.A. “Three-dimensional hybridization” with polyvalent DNA-gold nanoparticle conjugates. J. Am. Chem. Soc. 2008, 130, 12192–12200. [Google Scholar] [CrossRef]
- Hill, H.D.; Macfarlane, R.J.; Senesi, A.J.; Lee, B.; Park, S.Y.; Mirkin, C.A. Controlling the lattice parameters of gold nanoparticle FCC crystals with duplex DNA linkers. Nano Lett. 2008, 8, 2341–2344. [Google Scholar] [CrossRef]
- Macfarlane, R.J.; Lee, B.; Hill, H.D.; Senesi, A.J.; Seifert, S.; Mirkin, C.A. Assembly and organization processes in DNA-directed colloidal crystallization. PNAS 2009, 106, 10493–10498. [Google Scholar]
- Jones, M.R.; Macfarlane, R.J.; Lee, B.; Zhang, J.; Young, K.L.; Senesi, A.J.; Mirkin, C.A. DNA-nanoparticle superlattices formed from anisotropic building blocks. Nat. Mater. 2010, 9, 913–917. [Google Scholar] [CrossRef]
- Macfarlane, R.J.; Jones, M.R.; Senesi, A.J.; Young, K.L.; Lee, B.; Wu, J.; Mirkin, C.A. Establishing the design rules for DNA-mediated programmable colloidal crystallization. Angew. Chem. Int. Ed. Engl. 2010, 122, 4589–4592. [Google Scholar]
- Macfarlane, R.J.; Lee, B.; Jones, M.R.; Harris, N.; Schatz, G.C.; Mirkin, C.A. Nanoparticle superlattice engineering with DNA. Science 2011, 334, 204–208. [Google Scholar] [CrossRef]
- Maye, M.M.; Nykypanchuk, D.; van der Lelie, D.; Gang, O. A simple method for kinetic control of DNA-induced nanoparticle assembly. J. Am. Chem. Soc. 2006, 128, 14020–14021. [Google Scholar] [CrossRef]
- Maye, M.M.; Nykypanchuk, D.; van der Lelie, D.; Gang, O. DNA-regulated micro- and nanoparticle assembly. Small 2007, 3, 1678–1682. [Google Scholar] [CrossRef]
- Nykypanchuk, D.; Maye, M.M.; van der Lelie, D.; Gang, O. DNA-based approach for interparticle interaction control. Langmuir 2007, 23, 6305–6314. [Google Scholar] [CrossRef]
- Xiong, H.; van der Lelie, D.; Gang, O. DNA linker-mediated crystallization of nanocolloids. J. Am. Chem. Soc. 2008, 130, 2442–2443. [Google Scholar] [CrossRef]
- Maye, M.M.; Kumara, M.T.; Nykypanchuk, D.; Sherman, W.B.; Gang, O. Switching binary states of nanoparticle superlattices and dimer clusters by DNA strands. Nat. Nanotechnol. 2010, 5, 116–120. [Google Scholar] [CrossRef]
- Xiong, H.; Sfeir, M.Y.; Gang, O. Assembly, structure and optical response of three-dimensional dynamically tunable multicomponent superlattices. Nano Lett. 2010, 10, 4456–4462. [Google Scholar] [CrossRef]
- Nykypanchuk, D.; Maye, M.M.; van der Lelie, D.; Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nat. Lett. 2008, 451, 549–552. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Doyen, M.; Bartik, K.; Bruylants, G. UV-Vis and NMR study of the formation of gold nanoparticles by citrate reduction: Observation of gold-citrate aggregates. J. Colloid Interface Sci. 2013, 399, 1–5. [Google Scholar] [CrossRef]
- He, Y.Q.; Liu, S.P.; Kong, L.; Liu, Z.F. A study on the sizes and concentrations of gold nanoparticles by spectra of absorption, resonance Rayleigh scattering and resonance non-linear scattering. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005, 61, 2861–2866. [Google Scholar] [CrossRef]
- Bruylants, G.; Boccongelli, M.; Snoussi, K.; Bartik, K. Comparison of the thermodynamics and base-pair dynamics of a full LNA:DNA duplex and of the isosequential DNA:DNA duplex. Biochemistry 2009, 48, 8473–8482. [Google Scholar] [CrossRef]
- Cahen, P.; Luhmer, M.; Fontaine, C.; Morat, C.; Reisse, J.; Bartik, K. Study by 23Na-NMR, 1H-NMR, and ultraviolet spectroscopy of the thermal stability of an 11-basepair oligonucleotide. Biophys. J. 2000, 78, 1059–1069. [Google Scholar] [CrossRef]
- Cahen, P. Etude de l’atmosphère ionique et de la stabilité de la structure double brin d’oligonucléotides par RMN du 23Na. Ph.D. Thesis, Université Libre de Bruxelles, Bruxelles, Belgium, 28 January 2000. [Google Scholar]
- Braulin, W.H.; Bloomfield, V.A. 1H NMR study of the base-pairing reactions of d(GGAATTCC): Salt effects on the equilibria and kinetics of strand association. Biochemistry 1991, 30, 754–758. [Google Scholar] [CrossRef]
- Lang, B. Hybridization thermodynamics of DNA bound to gold nanoparticles. J. Chem. Thermodyn. 2010, 42, 1435–1440. [Google Scholar] [CrossRef]
- Largo, J.; Starr, F.W.; Sciortino, F. Self-assembling DNA dendrimers: A numerical study. Langmuir 2007, 23, 5896–5905. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Doyen, M.; Bartik, K.; Bruylants, G. DNA-Promoted Auto-Assembly of Gold Nanoparticles: Effect of the DNA Sequence on the Stability of the Assemblies. Polymers 2013, 5, 1041-1055. https://doi.org/10.3390/polym5031041
Doyen M, Bartik K, Bruylants G. DNA-Promoted Auto-Assembly of Gold Nanoparticles: Effect of the DNA Sequence on the Stability of the Assemblies. Polymers. 2013; 5(3):1041-1055. https://doi.org/10.3390/polym5031041
Chicago/Turabian StyleDoyen, Matthieu, Kristin Bartik, and Gilles Bruylants. 2013. "DNA-Promoted Auto-Assembly of Gold Nanoparticles: Effect of the DNA Sequence on the Stability of the Assemblies" Polymers 5, no. 3: 1041-1055. https://doi.org/10.3390/polym5031041