
Lymphoma: Immune Evasion Strategies

{kind=link}
Abstract
:1. Introduction
2. Surface Molecules
2.1. Antigen Presentation Machinery
2.2. Costimulatory and Checkpoint Molecules
2.3. Effector Molecules
2.4. Therapeutic Potential
3. Cellular Regulation
3.1. Regulatory T Cells
3.2. Tumor-Associated Macrophages
3.3. Myeloid-Derived Suppressor Cells
3.4. Stromal Cells
3.5. Therapeutic Potential
4. Soluble Factors
4.1. IDO, Galectins, and Prostaglandins
4.2. Decoy Proteins
4.3. Cytokines
4.4. Chemokines
4.5. Therapeutic Potential
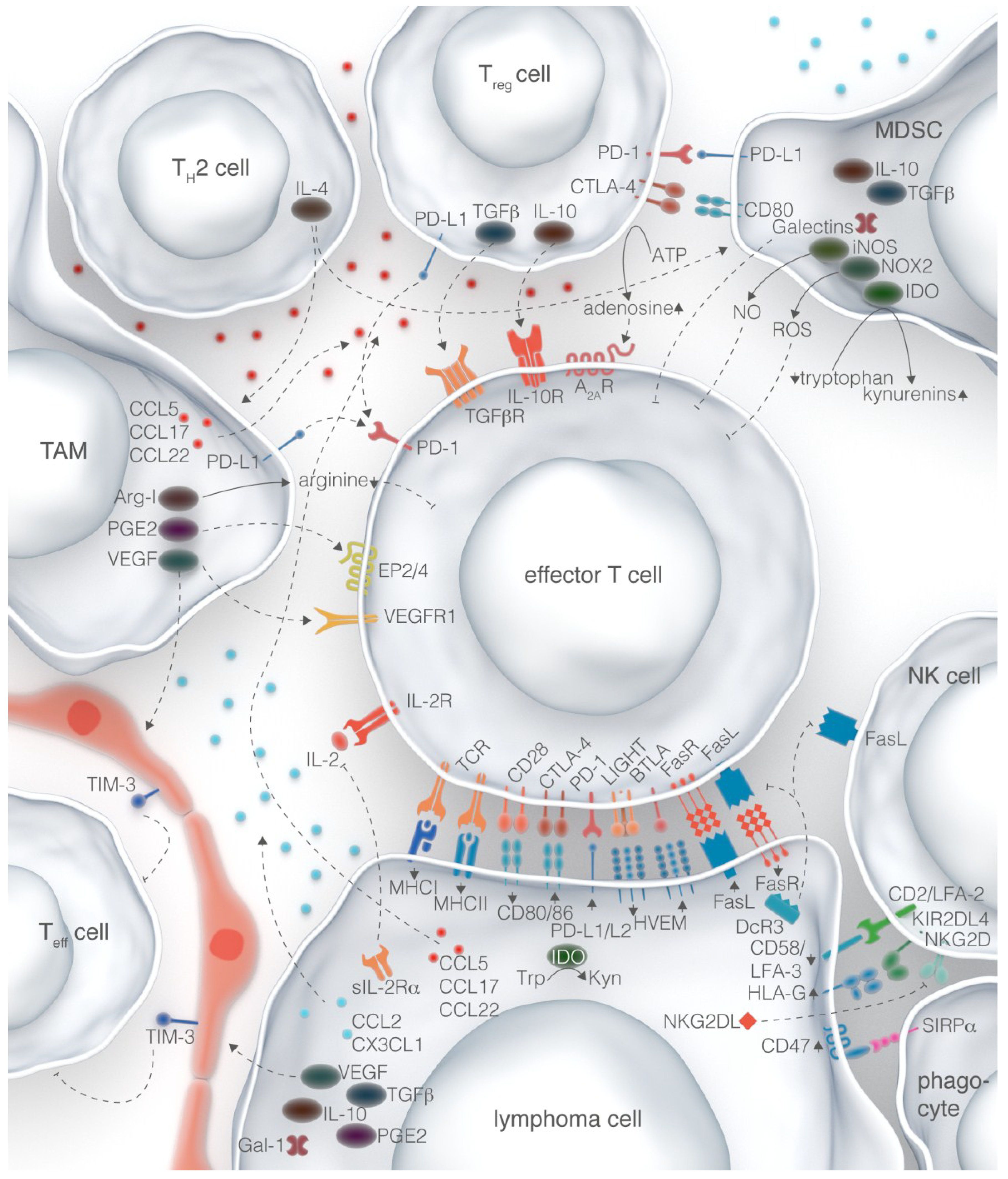
5. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.A.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [PubMed]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 2000, 74, 181–273. [Google Scholar] [PubMed]
- Riemersma, S.A.; Jordanova, E.S.; Schop, R.F.; Philippo, K.; Looijenga, L.H.; Schuuring, E.; Kluin, P.M. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood 2000, 96, 3569–3577. [Google Scholar] [PubMed]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined genetic inactivation of β2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015, 125, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Diepstra, A.; Poppema, S.; Boot, M.; Visser, L.; Nolte, I.M.; Niens, M.; Te Meerman, G.J.; van den Berg, A. HLA-G protein expression as a potential immune escape mechanism in classical Hodgkin’s lymphoma. Tissue Antigens 2008, 71, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Frisan, T.; Levitsky, V.; Masucci, M.G. Variations in proteasome subunit composition and enzymatic activity in B-lymphoma lines and normal B cells. Int. J. Cancer 2000, 88, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Diepstra, A.; van Imhoff, G.W.; Karim-Kos, H.E.; van den Berg, A.; Meermante, G.J.; Niens, M.; Nolte, I.M.; Bastiaannet, E.; Schaapveld, M.; Vellenga, E.; et al. HLA class II expression by Hodgkin Reed-Sternberg cells is an independent prognostic factor in classical Hodgkin’s lymphoma. J. Clin. Oncol. 2007, 25, 3101–3108. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.A.; Wright, G.; Rosenwald, A.R.; Jaramillo, M.A.; Grogan, T.M.; Miller, T.P.; Frutiger, Y.; Chan, W.C.; Gascoyne, R.D.; Ott, G.; et al. Loss of major histocompatibility class II gene and protein expression in primary mediastinal large B-cell lymphoma is highly coordinated and related to poor patient survival. Blood 2006, 108, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Rimsza, L.M.; Roberts, R.A.; Miller, T.P.; Unger, J.M.; LeBlanc, M.; Braziel, R.M.; Weisenberger, D.D.; Chan, W.C.; Muller-Hermelink, H.K.; Jaffe, E.S.; et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: A follow-up study from the Leukemia and Lymphoma molecular profiling project. Blood 2004, 103, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Dueñas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.T.; Vanpatten, K.A.; Fernandez, D.R.; Brunhoeber, P.; Garsha, K.E.; Glinsmann-Gibson, B.J.; Grogan, T.M.; Teruya-Feldstein, J.; Rimsza, L.M. Partial plasma cell differentiation as a mechanism of lost major histocompatibility complex class II expression in diffuse large B-cell lymphoma. Blood 2012, 119, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Elpek, K.G.; Lacelle, C.; Singh, N.P.; Yolcu, E.S.; Shirwan, H. CD4+CD25+ T regulatory cells dominate multiple immune evasion mechanisms in early but not late phases of tumor development in a B cell lymphoma model. J. Immunol. 2007, 178, 6840–6848. [Google Scholar] [CrossRef] [PubMed]
- Stopeck, A.T.; Gessner, A.; Miller, T.P.; Hersh, E.M.; Johnson, C.S.; Cui, H.; Frutiger, Y.; Grogan, T.M. Loss of B7.2 (CD86) and intracellular adhesion molecule 1 (CD54) expression is associated with decreased tumor-infiltrating T lymphocytes in diffuse B-cell large-cell lymphoma. Clin. Cancer Res. 2000, 6, 3904–3909. [Google Scholar] [PubMed]
- Delabie, J.; Ceuppens, J.L.; Vandenberghe, P.; de Boer, M.; Coorevits, L.; de Wolf-Peeters, C. The B7/BB1 antigen is expressed by Reed-Sternberg cells of Hodgkin’s disease and contributes to the stimulating capacity of Hodgkin’s disease-derived cell lines. Blood 1993, 82, 2845–2852. [Google Scholar] [PubMed]
- Suvas, S.; Singh, V.; Sahdev, S.; Vohra, H.; Agrewala, J.N. Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma. J. Biol. Chem. 2002, 277, 7766–7775. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; June, C.H. Putting the brakes on BTLA in T cell-mediated cancer immunotherapy. J. Clin. Investig. 2010, 120, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.-J.J.; Johnson, N.A.; Affleck, J.G.; Severson, T.; Steidl, C.; Ben-Neriah, S.; Schein, J.; Morin, R.D.; Moore, R.; Shah, S.P.; et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010, 70, 9166–9174. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Mallet, F.; Barbarat, B.; Schiano De Colella, J.-M.; Sainty, D.; Sweet, R.W.; Truneh, A.; Olive, D. Stimulation of non-Hodgkin’s lymphoma via HVEM: An alternate and safe way to increase Fas-induced apoptosis and improve tumor immunogenicity. Leukemia 2003, 17, 2500–2507. [Google Scholar] [CrossRef] [PubMed]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Gertner-Dardenne, J.; Fauriat, C.; Orlanducci, F.; Thibult, M.-L.; Pastor, S.; Fitzgibbon, J.; Bouabdallah, R.; Xerri, L.; Olive, D. The co-receptor BTLA negatively regulates human Vγ9Vδ2 T-cell proliferation: A potential way of immune escape for lymphoma cells. Blood 2013, 122, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Greaves, P.; Gribben, J.G. The role of B7 family molecules in hematologic malignancy. Blood 2013, 121, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Nishikori, M.; Kitawaki, T.; Sakai, T.; Hishizawa, M.; Tashima, M.; Kondo, T.; Ohmori, K.; Kurata, M.; Hayashi, T.; et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood 2008, 111, 3220–3224. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.A.; Feldman, A.L.; Wada, D.A.; Yang, Z.-Z.; Comfere, N.I.; Dong, H.; Kwon, E.D.; Novak, A.J.; Markovic, S.N.; Pittelkow, M.R.; et al. B7-H1 (PD-L1, CD274) suppresses host immunity in T-cell lymphoproliferative disorders. Blood 2009, 114, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.M.; Xu, M.L.; Yu, H.; Fletcher, C.D.M.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [PubMed]
- Andorsky, D.J.; Yamada, R.E.; Said, J.; Pinkus, G.S.; Betting, D.J.; Timmerman, J.M. Programmed death ligand 1 is expressed by non-hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin. Cancer Res. 2011, 17, 4232–4244. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Sterle, S.; Zotos, D.; Bernard, N.J.; Scherger, A.K.; Rödling, L.; Alsop, A.E.; Walker, J.; Masson, F.; Belz, G.T.; Corcoran, L.M.; et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat. Med. 2014, 20, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Tsurumi, H.; Goto, N.; Shimizu, M.; Kasahara, S.; Yamada, T.; Kanemura, N.; Hara, T.; Sawada, M.; Saio, M.; et al. Fas and Fas ligand expression on germinal center type-diffuse large B-cell lymphoma is associated with the clinical outcome. Eur. J. Haematol. 2006, 76, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Dutton, A.; O’Neil, J.D.; Milner, A.E.; Reynolds, G.M.; Starczynski, J.; Crocker, J.; Young, L.S.; Murray, P.G. Expression of the cellular FLICE-inhibitory protein (c-FLIP) protects Hodgkin’s lymphoma cells from autonomous Fas-mediated death. Proc. Natl. Acad. Sci. USA. 2004, 101, 6611–6616. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.M.; Scott, D.W. Distinct molecular mechanisms of Fas resistance in murine B lymphoma cells. J. Immunol. 2000, 165, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Lietz, A.; Anagnostopoulos, I.; Hummel, F.; Wiesner, B.; Janz, M.; Jundt, F.; Hirsch, B.; Jöhrens-Leder, K.; Vornlocher, H.-P.; et al. c-FLIP mediates resistance of Hodgkin/Reed-Sternberg cells to death receptor-induced apoptosis. J. Exp. Med. 2004, 199, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Zeytun, A.; Hassuneh, M.; Nagarkatti, M.; Nagarkatti, P.S. Fas-Fas ligand-based interactions between tumor cells and tumor-specific cytotoxic T lymphocytes: A lethal two-way street. Blood 1997, 90, 1952–1959. [Google Scholar] [PubMed]
- Verbeke, C.S.; Wenthe, U.; Grobholz, R.; Zentgraf, H. Fas ligand expression in Hodgkin lymphoma. Am. J. Surg. Pathol. 2001, 25, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47–SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Tang, C.; Pachynski, R.K.; Chin, R.; Majeti, R.; Weissman, I.L. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood 2011, 118, 4890–4901. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, Y.; Qian, J.; Sun, L.; Lu, Y.; Li, H.; Li, Y.; Yang, J.; Cai, Z.; Yi, Q. Toll-like receptor-4 signaling in mantle cell lymphoma: Effects on tumor growth and immune evasion. Cancer 2013, 119, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Clear, A.J.; Kelly, G.; Fatah, R.; Matthews, J.; Macdougall, F.; Lister, T.A.; Lee, A.M.; Calaminici, M.; Gribben, J.G. Follicular lymphoma cells induce T-cell immunologic synapse dysfunction that can be repaired with lenalidomide: Implications for the tumor microenvironment and immunotherapy. Blood 2009, 114, 4713–4720. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Görgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Investig. 2008, 118, 2427–2437. [Google Scholar] [PubMed]
- Görgün, G.; Ramsay, A.G.; Holderried, T.A.W.; Zahrieh, D.; Le Dieu, R.; Liu, F.; Quackenbush, J.; Croce, C.M.; Gribben, J.G. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 6250–6255. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.; Holderried, T.A.W.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: Establishing a reversible immune evasion mechanism in human cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Nagler, A.; Weller, E.A.; Devine, S.M.; Avigan, D.E.; Chen, Y.-B.; Kaminski, M.S.; Holland, H.K.; Winter, J.N.; Mason, J.R.; et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: Results of an international phase II trial. J. Clin. Oncol. 2013, 31, 4199–4206. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R.; Chu, F.; Zhang, M.; Fayad, L.E.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Ansell, S.M.; Lesokhin, A.M.; Halwani, A.; Millenson, M.M.; Schuster, S.J.; Timmerman, J.; Borrello, I.; Gutierrez, M.; Scott, E.C.; et al. Nivolumab in patients with relapsed or refractory hodgkin lymphoma—Preliminary safety, efficacy and biomarker results of a phase I study. Blood 2014, 124, 289. [Google Scholar]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Preliminary results of a phase I study of nivolumab (BMS-936558) in patients with relapsed or refractory lymphoid malignancies. Blood 2014, 124, 291. [Google Scholar]
- Moskowitz, C.H.; Ribrag, V.; Michot, J.-M.; Martinelli, G.; Zinzani, P.L.; Gutierrez, M.; de Maeyer, G.; Jacob, A.G.; Giallella, K.; Weimer Anderson, J.; et al. PD-1 blockade with the monoclonal antibody pembrolizumab (MK-3475) in patients with classical hodgkin lymphoma after brentuximab vedotin failure: Preliminary results from a phase 1b study (KEYNOTE-013). Blood 2014, 124, 290. [Google Scholar]
- O’Mahony, D.; Morris, J.C.; Quinn, C.; Gao, W.; Wilson, W.H.; Gause, B.; Pittaluga, S.; Neelapu, S.; Brown, M.; Fleisher, T.A.; et al. A pilot study of CTLA-4 blockade after cancer vaccine failure in patients with advanced malignancy. Clin. Cancer Res. 2007, 13, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Hurvitz, S.A.; Koenig, P.A.; LaPlant, B.R.; Kabat, B.F.; Fernando, D.; Habermann, T.M.; Inwards, D.J.; Verma, M.; Yamada, R.; et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2009, 15, 6446–6453. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Kim, H.T.; Costello, C.L.; Avigan, D.; Chen, Y.-B.; Armand, P.; Alyea, E.P.; Hedlund, J.; McSweeney, P.A.; Liguori, R.; et al. A multicenter phase I Study of CTLA-4 blockade with ipilimumab for relapsed hematologic malignancies after allogeneic hematopoietic cell transplantation. Blood 2014, 124, 3964. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, S.P.; Restifo, N.P. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.S.; Wright, G.; Tan, B.; Rosenwald, A.; Gascoyne, R.D.; Chan, W.C.; Fisher, R.I.; Braziel, R.M.; Rimsza, L.M.; Grogan, T.M.; et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 2004, 351, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Marshall, N.A.; Duncan, L.; Culligan, D.J.; Barker, R.N.; Vickers, M.A. Local and systemic induction of CD4+CD25+ regulatory T-cell population by non-Hodgkin lymphoma. Blood 2008, 111, 5359–5370. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Novak, A.J.; Stenson, M.J.; Witzig, T.E.; Ansell, S.M. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood 2006, 107, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.A.; Christie, L.E.; Munro, L.R.; Culligan, D.J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood 2004, 103, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Novak, A.J.; Ziesmer, S.C.; Witzig, T.E.; Ansell, S.M. Attenuation of CD8(+) T-cell function by CD4(+)CD25(+) regulatory T cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2006, 66, 10145–10152. [Google Scholar] [CrossRef] [PubMed]
- Brody, J.D.; Ai, W.Z.; Czerwinski, D.K.; Torchia, J.A.; Levy, M.; Advani, R.H.; Kim, Y.H.; Hoppe, R.T.; Knox, S.J.; Shin, L.K.; et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: A phase I/II study. J. Clin. Oncol. 2010, 28, 4324–4332. [Google Scholar] [CrossRef] [PubMed]
- Ai, W.Z.; Hou, J.-Z.; Zeiser, R.; Czerwinski, D.; Negrin, R.S.; Levy, R. Follicular lymphoma B cells induce the conversion of conventional CD4+ T cells to T-regulatory cells. Int. J. Cancer 2009, 124, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Novak, A.J.; Ziesmer, S.C.; Witzig, T.E.; Ansell, S.M. Malignant B cells skew the balance of regulatory T cells and TH17 cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2009, 69, 5522–5530. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Farinha, P.; Masoudi, H.; Skinnider, B.F.; Shumansky, K.; Spinelli, J.J.; Gill, K.; Klasa, R.; Voss, N.; Connors, J.M.; Gascoyne, R.D. Analysis of multiple biomarkers shows that lymphoma-associated macrophage (LAM) content is an independent predictor of survival in follicular lymphoma (FL). Blood 2005, 106, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Zaki, M.A.A.; Hori, Y.; Hashimoto, K.; Tsukaguchi, M.; Tatsumi, Y.; Ishikawa, J.; Tominaga, N.; Sakoda, H.; Take, H.; et al. Osaka lymphoma study group tumour-associated macrophages in diffuse large B-cell lymphoma: A study of the Osaka lymphoma study group. Histopathology 2012, 60, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Schulte, B.C.; Zhou, Y.; Haribhai, D.; Mackinnon, A.C.; Plaza, J.A.; Williams, C.B.; Hwang, S.T. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J. Investig. Dermatol. 2014, 134, 2814–2822. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Lerman, B.; Sakamaki, I.; Wei, G.; Cha, S.C.; Rao, S.S.; Qian, J.; Hailemichael, Y.; Nurieva, R.; Dwyer, K.C.; et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 2014, 20, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Renukaradhya, G.J.; Khan, M.A.; Vieira, M.; Du, W.; Gervay-Hague, J.; Brutkiewicz, R.R. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood 2008, 111, 5637–5645. [Google Scholar] [CrossRef] [PubMed]
- Butsch, R.; Lukas Waelti, S.; Schaerer, S.; Braun, J.; Korol, D.; Probst-Hensch, N.; Moch, H.; Kurrer, M. Intratumoral plasmacytoid dendritic cells associate with increased survival in patients with follicular lymphoma. Leuk. Lymphoma 2011, 52, 1230–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esche, C.; Subbotin, V.M.; Maliszewski, C.; Lotze, M.T.; Shurin, M.R. FLT3 ligand administration inhibits tumor growth in murine melanoma and lymphoma. Cancer Res. 1998, 58, 380–383. [Google Scholar] [PubMed]
- Zhang, Y.-L.; Wei, Y.-J.; Deng, Y.-C.; Wang, Y.-D.; Liu, C.-Z.; Su, L.; Yang, K.-G.; Chen, S.-S. Human Flt3 ligand from Pichia pastoris inhibits growth of lymphoma and colon adenocarcinoma in mice. J. Exp. Ther. Oncol. 2006, 5, 161–166. [Google Scholar] [PubMed]
- Fiore, F.; von Bergwelt-Baildon, M.S.; Drebber, U.; Beyer, M.; Popov, A.; Manzke, O.; Wickenhauser, C.; Baldus, S.E.; Schultze, J.L. Dendritic cells are significantly reduced in non-Hodgkin’s lymphoma and express less CCR7 and CD62L. Leuk. Lymphoma 2006, 47, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Gustafson, M.P.; Bulur, P.A.; Gastineau, D.A.; Witzig, T.E.; Dietz, A.B. Immunosuppressive CD14+HLA-DR(low)/-monocytes in B-cell non-Hodgkin lymphoma. Blood 2011, 117, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Bai, X.; Cao, Y.; Wu, J.; Huang, M.; Tang, D.; Tao, S.; Zhu, T.; Liu, Y.; Yang, Y.; et al. Lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J. Exp. Med. 2010, 207, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. JCO 2001, 19, 376–388. [Google Scholar]
- Dang, N.H.; Hagemeister, F.B.; Pro, B.; McLaughlin, P.; Romaguera, J.E.; Jones, D.; Samuels, B.; Samaniego, F.; Younes, A.; Wang, M.; et al. Phase II study of denileukin diftitox for relapsed/refractory B-cell non-Hodgkin’s lymphoma. JCO 2004, 22, 4095–4102. [Google Scholar] [CrossRef]
- Dang, N.H.; Fayad, L.; McLaughlin, P.; Romaguara, J.E.; Hagemeister, F.; Goy, A.; Neelapu, S.; Samaniego, F.; Walker, P.L.; Wang, M.; et al. Phase II trial of the combination of denileukin diftitox and rituximab for relapsed/refractory B-cell non-Hodgkin lymphoma. Br. J. Haematol. 2007, 138, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Tang, H.; Kurtin, P.J.; Koenig, P.A.; Nowakowski, G.S.; Nikcevich, D.A.; Nelson, G.D.; Yang, Z.; Grote, D.M.; Ziesmer, S.C.; et al. Denileukin diftitox in combination with rituximab for previously untreated follicular B-cell non-Hodgkin’s lymphoma. Leukemia 2012, 26, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Kuzel, T.M.; Li, S.; Eklund, J.; Foss, F.; Gascoyne, R.; Abramson, N.; Schwerkoske, J.F.; Weller, E.; Horning, S.J. Phase II study of denileukin diftitox for previously treated indolent non-Hodgkin lymphoma: Final results of E1497. Leuk. Lymphoma 2007, 48, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, I.; Kwak, L.W.; Cha, S.-C.; Yi, Q.; Lerman, B.; Chen, J.; Surapaneni, S.; Bateman, S.; Qin, H. Lenalidomide enhances the protective effect of a therapeutic vaccine and reverses immune suppression in mice bearing established lymphomas. Leukemia 2014, 28, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Vose, J.M.; Zinzani, P.L.; Reeder, C.B.; Buckstein, R.; Polikoff, J.A.; Bouabdallah, R.; Haioun, C.; Tilly, H.; Guo, P.; et al. An international phase II trial of single-agent lenalidomide for relapsed or refractory aggressive B-cell non-Hodgkin’s lymphoma. Ann. Oncol. 2011, 22, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Davis, R.E.; Rawal, S.; Nastoupil, L.; Hagemeister, F.B.; McLaughlin, P.; Kwak, L.W.; Romaguera, J.E.; Fanale, M.A.; Fayad, L.E.; et al. Safety and activity of lenalidomide and rituximab in untreated indolent lymphoma: An open-label, phase 2 trial. Lancet Oncol. 2014, 15, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Adams, M.; Carter, T.; Chen, R.; Muller, G.; Stirling, D.; Schafer, P.; Bartlett, J.B. lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin. Cancer Res. 2008, 14, 4650–4657. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.; Hernandez-Ilizaliturri, F.J.; Deeb, G.; Roth, M.; Vaughn, M.; Knight, J.; Wallace, P.; Czuczman, M.S. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br. J. Haematol. 2008, 140, 36–45. [Google Scholar] [PubMed]
- Nowakowski, G.S.; LaPlant, B.; Habermann, T.M.; Rivera, C.E.; Macon, W.R.; Inwards, D.J.; Micallef, I.N.; Johnston, P.B.; Porrata, L.F.; Ansell, S.M.; et al. Lenalidomide can be safely combined with R-CHOP (R2CHOP) in the initial chemotherapy for aggressive B-cell lymphomas: Phase I study. Leukemia 2011, 25, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Tilly, H.; Morschhauser, F.; Salles, G.; Casasnovas, R.-O.; Feugier, P.; Molina, T.J.; Jardin, F.; Terriou, L.; Haioun, C.; Coiffier, B. Phase 1b study of lenalidomide in combination with rituximab-CHOP (R2-CHOP) in patients with B-cell lymphoma. Leukemia 2013, 27, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Chiappella, A.; Tucci, A.; Castellino, A.; Pavone, V.; Baldi, I.; Carella, A.M.; Orsucci, L.; Zanni, M.; Salvi, F.; Liberati, A.M.; et al. Fondazione Italiana Linfomi Lenalidomide plus cyclophosphamide, doxorubicin, vincristine, prednisone and rituximab is safe and effective in untreated, elderly patients with diffuse large B-cell lymphoma: A phase I study by the Fondazione Italiana Linfomi. Haematologica 2013, 98, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, U.; Chiappella, A.; Franceschetti, S.; Carella, A.M.; Baldi, I.; Inghirami, G.; Spina, M.; Pavone, V.; Ladetto, M.; Liberati, A.M.; et al. Fondazione Italiana Linfomi Lenalidomide plus R-CHOP21 in elderly patients with untreated diffuse large B-cell lymphoma: Results of the REAL07 open-label, multicentre, phase 2 trial. Lancet Oncol. 2014, 15, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Hara, T.; Tsurumi, H.; Hoshi, M.; Kanemura, N.; Goto, N.; Kasahara, S.; Shimizu, M.; Ito, H.; Saito, K.; et al. Indoleamine 2,3-dioxygenase in tumor tissue indicates prognosis in patients with diffuse large B-cell lymphoma treated with R-CHOP. Ann. Hematol. 2011, 90, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [PubMed]
- Ling, W.; Zhang, J.; Yuan, Z.; Ren, G.; Zhang, L.; Chen, X.; Rabson, A.B.; Roberts, A.I.; Wang, Y.; Shi, Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014, 74, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Amé-Thomas, P.; Maby-El Hajjami, H.; Monvoisin, C.; Jean, R.; Monnier, D.; Caulet-Maugendre, S.; Guillaudeux, T.; Lamy, T.; Fest, T.; Tarte, K. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor B-cell growth: Role of stromal cells in follicular lymphoma pathogenesis. Blood 2007, 109, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Guilloton, F.; Caron, G.; Ménard, C.; Pangault, C.; Amé-Thomas, P.; Dulong, J.; de Vos, J.; Rossille, D.; Henry, C.; Lamy, T.; et al. Mesenchymal stromal cells orchestrate follicular lymphoma cell niche through the CCL2-dependent recruitment and polarization of monocytes. Blood 2012, 119, 2556–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juszczynski, P.; Ouyang, J.; Monti, S.; Rodig, S.J.; Takeyama, K.; Abramson, J.; Chen, W.; Kutok, J.L.; Rabinovich, G.A.; Shipp, M.A. The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2007, 104, 13134–13139. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Moll, G.; Smith, C.; Dua, U.; Lambley, E.; Ramuz, O.; Gill, D.; Marlton, P.; Seymour, J.F.; Khanna, R. Galectin-1 mediated suppression of Epstein-Barr virus specific T-cell immunity in classic Hodgkin lymphoma. Blood 2007, 110, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Murn, J.; Alibert, O.; Wu, N.; Tendil, S.; Gidrol, X. Prostaglandin E2 regulates B cell proliferation through a candidate tumor suppressor, Ptger. J. Exp. Med. 2008, 205, 3091–3103. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Driesen, J.; Classen, S.; Riley, J.L.; Debey, S.; Beyer, M.; Popov, A.; Zander, T.; Schultze, J.L. Prostaglandin E2 impairs CD4+ T cell activation by inhibition of lck: Implications in Hodgkin’s lymphoma. Cancer Res. 2006, 66, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Haraoka, S.; Sugihara, M.; Suzumiya, J.; Kawasaki, C.; Kanda, M.; Kikuchi, M. Amplification and expression of a decoy receptor for fas ligand (DcR3) in virus (EBV or HTLV-I) associated lymphomas. Cancer Lett. 2000, 160, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.M.-H.; Chen, P.-M.; Hsieh, S.-L.; Tzeng, C.-H.; Liu, J.-H.; Chiou, T.-J.; Wang, W.-S.; Yen, C.-C.; Gau, J.-P.; Yang, M.-H. Expression of a soluble decoy receptor 3 in patients with diffuse large B-cell lymphoma predicts clinical outcome. Int. J. Oncol. 2008, 33, 549–554. [Google Scholar] [PubMed]
- Hara, T.; Tsurumi, H.; Takemura, M.; Goto, H.; Yamada, T.; Sawada, M.; Takahashi, T.; Moriwaki, H. Serum-soluble fas level determines clinical symptoms and outcome of patients with aggressive non-Hodgkin’s lymphoma. Am. J. Hematol. 2000, 64, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef] [PubMed]
- Ambrosetti, A.; Nadali, G.; Vinante, F.; Carlini, S.; Veneri, D.; Todeschini, G.; Morosato, L.; de Sabata, D.; Chilosi, M.; Maggi, E. Serum levels of soluble interleukin-2 receptor in Hodgkin disease. Relationship with clinical stage, tumor burden, and treatment outcome. Cancer 1993, 72, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Damle, R.N.; Advani, S.H.; Gangal, S.G. Analysis of regulation of T-cell responses by soluble inhibitory factors from the sera of patients with Hodgkin's disease. Int. J. Cancer 1992, 50, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Voorzanger, N.; Touitou, R.; Garcia, E.; Delecluse, H.J.; Rousset, F.; Joab, I.; Favrot, M.C.; Blay, J.Y. Interleukin (IL)-10 and IL-6 are produced in vivo by non-Hodgkin's lymphoma cells and act as cooperative growth factors. Cancer Res. 1996, 56, 5499–5505. [Google Scholar] [PubMed]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, M.; Minard-Colin, V.; Matsushita, T.; Tedder, T.F. Regulatory B cell production of IL-10 inhibits lymphoma depletion during CD20 immunotherapy in mice. J. Clin. Invest. 2011, 121, 4268–4280. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Xiu, B.; Yates, N.R.; Secreto, F.J.; Hodge, L.S.; Witzig, T.E.; Novak, A.J.; Ansell, S.M. Soluble and membrane-bound TGF-β-mediated regulation of intratumoral T cell differentiation and function in B-cell non-Hodgkin lymphoma. PLoS ONE 2013, 8, e59456. [Google Scholar] [CrossRef] [PubMed]
- Torres-Poveda, K.; Bahena-Román, M.; Madrid-González, C.; Burguete-García, A.I.; Bermúdez-Morales, V.H.; Peralta-Zaragoza, O.; Madrid-Marina, V. Role of IL-10 and TGF-β1 in local immunosuppression in HPV-associated cervical neoplasia. World J. Clin. Oncol. 2014, 5, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Eggle, D.; Driesen, J.; Classen, S.; Riley, J.L.; Debey-Pascher, S.; Beyer, M.; Popov, A.; Zander, T.; Schultze, J.L. RNA fingerprints provide direct evidence for the inhibitory role of TGFbeta and PD-1 on CD4+ T cells in Hodgkin lymphoma. Blood 2007, 110, 3226–3233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-L.; Mourah, S.; Mounier, N.; Leboeuf, C.; Daneshpouy, M.E.; Legrès, L.; Meignin, V.; Oksenhendler, E.; Maignin, C.L.; Calvo, F.; et al. Vascular endothelial growth factor-A is expressed both on lymphoma cells and endothelial cells in angioimmunoblastic T-cell lymphoma and related to lymphoma progression. Lab. Investig. 2004, 84, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Doussis-Anagnostopoulou, I.A.; Talks, K.L.; Turley, H.; Debnam, P.; Tan, D.C.; Mariatos, G.; Gorgoulis, V.; Kittas, C.; Gatter, K.C. Vascular endothelial growth factor (VEGF) is expressed by neoplastic Hodgkin-Reed-Sternberg cells in Hodgkin’s disease. J. Pathol. 2002, 197, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Hazar, B.; Paydas, S.; Zorludemir, S.; Sahin, B.; Tuncer, I. Prognostic significance of microvessel density and vascular endothelial growth factor (VEGF) expression in non-Hodgkin’s lymphoma. Leuk. Lymphoma 2003, 44, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Vose, J.M.; Do, K.-A.; Johnson, M.M.; Manshouri, T.; Bociek, G.; Bierman, P.J.; O’Brien, S.M.; Kantarjian, H.M.; Armitage, J.O.; et al. Clinical relevance of circulating angiogenic factors in patients with non-Hodgkin’s lymphoma or Hodgkin’s lymphoma. Leuk. Res. 2004, 28, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Dincaslan, H.U.; Yavuz, G.; Unal, E.; Tacyildiz, N.; Ikinciogullari, A.; Dogu, F.; Guloglu, D.; Yuksek, N.; Ertem, U. Does serum soluble vascular endothelial growth factor levels have different importance in pediatric acute leukemia and malignant lymphoma patients? Pediatr. Hematol. Oncol. 2010, 27, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Salven, P.; Orpana, A.; Teerenhovi, L.; Joensuu, H. Simultaneous elevation in the serum concentrations of the angiogenic growth factors VEGF and bFGF is an independent predictor of poor prognosis in non-Hodgkin lymphoma: A single-institution study of 200 patients. Blood 2000, 96, 3712–3718. [Google Scholar] [PubMed]
- Fusetti, L.; Pruneri, G.; Gobbi, A.; Rabascio, C.; Carboni, N.; Peccatori, F.; Martinelli, G.; Bertolini, F. Human myeloid and lymphoid malignancies in the non-obese diabetic/severe combined immunodeficiency mouse model: Frequency of apoptotic cells in solid tumors and efficiency and speed of engraftment correlate with vascular endothelial growth factor production. Cancer Res. 2000, 60, 2527–2534. [Google Scholar] [PubMed]
- Wang, E.S.; Teruya-Feldstein, J.; Wu, Y.; Zhu, Z.; Hicklin, D.J.; Moore, M.A.S. Targeting autocrine and paracrine VEGF receptor pathways inhibits human lymphoma xenografts in vivo. Blood 2004, 104, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Lucania, G.; Aleandri, S.; Borgia, G.; Trivedi, P.; Cuomo, L.; Frati, L.; Faggioni, A. Suppression of dendritic cell differentiation through cytokines released by Primary Effusion Lymphoma cells. Immunol. Lett. 2008, 120, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Mazur, G.; Jaskuła, E.; Kryczek, I.; Dłubek, D.; Butrym, A.; Wróbel, T.; Lange, A.; Kuliczkowski, K. Proinflammatory chemokine gene expression influences survival of patients with non-Hodgkin’s lymphoma. Folia Histochem. Cytobiol. 2011, 49, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Corcione, A.; Ottonello, L.; Tortolina, G.; Facchetti, P.; Airoldi, I.; Guglielmino, R.; Dadati, P.; Truini, M.; Sozzani, S.; Dallegri, F.; et al. Stromal cell-derived factor-1 as a chemoattractant for follicular center lymphoma B cells. J. Natl. Cancer Inst. 2000, 92, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Arai, J.; Yasukawa, M.; Yakushijin, Y.; Miyazaki, T.; Fujita, S. Stromal cells in lymph nodes attract B-lymphoma cells via production of stromal cell-derived factor-1. Eur. J. Haematol. 2000, 64, 323–332. [Google Scholar]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Juremalm, M.; Olsson, N.; Backlin, C.; Sundström, C.; Nilsson, K.; Enblad, G.; Nilsson, G. Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg cells and its possible role in the recruitment of mast cells into lymphomatous tissue. Int. J. Cancer 2003, 107, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Molin, D.; Fischer, M.; Xiang, Z.; Larsson, U.; Harvima, I.; Venge, P.; Nilsson, K.; Sundström, C.; Enblad, G.; Nilsson, G. Mast cells express functional CD30 ligand and are the predominant CD30L-positive cells in Hodgkin’s disease. Br. J. Haematol. 2001, 114, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Lorenzon, D.; Cattaruzza, L.; Pinto, A.; Gloghini, A.; Carbone, A.; Colombatti, A. Expression of CCR5 receptors on Reed-Sternberg cells and Hodgkin lymphoma cell lines: Involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int. J. Cancer 2008, 122, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, A.; Visser, L.; Poppema, S. High expression of the CC chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic T-cell infiltratein Hodgkin’s lymphoma. Am. J. Pathol. 1999, 154, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Visser, L.; Roelofsen, H.; de Vries, M.; Diepstra, A.; van Imhoff, G.; van der Wal, T.; Luinge, M.; Alvarez-Llamas, G.; Vos, H.; et al. Proteomics analysis of Hodgkin lymphoma: Identification of new players involved in the cross-talk between HRS cells and infiltrating lymphocytes. Blood 2008, 111, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Malumbres, R.; Nechushtan, H.; Gentles, A.J.; Avisar, E.; Lossos, I.S. Novel IL-21 signaling pathway up-regulates c-Myc and induces apoptosis of diffuse large B-cell lymphomas. Blood 2010, 115, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yee, C. IL-21 mediated Foxp3 suppression leads to enhanced generation of antigen-specific CD8+ cytotoxic T lymphocytes. Blood 2008, 111, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Sarosiek, K.; Parvin, S.; Matthews, J.M.; Zhao, D.; Lossos, I.S. Interleukin-21 potently induces direct and indirect cytotoxicity of mantle cell lymphoma. Blood 2014, 124, 1776. [Google Scholar]
- Timmerman, J.M.; Byrd, J.C.; Andorsky, D.J.; Yamada, R.E.; Kramer, J.; Muthusamy, N.; Hunder, N.; Pagel, J.M. A phase I dose-finding trial of recombinant interleukin-21 and rituximab in relapsed and refractory low grade B-cell lymphoproliferative disorders. Clin. Cancer Res. 2012, 18, 5752–5760. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Geyer, S.M.; Maurer, M.J.; Kurtin, P.J.; Micallef, I.N.M.; Stella, P.; Etzell, P.; Novak, A.J.; Erlichman, C.; Witzig, T.E. Randomized phase II study of interleukin-12 in combination with rituximab in previously treated non-Hodgkin’s lymphoma patients. Clin. Cancer Res. 2006, 12, 6056–6063. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Niki, T.; Hirashima, M.; Novak, A.J.; Witzig, T.E.; Ansell, S.M. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J. Clin. Investig. 2012, 122, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Gluck, W.L.; Hurst, D.; Yuen, A.; Levine, A.M.; Dayton, M.A.; Gockerman, J.P.; Lucas, J.; Denis-Mize, K.; Tong, B.; Navis, D.; et al. Phase I studies of interleukin (IL)-2 and rituximab in B-cell non-hodgkin’s lymphoma: IL-2 mediated natural killer cell expansion correlations with clinical response. Clin. Cancer Res. 2004, 10, 2253–2264. [Google Scholar] [CrossRef] [PubMed]
- Eisenbeis, C.F.; Grainger, A.; Fischer, B.; Baiocchi, R.A.; Carrodeguas, L.; Roychowdhury, S.; Chen, L.; Banks, A.L.; Davis, T.; Young, D.; et al. Combination immunotherapy of B-cell non-Hodgkin’s lymphoma with rituximab and interleukin-2: A preclinical and phase I study. Clin. Cancer Res. 2004, 10, 6101–6110. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, J.W.; Neuberg, D.; Gribben, J.G.; Fisher, D.C.; Canning, C.; Koval, M.; Poor, C.M.; Green, L.M.; Daley, J.; Soiffer, R.; et al. Combination immunotherapy with rituximab and interleukin 2 in patients with relapsed or refractory follicular non-Hodgkin's lymphoma. Br. J. Haematol. 2002, 117, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Andritsos, L.; Byrd, J.C.; Jones, J.A.; Hewes, B.; Kipps, T.J.; Hsu, F.J.; Burger, J.A. Preliminary results from a phase I dose escalation study to determine the maximum tolerated dose of plerixafor in combination with rituximab in patients with relapsed chronic lymphocytic leukemia. ASH Annu. Meet. Abstr. 2010, 116, 2450. [Google Scholar]
- Bertolini, F.; Dell’Agnola, C.; Mancuso, P.; Rabascio, C.; Burlini, A.; Monestiroli, S.; Gobbi, A.; Pruneri, G.; Martinelli, G. CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin’s lymphoma. Cancer Res. 2002, 62, 3106–3112. [Google Scholar] [PubMed]
- Hu, Y.; Gale, M.; Shields, J.; Garron, C.; Swistak, M.; Nguyen, T.-H.; Jacques, G.; Fogle, R.; Siders, W.; Kaplan, J. Enhancement of the anti-tumor activity of therapeutic monoclonal antibodies by CXCR4 antagonists. Leuk. Lymphoma 2012, 53, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Gregory, S.A.; Christos, P.; Martin, P.; Furman, R.R.; Coleman, M.; Leonard, J.P. Long-term follow-up of R-CHOP with bevacizumab as initial therapy for mantle cell lymphoma: Clinical and correlative results. Clin. Lymphoma Myeloma Leuk. 2014, 14, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Pfreundschuh, M.; Trnĕný, M.; Sehn, L.H.; Catalano, J.; Csinady, E.; Moore, N.; Coiffier, B.; MAIN Study Investigators. R-CHOP with or without bevacizumab in patients with previously untreated diffuse large B-cell lymphoma: Final MAIN study outcomes. Haematologica 2014, 99, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Stopeck, A.T.; Unger, J.M.; Rimsza, L.M.; LeBlanc, M.; Farnsworth, B.; Iannone, M.; Glenn, M.J.; Fisher, R.I.; Miller, T.P. A phase 2 trial of standard-dose cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP) and rituximab plus bevacizumab for patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: SWOG. Blood 2012, 120, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.; Zent, C.; Byrd, J.; Erlichman, C.; Laplant, B.; Ghosh, A.; Call, T.; Villalona-Calero, M.; Jelinek, D.; Bowen, D.; et al. Phase II trials of single-agent anti-VEGF therapy for patients with chronic lymphocytic leukemia. Leuk. Lymphoma 2010, 51, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- Stopeck, A.T.; Unger, J.M.; Rimsza, L.M.; Bellamy, W.T.; Iannone, M.; Persky, D.O.; LeBlanc, M.; Fisher, R.I.; Miller, T.P. A phase II trial of single agent bevacizumab in patients with relapsed, aggressive non-Hodgkin lymphoma: Southwest oncology group study. Leuk. Lymphoma 2009, 50, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Ganjoo, K.; Hong, F.; Horning, S.J.; Gascoyne, R.D.; Natkunam, Y.; Swinnen, L.J.; Habermann, T.M.; Kahl, B.S.; Advani, R.H. Bevacizumab and cyclosphosphamide, doxorubicin, vincristine and prednisone in combination for patients with peripheral T-cell or natural killer cell neoplasms: An eastern cooperative Oncology Group study (E2404). Leuk. Lymphoma 2014, 55, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Greco, F.A.; Raefsky, E.L.; Thompson, D.S.; Lunin, S.; Reeves, J.; White, L.; Quinn, R.; DeBusk, L.M.; Flinn, I.W. Rituximab with or without bevacizumab for the treatment of patients with relapsed follicular lymphoma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 277–283. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upadhyay, R.; Hammerich, L.; Peng, P.; Brown, B.; Merad, M.; Brody, J.D. Lymphoma: Immune Evasion Strategies. Cancers 2015, 7, 736-762. https://doi.org/10.3390/cancers7020736
Upadhyay R, Hammerich L, Peng P, Brown B, Merad M, Brody JD. Lymphoma: Immune Evasion Strategies. Cancers. 2015; 7(2):736-762. https://doi.org/10.3390/cancers7020736
Chicago/Turabian StyleUpadhyay, Ranjan, Linda Hammerich, Paul Peng, Brian Brown, Miriam Merad, and Joshua D. Brody. 2015. "Lymphoma: Immune Evasion Strategies" Cancers 7, no. 2: 736-762. https://doi.org/10.3390/cancers7020736