A Phase I Study of the Combination of Temsirolimus with Irinotecan for Metastatic Sarcoma

Abstract
:1. Introduction
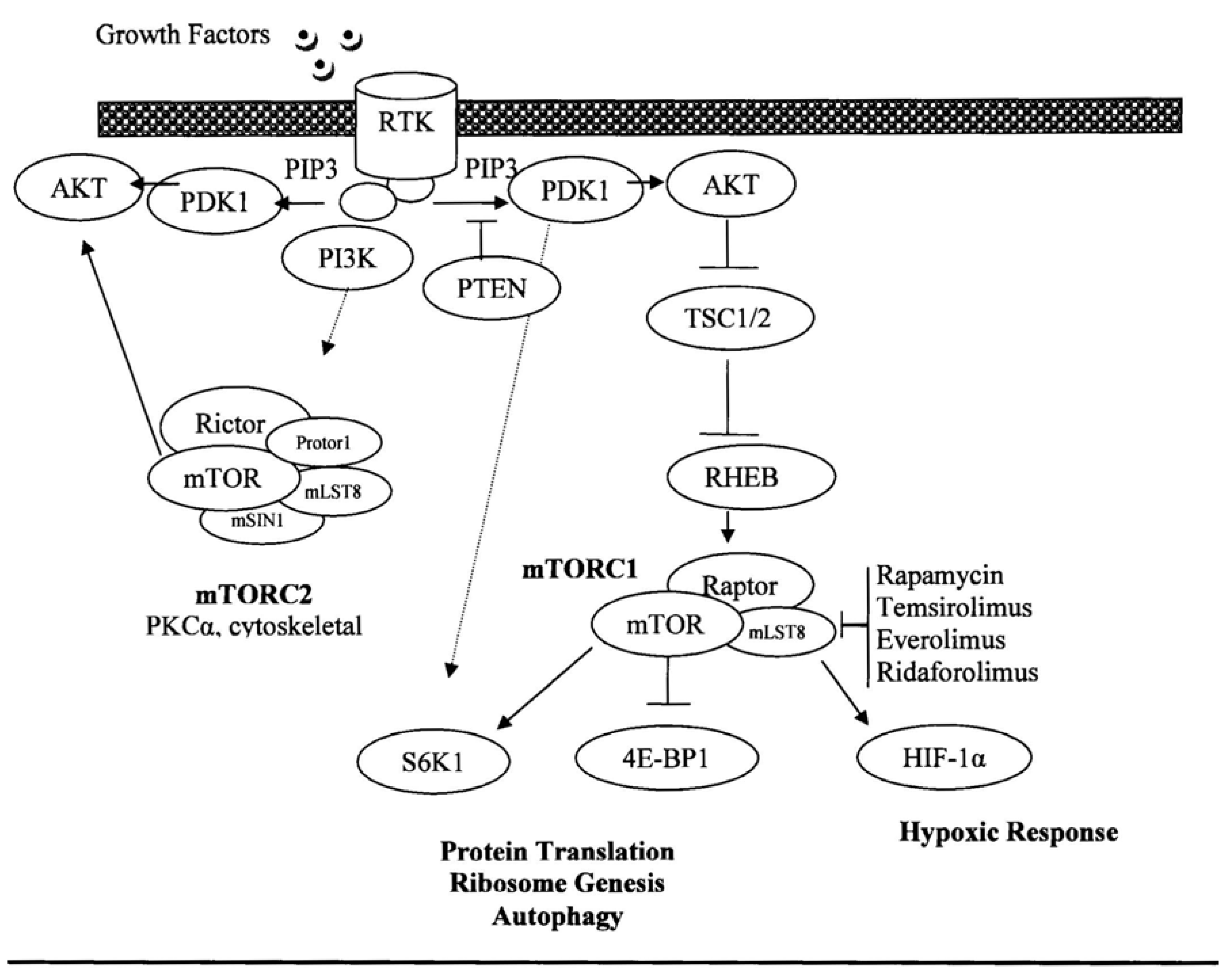
2. Experimental Section
3. Results and Discussion

{kind=link}
| Median Age | 57 years (range, 26–72) | |
| Performance Status | 0 (0–2) | |
| Races/Ethnicities | Non-Hispanic White | 8 |
| Hispanic White | 7 | |
| Black | 2 | |
| Male/Female | ||
| Histology | Undifferentiated sarcoma | 4/1 |
| Leiomyosarcoma | 0/4 | |
| Liposarcoma | 1/2 | |
| Myxofibrosarcoma | 3/0 | |
| Peripheral Nerve Sheath Tumor | 1/0 | |
| Extraosseous osteosarcoma | 1/0 | |
| `Prior Therapies | Surgery | 14 |
| Radiotherapy | 8 | |
| One prior chemotherapy | 13 | |
| More than one chemotherapy | 4 |
| Adverse Event | Cohort | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total/cohort | Percentage for all patients |
|---|---|---|---|---|---|---|---|
| Anemia | Arm B level 1 (N = 3) | 24% | |||||
| Arm A level 1 (N = 6) | 1 | 1 | |||||
| Arm A level 2 (N = 8) | 3 | 3 | |||||
| Neutropenia | Arm B level 1 (N = 3) | 1 | 1 | 2 | 35% | ||
| Arm A level 1 (N = 6) | 1 | 1 | 2 | ||||
| Arm A level 2 (N = 8) | 1 | 1 | 2 | ||||
| Thrombocytopenia | Arm B level 1 (N = 3) | 1 | 1 | 29% | |||
| Arm A level 1 (N = 6) | 1 | 1 | 2 | ||||
| Arm A level 2 (N = 8) | 2 | 2 | |||||
| Fever | Arm B level 1 (N = 3) | 1 | 12% | ||||
| Arm A level 1 (N = 6) | 1 | 1 § | 1 | ||||
| Arm A level 2 (N = 8) | |||||||
| Abdominal pain | Arm B level 1 (N = 3) | 1 | 1 | 12% | |||
| Arm A level 1 (N = 6) | 1 | 1 | |||||
| Arm A level 2 (N = 8) | |||||||
| Anorexia | Arm B level 1 (N = 3) | 2 | 2 | 12% | |||
| Arm A level 1 (N = 6) | |||||||
| Arm A level 2 (N = 8) | |||||||
| Diarrhea | Arm B level 1 (N = 3) | 1 | 1 | 2 | 35% | ||
| Arm A level 1 (N = 6) | 1 | 1 | |||||
| Arm A level 2 (N = 8) | 2 | 1 | 3 | ||||
| Nausea | Arm B level 1 (N = 3) | 2 | 2 | 35% | |||
| Arm A level 1 (N = 6) | 2 | 2 | |||||
| Arm A level 2 (N = 8) | 1 | 1 | 2 | ||||
| Vomiting | Arm B level 1 (N = 3) | 1 | 1 | 1 | 3 | 24% | |
| Arm A level 1 (N = 6) | |||||||
| Arm A level 2 (N = 8) | 1 | 1 | |||||
| Increased transaminases | Arm B level 1 (N = 3) | 6% | |||||
| Arm A level 1 (N = 6) | |||||||
| Arm A level 2 (N = 8) | 1 | 1 | |||||
| Mucositis | Arm B level 1 (N = 3) | 12% | |||||
| Arm A level 1 (N = 6) | 1 | 1 | 2 | ||||
| Arm A level 2 (N = 8) | |||||||
| Fatigue | Arm B level 1 (N = 3) | 1 | 1 | 2 | 35% | ||
| Arm A level 1 (N = 6) | 2 | 1 | 3 | ||||
| Arm A level 2 (N = 8) | 1 | 1 | |||||
| Headache | Arm B level 1 (N = 3) | 1 | 1 | 18% | |||
| Arm A level 1 (N = 6) | 1 | 1 | |||||
| Arm A level 2 (N = 8) | 1 | 1 | |||||
| Rash | Arm B level 1 (N = 3) | 2 | 2 | 29% | |||
| Arm A level 1 (N = 6) | |||||||
| Arm A level 2 (N = 8) | 2 | 1 | 3 | ||||
| Hyperglycemia | Arm B level 1 (N = 3) | 1 | 1 | 12% | |||
| Arm A level 1 (N = 6) | 1 | 1 | |||||
| Arm A level 2 (N = 8) | |||||||
| Muscle weakness | Arm B level 1 (N = 3) | 1 | 1 | 6% | |||
| Arm A level 1 (N = 6) | |||||||
| Arm A level 2 (N = 8) | |||||||
| Sensory neuropathy | Arm B level 1 (N = 3) | 6% | |||||
| Arm A level 1 (N = 6) | |||||||
| Arm A level 2 (N = 8) | 1 | 1 | |||||
| Thrombosis | Arm B level 1 (N = 3) | 6% | |||||
| Arm A level 1 (N = 6) | 1 ↑ | 1 | |||||
| Arm A level 2 (N = 8) | |||||||
| Not recovering counts on re-treatment day or inability to receive a full cycle of treatment on time | Arm B level 1 (N = 3) | 3 | 100% | ||||
| Arm A level 1 (N = 6) | 1 | 18% | |||||
| Arm A level 2 (N = 8) |
4. Conclusions
Acknowledgments
References
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef]
- Cully, M.; Downward, J. Translational responses to growth factors and stress. Biochem. Soc. Trans. 2009, 37, 284–288. [Google Scholar] [CrossRef]
- Sleijfer, S.; van der Graaf, W.T.; Blay, J.Y. Angiogenesis inhibition in non-GIST soft tissue sarcomas. Oncologist 2008, 13, 1193–1200. [Google Scholar] [CrossRef]
- Falcon, B.L.; Barr, S.; Gokhale, P.C.; Chou, J.; Fogarty, J.; Depeille, P.; Miglarese, M.; Epstein, D.M.; McDonald, D.M. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011, 71, 1573–1583. [Google Scholar] [CrossRef]
- Levine, A.J.; Feng, Z.; Mak, T.W.; You, H.; Jin, S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006, 20, 267–275. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hudes, G.R.; Curti, B.D.; McDermott, D.F.; Escudier, B.J.; Negrier, S.; Duclos, B.; Moore, L.; O’Toole, T.; Boni, J.P.; et al. Phase I/II trial of temsirolimus combined with interferon alfa for advanced renal cell carcinoma. J. Clin. Oncol. 2007, 25, 3958–3964. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grunwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Horsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Dickson, M.A.; Schwartz, G.K.; Antonescu, C.R.; Kwiatkowski, D.J.; Malinowska, I.A. Extrarenal perivascular epithelioid cell tumors (PEComas) respond to mTOR inhibition: Clinical and molecular correlates. Int. J. Cancer 2013, 132, 1711–1717. [Google Scholar] [CrossRef]
- Dabora, S.L.; Franz, D.N.; Ashwal, S.; Sagalowsky, A.; DiMario, F.J., Jr.; Miles, D.; Cutler, D.; Krueger, D.; Uppot, R.N.; Rabenou, R.; et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: Kidney angiomyolipomas and other tumors regress and VEGF-D levels decrease. PLoS One 2011, 6, e23379. [Google Scholar] [CrossRef]
- Okuno, S.; Bailey, H.; Mahoney, M.R.; Adkins, D.; Maples, W.; Fitch, T.; Ettinger, D.; Erlichman, C.; Sarkaria, J.N. A phase 2 study of temsirolimus (CCI-779) in patients with soft tissue sarcomas: A study of the Mayo phase 2 consortium (P2C). Cancer 2011, 117, 3468–3475. [Google Scholar]
- Chawla, S.P.; Staddon, A.P.; Baker, L.H.; Schuetze, S.M.; Tolcher, A.W.; D’Amato, G.Z.; Blay, J.Y.; Mita, M.M.; Sankhala, K.K.; Berk, L.; et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J. Clin. Oncol. 2012, 30, 78–84. [Google Scholar] [CrossRef]
- Chawla, S.; Blay, J.; Ray-Coquard, L.; Cesne, A.; Staddon, A.; Milhem, M.; Penel, N.; Riedel, R.; Nguyen, B.; Cranmer, L.; et al. Results of the phase III, placebo-controlled trial (SUCCEED) evaluating the mTOR inhibitor ridaforolimus (R) as maintenance therapy in advanced sarcoma patients (pts) following clinical benefit from prior standard cytotoxic chemotherapy. J. Clin. Oncol. 2011, 29, A10005. [Google Scholar]
- Creel, P.A. Management of mTOR inhibitor side effects. Clin. J. Oncol. Nurs. 2009, 13, 19–23. [Google Scholar] [CrossRef]
- Alagoz, M.; Gilbert, D.C.; El-Khamisy, S.; Chalmers, A.J. DNA repair and resistance to topoisomerase I inhibitors: Mechanisms, biomarkers and therapeutic targets. Curr. Med. Chem. 2012, 19, 3874–3885. [Google Scholar] [CrossRef]
- Jackson, S.P. The DNA-damage response: New molecular insights and new approaches to cancer therapy. Biochem. Soc. Trans. 2009, 37, 483–494. [Google Scholar] [CrossRef]
- Casey, D.A.; Wexler, L.H.; Merchant, M.S.; Chou, A.J.; Merola, P.R.; Price, A.P.; Meyers, P.A. Irinotecan and temozolomide for Ewing sarcoma: The Memorial Sloan-Kettering experience. Pediatr. Blood Cancer 2009, 53, 1029–1034. [Google Scholar] [CrossRef]
- Pappo, A.S.; Lyden, E.; Breitfeld, P.; Donaldson, S.S.; Wiener, E.; Parham, D.; Crews, K.R.; Houghton, P.; Meyer, W.H. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: The Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 362–369. [Google Scholar] [CrossRef]
- Vassal, G.; Couanet, D.; Stockdale, E.; Geoffray, A.; Geoerger, B.; Orbach, D.; Pichon, F.; Gentet, J.C.; Picton, S.; Bergeron, C.; et al. Phase II trial of irinotecan in children with relapsed or refractory rhabdomyosarcoma: A joint study of the French Society of Pediatric Oncology and the United Kingdom Children’s Cancer Study Group. J. Clin. Oncol. 2007, 25, 356–361. [Google Scholar] [CrossRef]
- Bisogno, G.; Riccardi, R.; Ruggiero, A.; Arcamone, G.; Prete, A.; Surico, G.; Provenzi, M.; Bertolini, P.; Paolucci, P.; Carli, M. Phase II study of a protracted irinotecan schedule in children with refractory or recurrent soft tissue sarcoma. Cancer 2006, 106, 703–707. [Google Scholar] [CrossRef]
- Eckhardt, S.G. Irinotecan: A review of the initial phase I trials. Oncology 1998, 12, 31–38. [Google Scholar]
- Pencreach, E.; Guerin, E.; Nicolet, C.; Lelong-Rebel, I.; Voegeli, A.C.; Oudet, P.; Larsen, A.K.; Gaub, M.P.; Guenot, D. Marked activity of irinotecan and rapamycin combination toward colon cancer cells in vivo and in vitro is mediated through cooperative modulation of the mammalian target of rapamycin/hypoxia-inducible factor-1alpha axis. Clin. Cancer Res. 2009, 15, 1297–1307. [Google Scholar] [CrossRef]
- Lee, S.J.; Gounder, M.; Rubin, E.H.; Li, J.M.; Gu, Z.; Thalasila, A.; Loyer, E.; Kudelka, A.P.; Verschraegen, C.F. Optimal modeling for phase I design of a two drug combination-results of a phase I study of cisplatin with 9-nitrocamptothecin. Invest. New Drugs 2008, 26, 541–551. [Google Scholar] [CrossRef]
- Back, J.H.; Rezvani, H.R.; Zhu, Y.; Guyonnet-Duperat, V.; Athar, M.; Ratner, D.; Kim, A.L. Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1. J. Biol. Chem. 2011, 286, 19100–19108. [Google Scholar]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Boni, J.P.; Hug, B.; Leister, C.; Sonnichsen, D. Intravenous temsirolimus in cancer patients: Clinical pharmacology and dosing considerations. Semin. Oncol. 2009, 36, S18–S25. [Google Scholar] [CrossRef]
- Klumpen, H.J.; Beijnen, J.H.; Gurney, H.; Schellens, J.H. Inhibitors of mTOR. Oncologist 2010, 15, 1262–1269. [Google Scholar] [CrossRef]
- Dilling, M.B.; Dias, P.; Shapiro, D.N.; Germain, G.S.; Johnson, R.K.; Houghton, P.J. Rapamycin selectively inhibits the growth of childhood rhabdomyosarcoma cells through inhibition of signaling via the type I insulin-like growth factor receptor. Cancer Res. 1994, 54, 903–907. [Google Scholar]
- Ogawa, T.; Tokuda, M.; Tomizawa, K.; Matsui, H.; Itano, T.; Konishi, R.; Nagahata, S.; Hatase, O. Osteoblastic differentiation is enhanced by rapamycin in rat osteoblast-like osteosarcoma (ROS 17/2.8) cells. Biochem. Biophys. Res. Commun. 1998, 249, 226–230. [Google Scholar] [CrossRef]
- Mateo-Lozano, S.; Tirado, O.M.; Notario, V. Rapamycin induces the fusion-type independent downregulation of the EWS/FLI-1 proteins and inhibits Ewing’s sarcoma cell proliferation. Oncogene 2003, 22, 9282–9287. [Google Scholar] [CrossRef]
- Schuetze, S.; Baker, L.; Maki, R. Sirolimus reduced tumor-related morbidity and resulted in biochemical and radiographic response in patients with progressive sarcoma. J. Clin. Oncol. 2006, 24, A9503. [Google Scholar]
- Mita, M.; Sankhala, K.; Abdel-Karim, I.; Mita, A.; Giles, F. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin. Investig. Drugs 2008, 17, 1947–1954. [Google Scholar] [CrossRef]
- Garrido-Laguna, I.; Janku, F. Ridaforolimus in advanced sarcomas: A leap forward or missed opportunity? J. Clin. Oncol. 2012, 30, 892–893. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Zhao, L.; Chugh, R.; Thomas, D.G.; Lucas, D.R.; Metko, G.; Zalupski, M.M.; Baker, L.H. Results of a phase II study of sirolimus and cyclophosphamide in patients with advanced sarcoma. Eur. J. Cancer 2012, 48, 1347–1353. [Google Scholar] [CrossRef]
- Gibault, L.; Ferreira, C.; Perot, G.; Audebourg, A.; Chibon, F.; Bonnin, S.; Lagarde, P.; Vacher-Lavenu, M.C.; Terrier, P.; Coindre, J.M.; et al. From PTEN loss of expression to RICTOR role in smooth muscle differentiation: Complex involvement of the mTOR pathway in leiomyosarcomas and pleomorphic sarcomas. Mod. Pathol. 2012, 25, 197–211. [Google Scholar]
- Italiano, A.; Kind, M.; Stoeckle, E.; Jones, N.; Coindre, J.M.; Bui, B. Temsirolimus in advanced leiomyosarcomas: Patterns of response and correlation with the activation of the mammalian target of rapamycin pathway. Anticancer Drugs 2011, 22, 463–467. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Verschraegen, C.F.; Movva, S.; Ji, Y.; Schmit, B.; Quinn, R.H.; Liem, B.; Bocklage, T.; Shaheen, M. A Phase I Study of the Combination of Temsirolimus with Irinotecan for Metastatic Sarcoma. Cancers 2013, 5, 418-429. https://doi.org/10.3390/cancers5020418
Verschraegen CF, Movva S, Ji Y, Schmit B, Quinn RH, Liem B, Bocklage T, Shaheen M. A Phase I Study of the Combination of Temsirolimus with Irinotecan for Metastatic Sarcoma. Cancers. 2013; 5(2):418-429. https://doi.org/10.3390/cancers5020418
Chicago/Turabian StyleVerschraegen, Claire F., Sujana Movva, Yongli Ji, Berndt Schmit, Robert H. Quinn, Ben Liem, Therese Bocklage, and Monte Shaheen. 2013. "A Phase I Study of the Combination of Temsirolimus with Irinotecan for Metastatic Sarcoma" Cancers 5, no. 2: 418-429. https://doi.org/10.3390/cancers5020418
APA StyleVerschraegen, C. F., Movva, S., Ji, Y., Schmit, B., Quinn, R. H., Liem, B., Bocklage, T., & Shaheen, M. (2013). A Phase I Study of the Combination of Temsirolimus with Irinotecan for Metastatic Sarcoma. Cancers, 5(2), 418-429. https://doi.org/10.3390/cancers5020418