Succinate Dehydrogenase B Subunit Immunohistochemical Expression Predicts Aggressiveness in Well Differentiated Neuroendocrine Tumors of the Ileum


Abstract
:1. Introduction
2. Results and Discussion

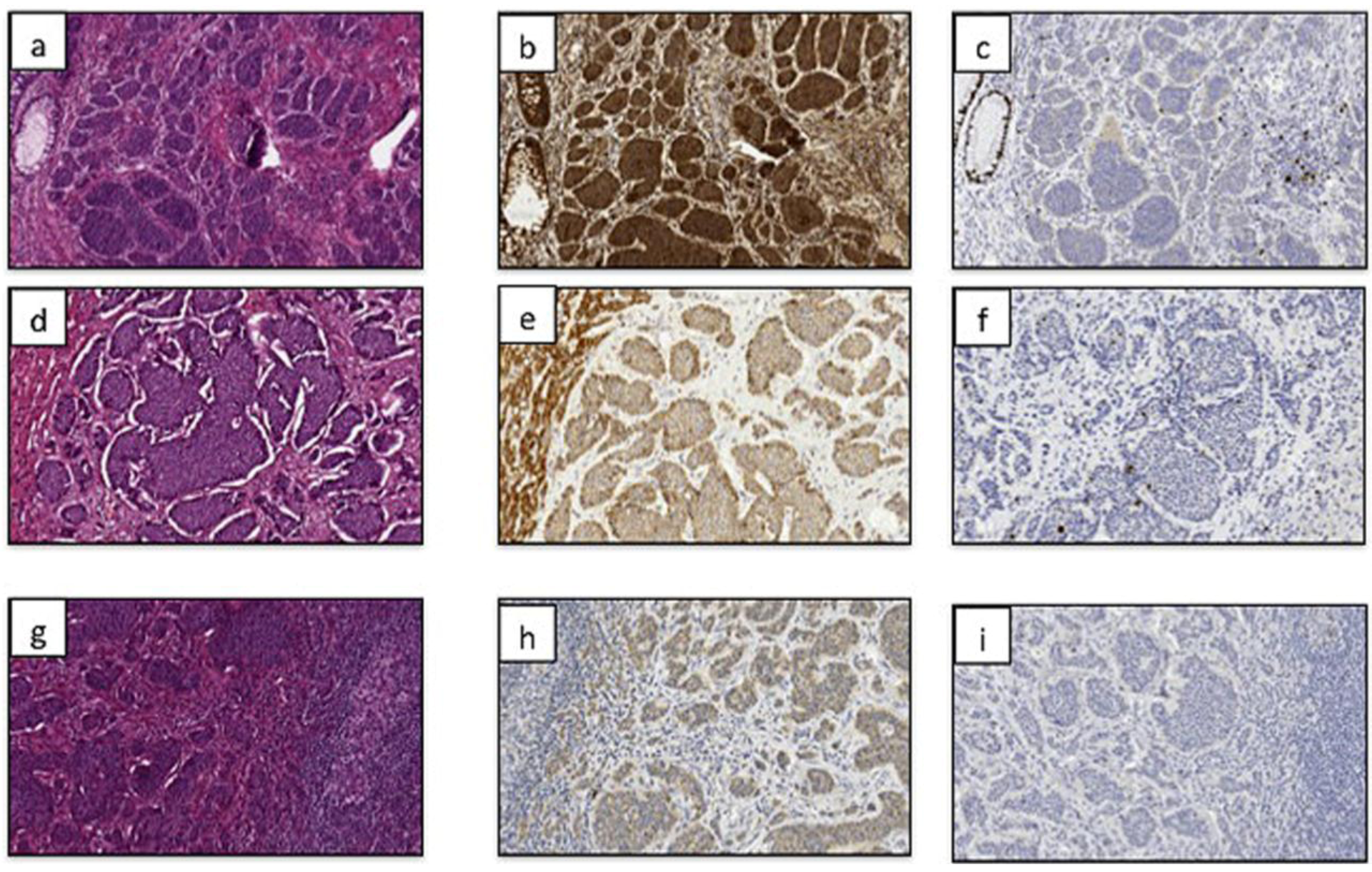
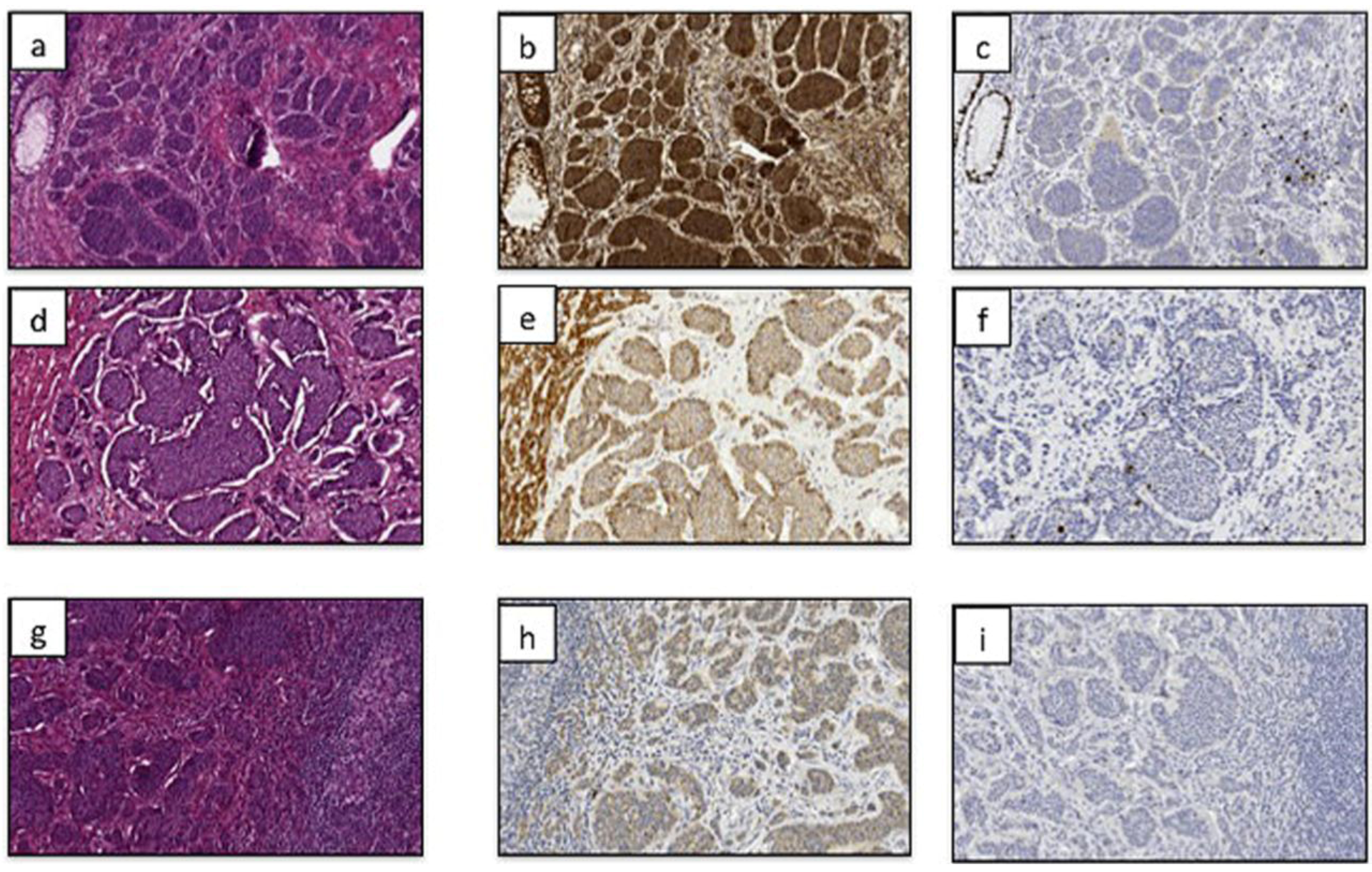{kind=link}
{kind=link}
| Variable | NFT | FT | p Value | |
|---|---|---|---|---|
| Age (Years) | <50 | 8 | 4 | 0.106 |
| 51–70 | 7 | 9 | ||
| >70 | 0 | 3 | ||
| Gender | Male | 13 | 12 | 0.653 |
| Female | 2 | 4 | ||
| Outcome | DOD | 5 | 8 | |
| AW | 0 | 0 | 0.3480 | |
| AWD | 7 | 3 | ||
| A.NED | 3 | 3 | ||
| Medical Treatment | SMS | 12 | 13 | |
| CT | 2 | 3 | 0.653 | |
| SMS+CT | 0 | 0 | ||
| NO | 1 | 0 | ||
| Liver Tumor Load | H1 | 13 | 1 | 0.0000026 |
| H2 | 1 | 1 | ||
| H3 | 1 | 14 | ||
| Basal CgA ng/mL | <200 | 10 | 3 | 0.0113 |
| >200 | 5 | 13 | ||
| All Measures | SDHB intensity | p Value | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | |||
| All measures | 39 | 19 | 9 | 11 | |
| SDHB expression | |||||
| 1–25% | 5 | 4 | 0 | 1 | |
| 26–50% | 9 | 6 | 2 | 1 | |
| 51–75% | 15 | 8 | 4 | 3 | 0.076 (Fisher exact) |
| 76–100% | 10 | 1 | 3 | 6 | 0.007 (trend) |
| SDHBExpression | SITE | Ki-67 labeling index | ||||
|---|---|---|---|---|---|---|
| Primary Tumor | Metastases | p Value | ≤1.3% | ≥1.3% | p Value | |
| 1–25% | 2 | 3 | 1 | 4 | ||
| 26–50% | 2 | 7 | 0.013 (trend) | 3 | 6 | 0.038 |
| 51–75% | 7 | 8 | 8 | 7 | ||
| 76–100% | 9 | 1 | 7 | 3 | ||
| SDHB Intensity | ||||||
| 1+ | 1 | 18 | 2 | 17 | ||
| 2+ | 8 | 1 | <0.0001(trend) | 7 | 2 | <0.0001(trend) |
| 3+ | 11 | 0 | 10 | 1 | ||
| HR (95% CI) | p Value | |
|---|---|---|
| CgA level | ||
| <200 | 1.00 | |
| >201 | 8.22 (1.02–66.4) | 0.048 |
| SHDB intensity | ||
| 1 | 1.00 | |
| 2–3 | 1.38 (0.49–3.91) | 0.55 |
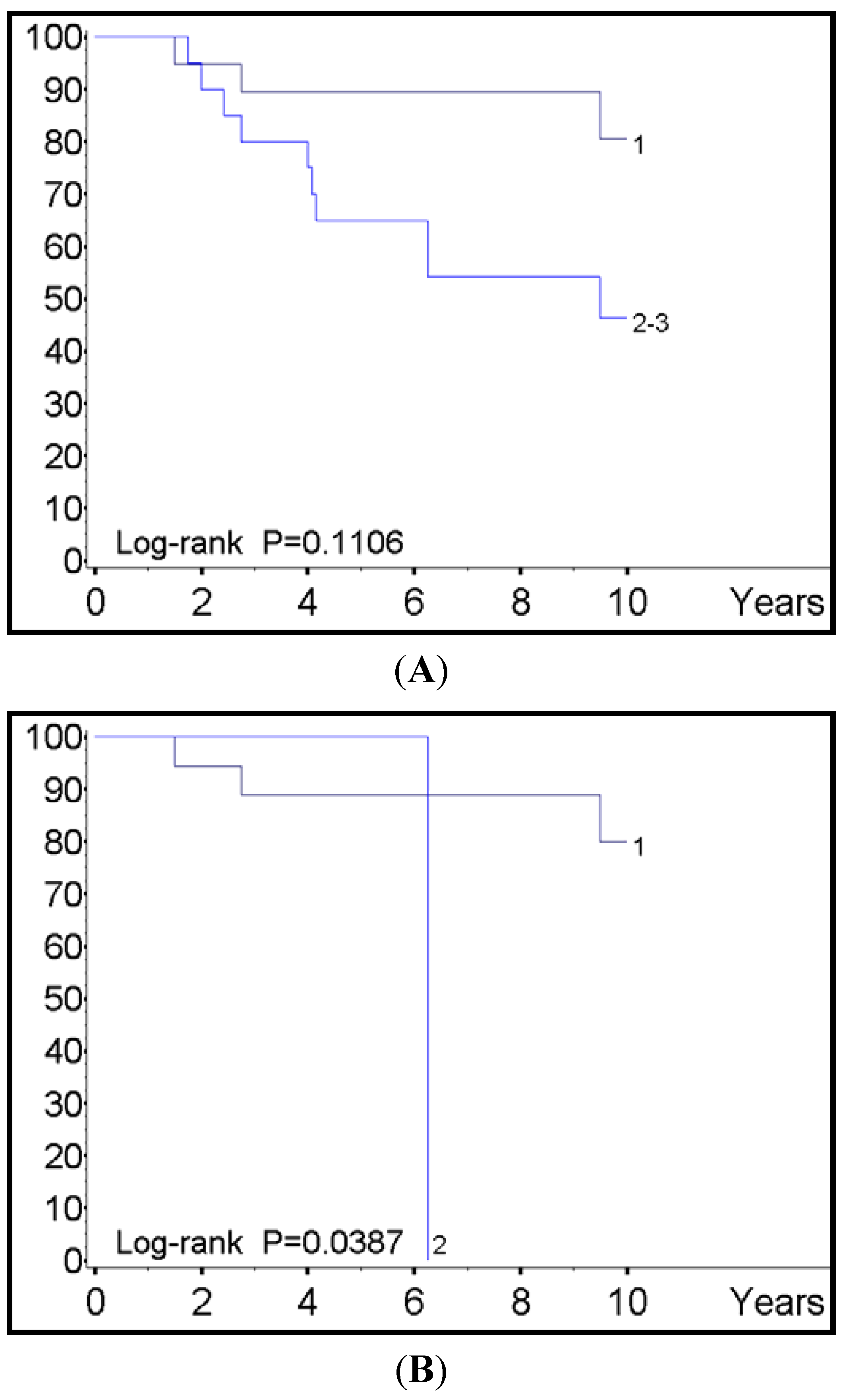
3. Experimental Section
3.1. Patients
3.2. Tumors Specimens, Immunohistochemical Methods and Scoring of Data
3.3. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar]
- Oberg, K. Neuroendocrine gastrointestinal tumors—A condensed overview of diagnosis and treatment. Ann. Oncol. 1999, 10, S3–S8. [Google Scholar]
- Kaltsas, G.A.; Besser, G.M.; Grossman, A.B. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr. Rev. 2004, 25, 458–511. [Google Scholar] [CrossRef]
- Niederle, M.B.; Hackl, M.; Kaserer, K.; Niederle, B. Gastroenteropancreatic neuroendocrine tumours: The current incidence and staging based on the WHO and European Neuroendocrine Tumour Society classification: An analysis based on prospectively collected parameters. Endocr. Relat. Cancer 2010, 17, 909–918. [Google Scholar] [CrossRef]
- Modlin, I.M.; Latich, I.; Kidd, M.; Zikusoka, M.; Eick, G. Therapeutic options for gastrointestinal carcinoids. Clin. Gastroenterol. Hepatol. 2006, 4, 526–547. [Google Scholar] [CrossRef]
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle, F.G.; Kaltsas, G.A.; Krenning, E.P.; et al. Sundin, Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Ahlman, H.; Nilsson, O.; McNicol, A.M.; Ruszniewski, P.; Niederle, B.; Ricke, J.; Jensen, R.; Kos-Kudla, B.; Oberg, K.; O’Connor, J.M.; et al. Poorly-differentiated endocrine carcinomas of midgut and hindgut origin. Neuroendocrinology 2008, 87, 40–46. [Google Scholar] [CrossRef]
- Nilsson, O.; van Cutsem, E.; Delle, F.G.; Yao, J.C.; Pavel, M.E.; McNicol, A.M.; Sevilla Garcia, M.I.; Knapp, W.H.; Kelestimur, F.; Sauvanet, A.; et al. Poorly differentiated carcinomas of the foregut (gastric, duodenal and pancreatic). Neuroendocrinology 2006, 84, 212–215. [Google Scholar] [CrossRef]
- Ballian, N.; Loeffler, A.G.; Rajamanickam, V.; Norstedt, P.A.; Weber, S.M.; Cho, C.S. A simplified prognostic system for resected pancreatic neuroendocrine neoplasms. HPB (Oxford) 2009, 11, 422–428. [Google Scholar]
- Couvelard, A.; O’Toole, D.; Turley, H.; Leek, R.; Sauvanet, A.; Degott, C.; Ruszniewski, P.; Belghiti, J.; Harris, A.L.; Gatter, K.; et al. Microvascular density and hypoxia-inducible factor pathway in pancreatic endocrine tumours: Negative correlation of microvascular density and VEGF expression with tumour progression. Br. J. Cancer 2005, 92, 94–101. [Google Scholar] [CrossRef]
- Rinke, A.; Muller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Blaker, M.; et al. Placebocontrolled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar]
- Moertel, C.G.; Lefkopoulo, M.; Lipsitz, S.; Hahn, R.G.; Klaassen, D. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N. Engl. J. Med. 1992, 326, 519–523. [Google Scholar] [CrossRef]
- Moertel, C.G.; Kvols, L.K.; O’Connell, M.J.; Rubin, J. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer 1991, 68, 227–232. [Google Scholar] [CrossRef]
- Kulke, M.H.; Siu, L.L.; Tepper, J.E.; Fisher, G.; Jaffe, D.; Haller, D.G.; Ellis, L.M.; Benedetti, J.K.; Bergsland, E.K.; Hobday, T.J.; et al. Future directions in the treatment of neuroendocrine tumors: Consensus report of the National Cancer Institute Tumor clinical trials planning meeting. J. Clin. Oncol. 2001, 29, 934–943. [Google Scholar]
- Lenders, J.W.; Eisenhofer, G.; Mannelli, M.; Pacak, K. Phaeochromocytoma. Lancet 2005, 366, 665–675. [Google Scholar]
- Pasini, B.; Stratakis, C.A. SDH mutations in tumorigenesis and inherited endocrine tumours: Lesson from the phaeochromocytoma-paraganglioma syndromes. J. Intern. Med. 2009, 266, 19–42. [Google Scholar] [CrossRef]
- Gimm, O.; Armanios, M.; Dziema, H.; Neumann, H.P.; Eng, C. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res. 2000, 60, 6822–6825. [Google Scholar]
- Astuti, D.; Douglas, F.; Lennard, T.W.; Aligianis, I.A.; Woodward, E.R.; Evans, D.G.; Eng, C.; Latif, F.; Maher, E.R. Germline SDHD mutation in familial phaeochromocytoma. Lancet 2001, 357, 1181–1182. [Google Scholar]
- Van Nederveen, F.H.; Korpershoek, E.; Lenders, J.W.; de Krijger, R.R.; Dinjens, W.N. Somatic SDHB mutation in an extraadrenal pheochromocytoma. N. Engl. J. Med. 2007, 357, 306–308. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Mourad, J.J.; Plouin, P.F.; Corvol, P.; Rotig, A.; Jeunemaitre, X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am. J. Hum. Genet. 2001, 69, 1186–1197. [Google Scholar] [CrossRef]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Kerlan, V.; Plouin, P.F.; Rotig, A.; Jeunemaitre, X. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar]
- Pasini, B.; McWhinney, S.R.; Bei, T.; Matyakhina, L.; Stergiopoulos, S.; Muchow, M.; Boikos, S.A.; Ferrando, B.; Pacak, K.; Assie, G.; et al. Clinical and molecular genetics of patientswith the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008, 16, 79–88. [Google Scholar]
- Levitas, A.; Muhammad, E.; Harel, G.; Saada, A.; Caspi, V.C.; Manor, E.; Beck, J.C.; Sheffield, V.; Parvari, R. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur. J. Hum. Genet. 2010, 18, 1160–1165. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Carney, J.A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar]
- Ricketts, C.J.; Forman, J.R.; Rattenberry, E.; Bradshaw, N.; Lalloo, F.; Izatt, L.; Cole, T.R.; Armstrong, R.; Kumar, V.K.; Morrison, P.J.; et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum. Mutat. 2010, 31, 41–51. [Google Scholar] [CrossRef]
- Vanharanta, S.; Buchta, M.; McWhinney, S.R.; Virta, S.K.; Peczkowska, M.; Morrison, C.D.; Lehtonen, R.; Januszewicz, A.; Jarvinen, H.; Juhola, M.; et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am. J. Hum. Genet. 2004, 74, 153–159. [Google Scholar] [CrossRef]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef]
- Schimke, R.N.; Collins, D.L.; Stolle, C.A. Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am. J. Med. Genet. 2010, 152A, 1531–1535. [Google Scholar]
- Zantour, B.; Guilhaume, B.; Tissier, F.; Louvel, A.; Jeunemaitre, X.; Gimenez-Roqueplo, A.P.; Bertagna, X. A thyroid nodule revealing a paraganglioma in a patient with a new germline mutation in the succinate dehydrogenase B gene. Eur. J. Endocrinol. 2004, 151, 433–438. [Google Scholar] [CrossRef]
- Galera-Ruiz, H.; Gonzalez-Campora, R.; Rey-Barrera, M.; Rollon-Mayordomo, A.; Garcia-Escudero, A.; Fernandez-Santos, J.M.; DeMiguel, M.; Galera-Davidson, H. W43X SDHD mutation in sporadic head and neck paraganglioma. Anal. Quant. Cytol. Histol. 2008, 30, 119–123. [Google Scholar]
- Astuti, D.; Morris, M.; Krona, C.; Abel, F.; Gentle, D.; Martinsson, T.; Kogner, P.; Neumann, H.P.; Voutilainen, R.; Eng, C.; et al. Investigation of the role of SDHB inactivation in sporadic phaeochromocytoma and neuroblastoma. Br. J. Cancer 2004, 91, 1835–1841. [Google Scholar] [CrossRef]
- Grau, E.; Oltra, S.; Orellana, C.; Hernandez-Marti, M.; Castel, V.; Martinez, F. There is no evidence that the SDHB gene is involved in neuroblastoma development. Oncol. Res. 2005, 15, 393–398. [Google Scholar]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Pollard, P.; Wortham, N.; Barclay, E.; Alam, A.; Elia, G.; Manek, S.; Poulsom, R.; Tomlinson, I. Evidence of increased microvessel density and activation of the hypoxia pathway in tumours from the hereditary leiomyomatosis and renal cell cancer syndrome. J. Pathol. 2005, 205, 41–49. [Google Scholar] [CrossRef]
- Dahia, P.L.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, 72–80. [Google Scholar]
- Lopez-Jimenez, E.; Gomez-Lopez, G.; Leandro-Garcia, L.J.; Munoz, I.; Schiavi, F.; Montero-Conde, C.; de Cubas, A.A.; Ramires, R.; Landa, I.; Leskela, S.; et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol. Endocrinol. 2010, 24, 2382–2391. [Google Scholar] [CrossRef]
- Favier, J.; Briere, J.J.; Burnichon, N.; Riviere, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; de Reynies, A.; Bertherat, J.; Badoual, C.; et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar]
- Gill, A.J.; Benn, D.E.; Chou, A.; Clarkson, A.; Muljono, A.; Meyer-Rochow, G.Y.; Richardson, A.L.; Sidhu, S.B.; Robinson, B.G.; Clifton-Bligh, R.J. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromo-cytoma syndromes. Hum. Pathol. 2010, 34, 636–644. [Google Scholar]
- Gill, A.J.; Chou, A.; Vilain, R.; Clarkson, A.; Lui, M.; Jin, R.; Tobias, V.; Samra, J.; Goldstein, D.; Smith, C.; et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am. J. Surg. Pathol. 2010, 34, 805–814. [Google Scholar]
- Cervera, A.M.; Apostolova, N.; Crespo, F.L.; Mata, M.; McCreath, K.J. Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Res. 2008, 68, 4058–4067. [Google Scholar]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, amitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar]
- Burnichon, N.; Briere, J.J.; Libe, R.; Vescovo, L.; Riviere, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Benit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef]
- Amar, L.; Baudin, E.; Burnichon, N.; Peyrard, S.; Silvera, S.; Bertherat, J.; Bertagna, X.; Schlumberger, M.; Jeunemaitre, X.; Gimenez-Roqueplo, A.P.; et al. Succinate dehy-drogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 3822–3828. [Google Scholar] [CrossRef]
- Capella, C.; Heitz, P.U.; Hofler, H.; Solcia, E.; Kloppel, G. Revised classification of neuroendocrine tumours of the lungg, pancreas and gut. Virchows Arch. 1995, 425, 547–560. [Google Scholar]
- Suzuki, H.; Christofides, N.D.; Ghiglione, M.; Ferri, G.L.; Chretien, M.; Seidah, N.G.; Polak, J.M.; Bloom, S.R. Distribution of a novel pituitary protein (7B2) in mammalian gastrointestinal tract and pancreas. Dig. Dis. Sci. 1998, 33, 718–723. [Google Scholar]
- Al-Khafaji, B.; Noffsinger, A.E.; Miller, M.A.; DeVoe, G.; Stemmermann, G.N.; Fenoglio-Preiser, C. Immunohistologic analysis of gastrointestinal and pulmonary carcinoid tumors. Hum. Pathol. 1998, 29, 992–999. [Google Scholar] [CrossRef]
- Barbareschi, M.; Girlando, S.; Mauri, F.A.; Arrigoni, G.; Laurino, L.; Dalla Palma, P.; Doglioni, C. Tumour suppressor gene products, proliferation, and differentiation markers in lung neuroendocrine neoplasms. J. Pathol. 1992, 166, 343–350. [Google Scholar] [CrossRef]
- Cunningham, R.T.; Pogue, K.M.; Curry, W.J.; Johnston, C.F.; Sloan, J.M.; Buchanan, K.D. Immunostaining for vasostatin I distinguishes between ileal and lung carcinoids. J. Pathol. 1999, 187, 321–325. [Google Scholar] [CrossRef]
- Pelosi, G.; Bresaola, E.; Bogina, G.; Pasini, F.; Rodella, S.; Castelli, P.; Iacono, C.; Serio, G.; Zamboni, G. Endocrine tumors of the pancreas: Ki-67 immunoreactivity on paraffin sections is an independent predictor for malignancy: A comparative study with proliferating-cell nuclear antigen and progesterone receptor protein immunostaining, mitotic index, and other clinicopathologic variables. Hum. Pathol. 1996, 27, 1124–1134. [Google Scholar] [CrossRef]
- Tonnies, H.; Toliat, M.R.; Ramel, C.; Pape, U.F.; Neitzel, H.; Berger, W.; Wiedenmann, B. Analysis of sporadic neuroendocrine tumours of the enteropancreatic system by comparative genomic hybridisation. Gut 2001, 48, 536–541. [Google Scholar] [CrossRef]
- Panzuto, F.; Nasoni, S.; Falconi, M.; Corleto, V.D.; Capurso, G.; Cassetta, S.; Di Fonzo, M.; Tornatore, V.; Milione, M.; Angeletti, S.; et al. Prognostic factors and survival in endocrine tumour patients: Comparison between gastrointestinal and pancreatic localization. Endocr. Relat. Cancer 2005, 12, 1083–1092. [Google Scholar] [CrossRef]
- Rigaud, G.; Missiaglia, E.; Moore, P.S.; Zamboni, G.; Falconi, M.; Talamini, G.; Pesci, A.; Baron, A.; Lissandrini, D.; Rindi, G.; et al. High resolution allelotype of nonfunctional pancreatic endocrine tumours: Identification of two molecular subgroups with clinical implications. Cancer Res. 2001, 61, 285–292. [Google Scholar]
- Furlan, D.; Cerutti, R.; Uccella, S.; La Rosa, S.; Rigoli, E.; Genasetti, A.; Capella, C. Different molecular profiles characterize well-differentiated endocrine tumours and poorly differentiated endocrine carcinomas of the gastroenteropancreatic tract. Clin. Cancer Res. 2004, 10, 947–957. [Google Scholar]
- Honegger, J.; Prettin, C.; Feuerhake, F.; Petrick, M.; Schulte-Mönting, J.; Reincke, M. Expression of Ki-67 antigen in nonfunctioning pituitary adenomas: Correlation with growth velocity and invasiveness. J. Neurosurg. 2003, 99, 674–679. [Google Scholar] [CrossRef]
- Mazzaferro, V.; Bhoori, S.; Sposito, C.; Bongini, M.; Langer, M.; Miceli, R.; Mariani, L. Milan criteria in liver transplantation for hepatocellular carcinoma: An evidence-based analysis of 15 years of experience. Liver Transpl. 2011, 17, S44–S57. [Google Scholar] [CrossRef]
- Belli, L.S.; Caccamo, L.; Mazzaferro, V.; Silini, E.; Alberti, A.; Melada, E.; Regalia, E.; Gridelli, B.; Rubino, A.; Gennari, L.; et al. Milan multicenter experience in liver transplantation for hepatitis C-related cirrhosis: Report of 105 cases. Transplant. Proc. 1994, 26, 3582–3584. [Google Scholar]
- World Health Organization Classification of Tumours of the Digestive System, 4th; Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. (Eds.) International Agency for Research on Cancer (IARC): Lyon, France, 2010.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Milione, M.; Pusceddu, S.; Gasparini, P.; Melotti, F.; Maisonneuve, P.; Mazzaferro, V.; De Braud, F.G.; Pelosi, G. Succinate Dehydrogenase B Subunit Immunohistochemical Expression Predicts Aggressiveness in Well Differentiated Neuroendocrine Tumors of the Ileum. Cancers 2012, 4, 808-820. https://doi.org/10.3390/cancers4030808
Milione M, Pusceddu S, Gasparini P, Melotti F, Maisonneuve P, Mazzaferro V, De Braud FG, Pelosi G. Succinate Dehydrogenase B Subunit Immunohistochemical Expression Predicts Aggressiveness in Well Differentiated Neuroendocrine Tumors of the Ileum. Cancers. 2012; 4(3):808-820. https://doi.org/10.3390/cancers4030808
Chicago/Turabian StyleMilione, Massimo, Sara Pusceddu, Patrizia Gasparini, Flavia Melotti, Patrick Maisonneuve, Vincenzo Mazzaferro, Filippo G. De Braud, and Giuseppe Pelosi. 2012. "Succinate Dehydrogenase B Subunit Immunohistochemical Expression Predicts Aggressiveness in Well Differentiated Neuroendocrine Tumors of the Ileum" Cancers 4, no. 3: 808-820. https://doi.org/10.3390/cancers4030808