NF-κB in T-cell Acute Lymphoblastic Leukemia: Oncogenic Functions in Leukemic and in Microenvironmental Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
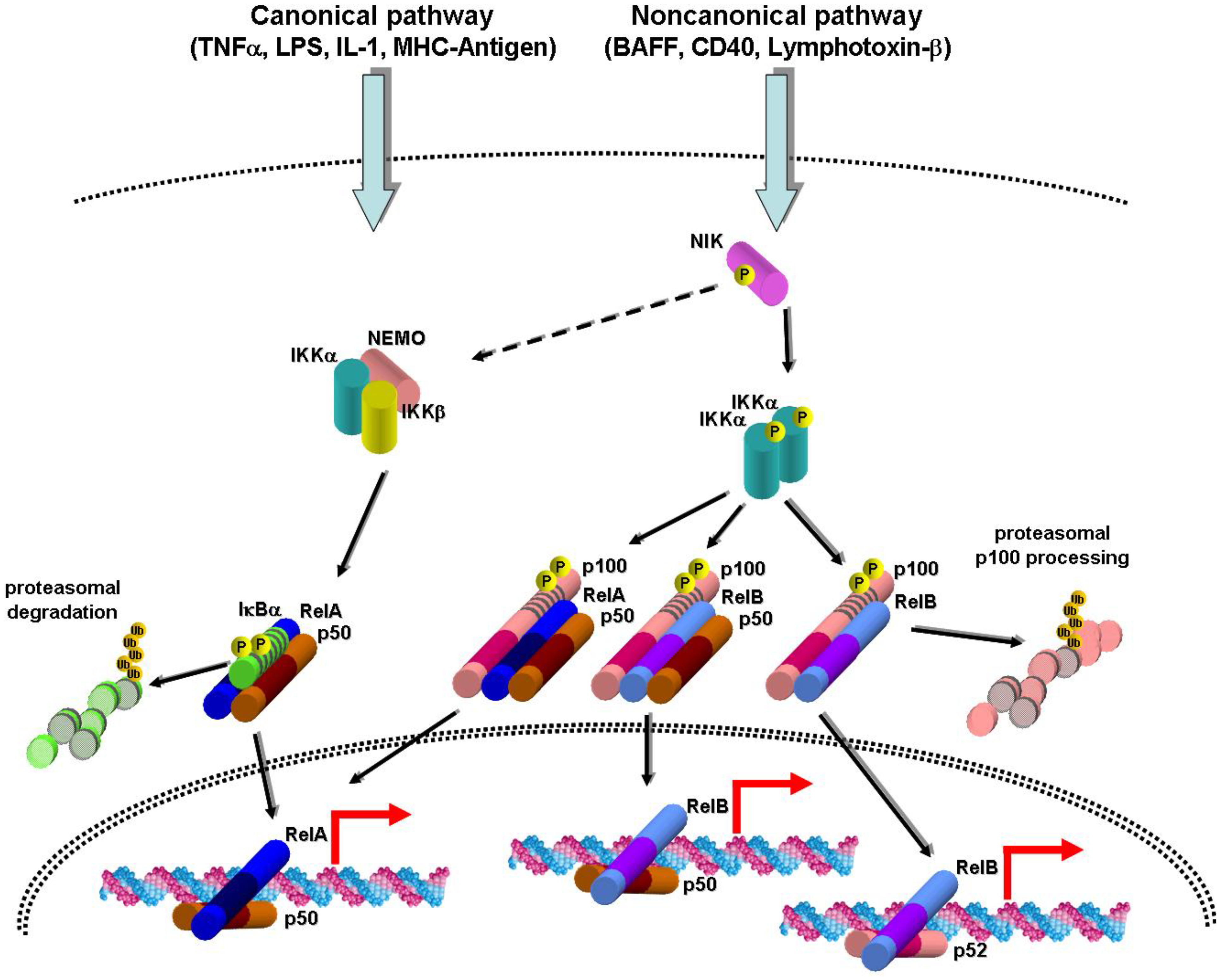
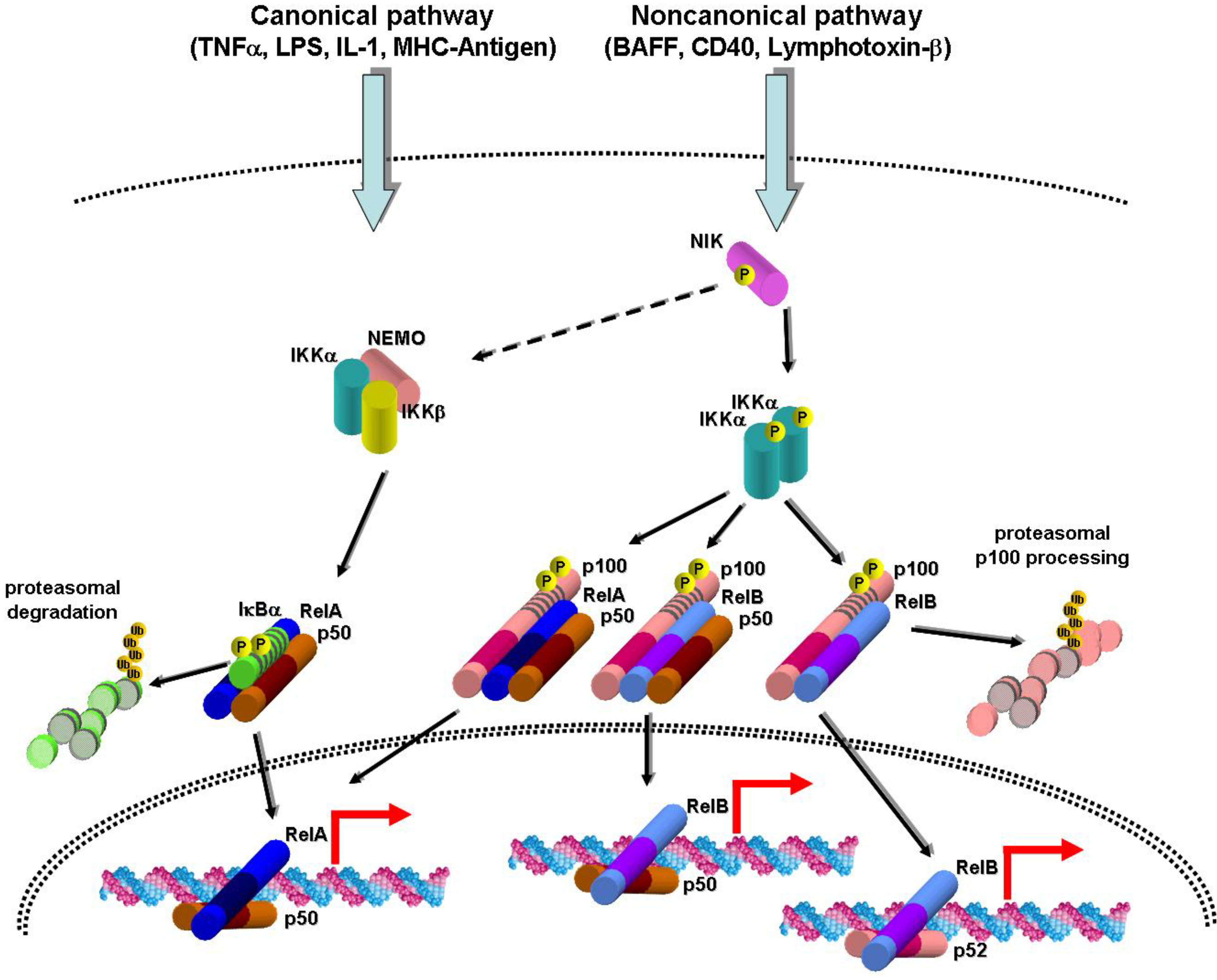
2. Canonical and Noncanonical NF-κB Activation Pathways
3. NF-κB Activation in Leukemia and Lymphoma
4. Molecular Pathogenesis of T-cell Acute Lymphoblastic Leukemia
5. NF-κB Activation in Human T-cell Acute Lymphoblastic Leukemia and T-cell Leukemia/Lymphoma Mouse Models
6. NF-κB Inhibition in Human and Murine Leukemic T Cells
7. Paracrine/Autocrine Mechanisms of NF-κB Activation in T-ALL
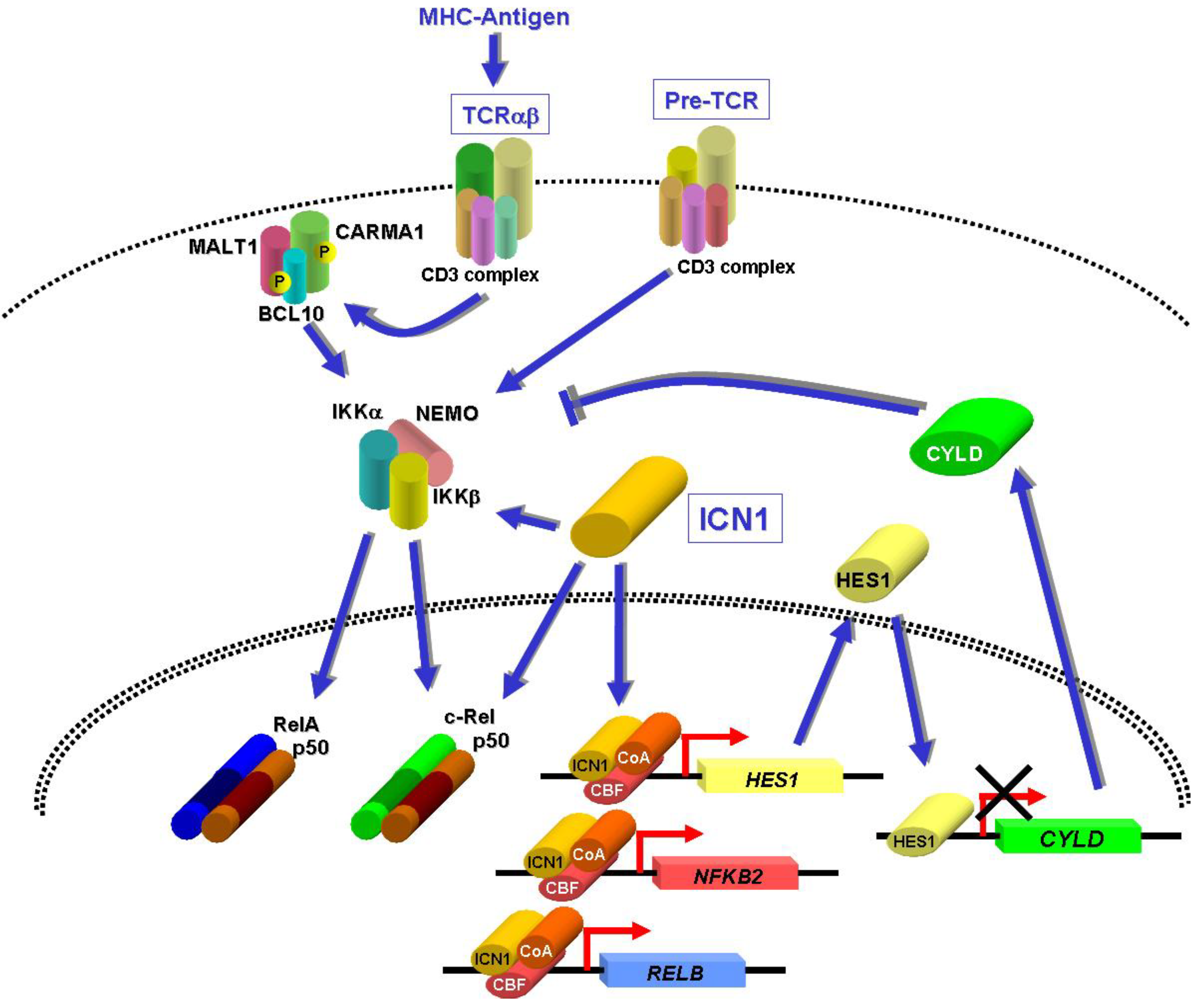
8. NF-κB Activation by NOTCH1 in T-ALL
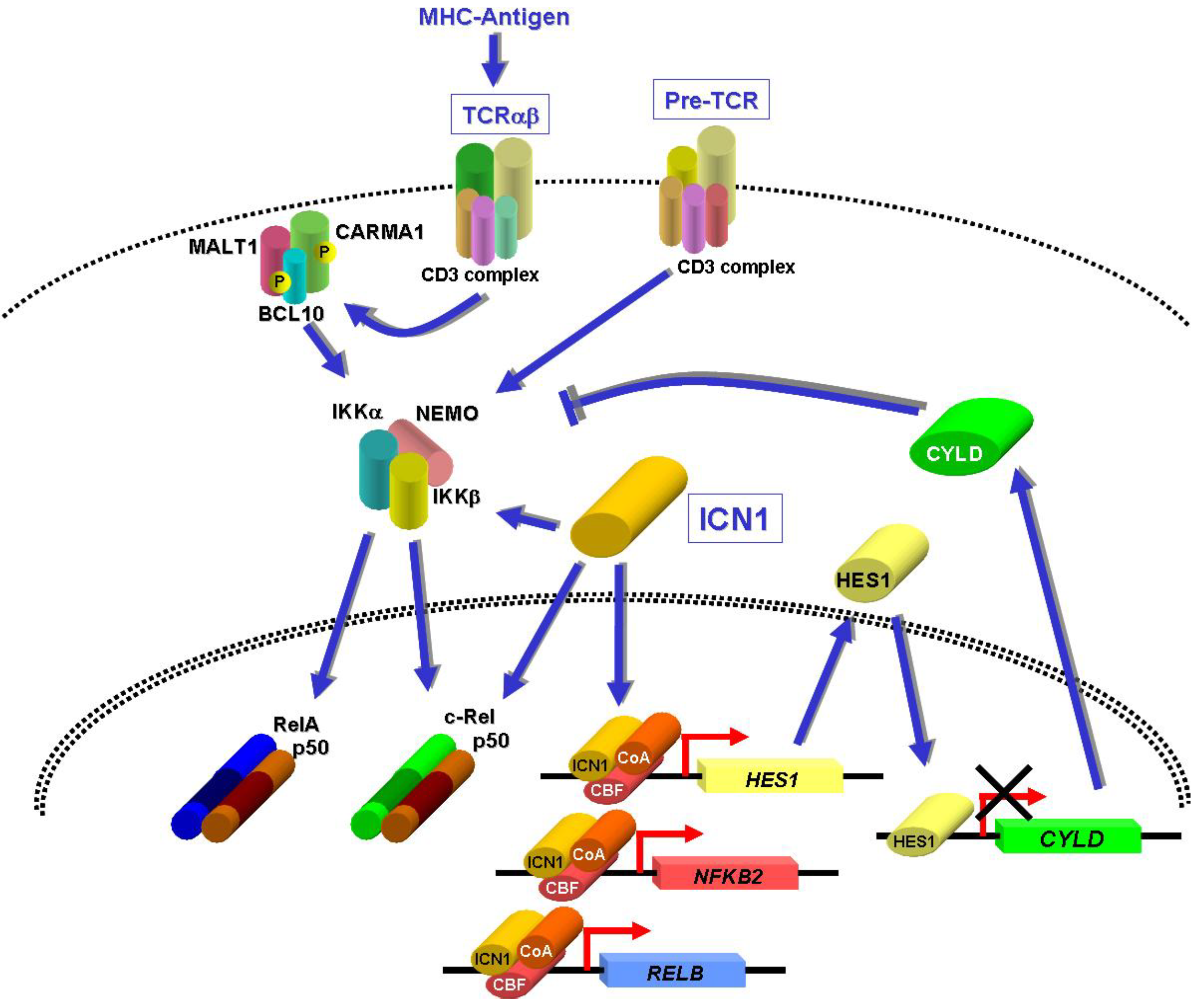
9. NF-κB Activation by Other Oncoproteins?
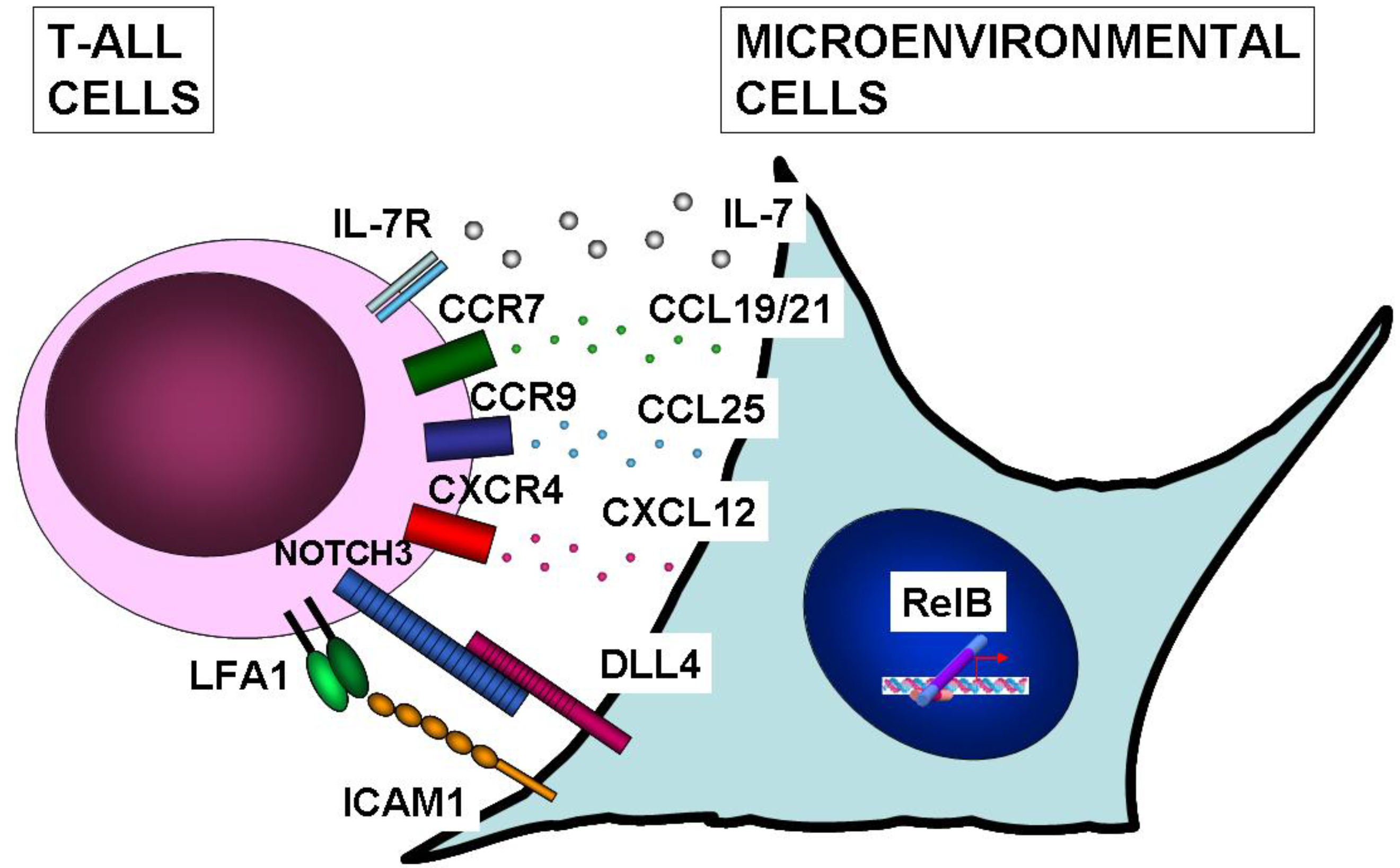
10. NF-κB Function in Microenvironmental Cells of T-ALL
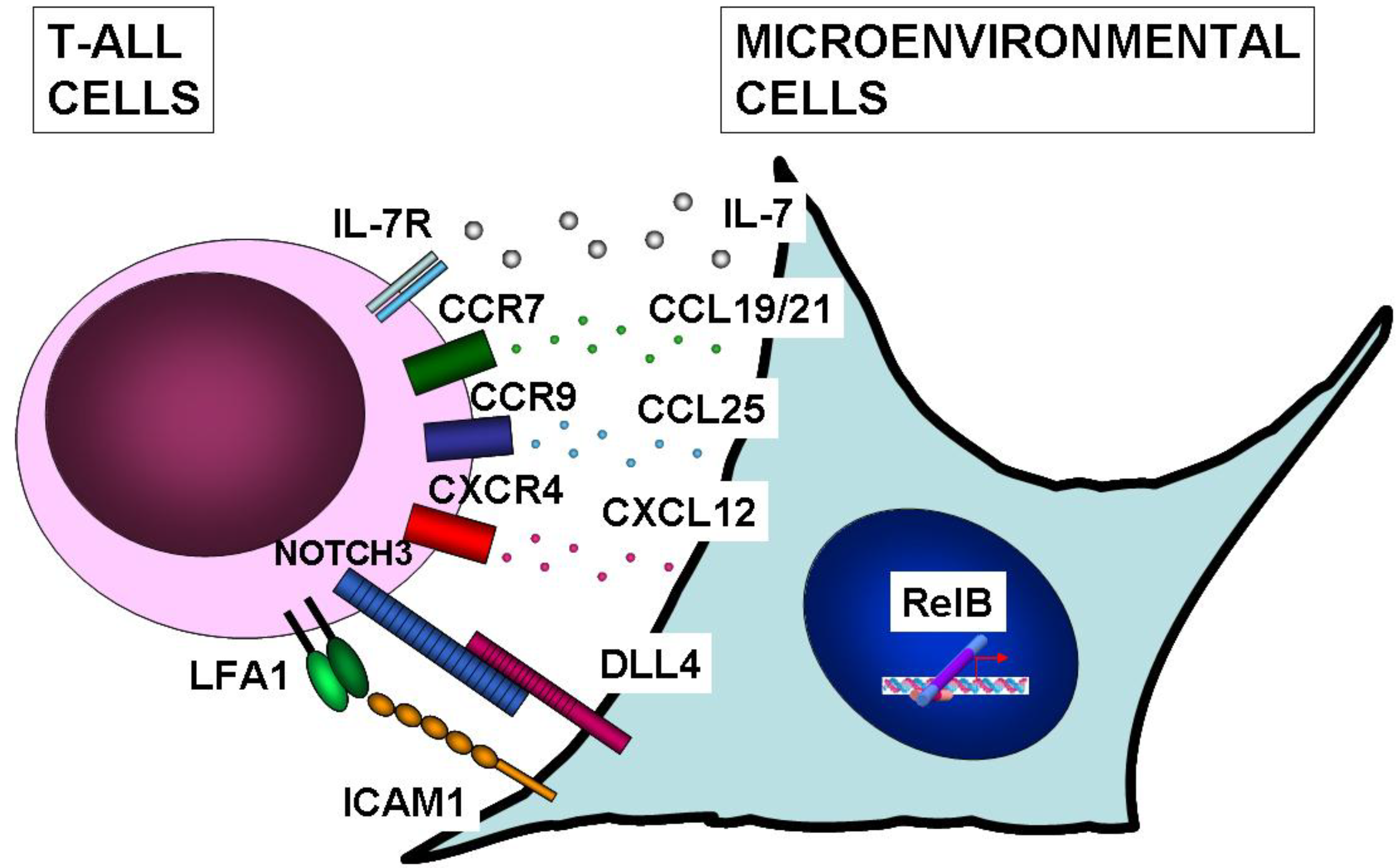
11. NF-κB Inhibition as a Therapeutic Strategy for T-ALL?
12. Conclusions
Acknowledgements
References
- Courtois, G.; Gilmore, T.D. Mutations in the NF-κB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef]
- Bassères, D.S.; Baldwin, A.S. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Hu, Y.; Baud, V.; Delhase, M.; Zhang, P.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKα subunit of IκB kinase. Science 1999, 284, 316–320. [Google Scholar] [CrossRef]
- Li, Q.; Van Antwerp, D.; Mercurio, F.; Lee, K.F.; Verma, I.M. Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science 1999, 284, 321–325. [Google Scholar] [CrossRef]
- Li, Q.; Lu, Q.; Hwang, J.Y.; Büscher, D.; Lee, K.F.; Izpisua-Belmonte, J.C.; Verma, I.M. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999, 13, 1322–1328. [Google Scholar] [CrossRef]
- Li, Z. W.; Chu, W.; Hu, Y.; Delhase, M.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. The IKKβ subunit of IκB kinase (IKK) is essential for nuclear factor κB activation and prevention of apoptosis. J. Exp. Med. 1999, 189, 1839–1845. [Google Scholar] [CrossRef]
- Rudolph, D.; Yeh, W.C.; Wakeham, A.; Rudolph, B.; Nallainathan, D.; Potter, J.; Elia, A.J.; Mak, T.W. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev. 2000, 14, 854–862. [Google Scholar]
- Takeda, K.; Takeuchi, O.; Tsujimura, T.; Itami, S.; Adachi, O.; Kawai, T.; Sanjo, H.; Yoshikawa, K.; Terada, N.; Akira, S. Limb and skin abnormalities in mice lacking IKKα. Science 1999, 284, 313–316. [Google Scholar] [CrossRef]
- Liao, G.; Zhang, M.; Harhaj, E.W.; Sun, S. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 2004, 279, 26243–26250. [Google Scholar] [CrossRef]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.; Mak, T.W.; Korneluk, R.G.; Cheng, G. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.; Keats, J.J.; Wang, H.; Vignali, D.A.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef]
- Xiao, G.; Fong, A.; Sun, S. Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα-mediated phosphorylation. J. Biol. Chem. 2004, 279, 30099–30105. [Google Scholar] [CrossRef]
- Derudder, E.; Dejardin, E.; Pritchard, L.L.; Green, D.R.; Korner, M.; Baud, V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-α and lymphotoxin-β receptor activation: Critical roles for p100. J. Biol. Chem. 2003, 278, 23278–23284. [Google Scholar]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O'Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; Hoffmann, A. A fourth IκB protein within the NF-κB signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef]
- Müller, J.R.; Siebenlist, U. Lymphotoxin β receptor induces sequential activation of distinct NF-κB factors via separate signaling pathways. J. Biol. Chem. 2003, 278, 12006–12012. [Google Scholar] [CrossRef]
- Yilmaz, Z.B.; Weih, D.S.; Sivakumar, V.; Weih, F. RelB is required for Peyer's patch development: Differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003, 22, 121–130. [Google Scholar] [CrossRef]
- Bren, G.D.; Solan, N.J.; Miyoshi, H.; Pennington, K.N.; Pobst, L.J.; Paya, C.V. Transcription of the RelB gene is regulated by NF-κB. Oncogene 2001, 20, 7722–7733. [Google Scholar] [CrossRef]
- Liptay, S.; Schmid, R.M.; Nabel, E.G.; Nabel, G.J. Transcriptional regulation of NF-κB2: Evidence for κB-mediated positive and negative autoregulation. Mol. Cell. Biol. 1994, 14, 7695–7703. [Google Scholar]
- Basak, S.; Shih, V.F.; Hoffmann, A. Generation and activation of multiple dimeric transcription factors within the NF-κB signaling system. Mol. Cell. Biol. 2008, 28, 3139–3150. [Google Scholar] [CrossRef]
- Razani, B.; Zarnegar, B.; Ytterberg, A.J.; Shiba, T.; Dempsey, P.W.; Ware, C.F.; Loo, J.A.; Cheng, G. Negative feedback in noncanonical NF-κB signaling modulates NIK stability through IKKα-mediated phosphorylation. Sci. Signal. 2010, 3, ra41. [Google Scholar] [CrossRef]
- Ramakrishnan, P.; Wang, W.; Wallach, D. Receptor-specific signaling for both the alternative and the canonical NF-κB activation pathways by NF-κB-inducing kinase. Immunity 2004, 21, 477–489. [Google Scholar] [CrossRef]
- Zarnegar, B.; Yamazaki, S.; He, J.Q.; Cheng, G. Control of canonical NF-κB activation through the NIK-IKK complex pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 3503–3508. [Google Scholar] [CrossRef]
- Jost, P.J.; Ruland, J. Aberrant NF-κB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar]
- Packham, G. The role of NF-κB in lymphoid malignancies. Br. J. Haematol. 2008, 143, 3–15. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; Dave, S.; Hurt, E.M.; Tan, B.; Zhao, H.; Stephens, O.; Santra, M.; Williams, D.R.; Dang, L.; Barlogie, B.; Shaughnessy, J.D.; Kuehl, W.M.; Staudt, L.M. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.; Van Wier, S.; Tiedemann, R.; Shi, C.; Sebag, M.; Braggio, E.; Henry, T.; Zhu, Y.; Fogle, H.; Price-Troska, T.; Ahmann, G.; Mancini, C.; Brents, L.A.; Kumar, S.; Greipp, P.; Dispenzieri, A.; Bryant, B.; Mulligan, G.; Bruhn, L.; Barrett, M.; Valdez, R.; Trent, J.; Stewart, A.K.; Carpten, J.; Bergsagel, P.L. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef]
- Hideshima, T.; Neri, P.; Tassone, P.; Yasui, H.; Ishitsuka, K.; Raje, N.; Chauhan, D.; Podar, K.; Mitsiades, C.; Dang, L.; Munshi, N.; Richardson, P.; Schenkein, D.; Anderson, K.C. MLN120B, a novel IκB kinase β inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin. Cancer Res. 2006, 12, 5887–5894. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; Bhagat, G.; Chadburn, A.; Dalla-Favera, R.; Pasqualucci, L. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; Asakura, Y.; Muto, S.; Tamura, A.; Iio, M.; Akatsuka, Y.; Hayashi, Y.; Mori, H.; Igarashi, T.; Kurokawa, M.; Chiba, S.; Mori, S.; Ishikawa, Y.; Okamoto, K.; Tobinai, K.; Nakagama, H.; Nakahata, T.; Yoshino, T.; Kobayashi, Y.; Ogawa, S. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; Ott, G.; Muller-Hermelink, H.K.; Gascoyne, R.D.; Connors, J.M.; Rimsza, L.M.; Campo, E.; Jaffe, E.S.; Delabie, J.; Smeland, E.B.; Fisher, R.I.; Chan, W.C.; Staudt, L.M. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef]
- Horie, R.; Watanabe, T.; Morishita, Y.; Ito, K.; Ishida, T.; Kanegae, Y.; Saito, I.; Higashihara, M.; Mori, S.; Kadin, M.E.; Watanabe, T. Ligand-independent signaling by overexpressed CD30 drives NF-κB activation in Hodgkin-Reed-Sternberg cells. Oncogene 2002, 21, 2493–2503. [Google Scholar] [CrossRef]
- Annunziata, C.M.; Safiran, Y.J.; Irving, S.G.; Kasid, U.N.; Cossman, J. Hodgkin disease: Pharmacologic intervention of the CD40-NF-κB pathway by a protease inhibitor. Blood 2000, 96, 2841–2848. [Google Scholar]
- Endo, T.; Nishio, M.; Enzler, T.; Cottam, H.B.; Fukuda, T.; James, D.F.; Karin, M.; Kipps, T.J. BAFF and APRIL support chronic lymphocytic leukemia B-cell survival through activation of the canonical NF-κB pathway. Blood 2007, 109, 703–710. [Google Scholar] [CrossRef]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef]
- Novak, A.J.; Grote, D.M.; Stenson, M.; Ziesmer, S.C.; Witzig, T.E.; Habermann, T.M.; Harder, B.; Ristow, K.M.; Bram, R.J.; Jelinek, D.F.; Gross, J.A.; Ansell, S.M. Expression of BLyS and its receptors in B-cell non-Hodgkin lymphoma: Correlation with disease activity and patient outcome. Blood 2004, 104, 2247–2253. [Google Scholar] [CrossRef]
- Cilloni, D.; Messa, F.; Arruga, F.; Defilippi, I.; Morotti, A.; Messa, E.; Carturan, S.; Giugliano, E.; Pautasso, M.; Bracco, E.; Rosso, V.; Sen, A.; Martinelli, G.; Baccarani, M.; Saglio, G. The NF-κB pathway blockade by the IKK inhibitor PS1145 can overcome imatinib resistance. Leukemia 2006, 20, 61–67. [Google Scholar] [CrossRef]
- Gatto, S.; Scappini, B.; Pham, L.; Onida, F.; Milella, M.; Ball, G.; Ricci, C.; Divoky, V.; Verstovsek, S.; Kantarjian, H.M.; Keating, M.J.; Cortes-Franco, J.E.; Beran, M. The proteasome inhibitor PS-341 inhibits growth and induces apoptosis in Bcr/Abl-positive cell lines sensitive and resistant to imatinib mesylate. Haematologica 2003, 88, 853–863. [Google Scholar]
- Duncan, E.A.; Goetz, C.A.; Stein, S.J.; Mayo, K.J.; Skaggs, B.J.; Ziegelbauer, K.; Sawyers, C.L.; Baldwin, A.S. IκB kinase β inhibition induces cell death in Imatinib-resistant and T315I Dasatinib-resistant BCR-ABL+ cells. Mol. Cancer Ther. 2008, 7, 391–397. [Google Scholar] [CrossRef]
- Besançon, F.; Atfi, A.; Gespach, C.; Cayre, Y.E.; Bourgeade, M.F. Evidence for a role of NF-κB in the survival of hematopoietic cells mediated by interleukin 3 and the oncogenic TEL/platelet-derived growth factor receptor β fusion protein. Proc. Natl. Acad. Sci. USA 1998, 95, 8081–8086. [Google Scholar]
- Han, X.; Bueso-Ramos, C.E. Precursor T-cell acute lymphoblastic leukemia/lymphoblastic lymphoma and acute biphenotypic leukemias. Am. J. Clin. Pathol. 2007, 127, 528–544. [Google Scholar] [CrossRef]
- Schraders, M.; van Reijmersdal, S.V.; Kamping, E.J.; van Krieken, J.H.J.M.; van Kessel, A.G.; Groenen, P.J.T.A.; Hoogerbrugge, P.M.; Kuiper, R.P. High-resolution genomic profiling of pediatric lymphoblastic lymphomas reveals subtle differences with pediatric acute lymphoblastic leukemias in the B-lineage. Cancer Genet. Cytogenet 2009, 191, 27–33. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Marynen, P.; Cools, J. Genetic insights in the pathogenesis of T-cell acute lymphoblastic leukemia. Haematologica 2005, 90, 1116–1127. [Google Scholar]
- Aifantis, I.; Raetz, E.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390. [Google Scholar] [CrossRef]
- Graux, C.; Cools, J.; Michaux, L.; Vandenberghe, P.; Hagemeijer, A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: From thymocyte to lymphoblast. Leukemia 2006, 20, 1496–1510. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Pieters, R.; Beverloo, H.B.; Meijerink, J.P.P. Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2008, 143, 153–168. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Cayuela, J.M.; Madani, A.; Sanhes, L.; Stern, M.H.; Sigaux, F. Multiple tumor-suppressor gene 1 inactivation is the most frequent genetic alteration in T-cell acute lymphoblastic leukemia. Blood 1996, 87, 2180–2186. [Google Scholar]
- Flex, E.; Petrangeli, V.; Stella, L.; Chiaretti, S.; Hornakova, T.; Knoops, L.; Ariola, C.; Fodale, V.; Clappier, E.; Paoloni, F.; Martinelli, S.; Fragale, A.; Sanchez, M.; Tavolaro, S.; Messina, M.; Cazzaniga, G.; Camera, A.; Pizzolo, G.; Tornesello, A.; Vignetti, M.; Battistini, A.; Cavé, H.; Gelb, B.D.; Renauld, J.; Biondi, A.; Constantinescu, S.N.; Foà, R.; Tartaglia, M. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 2008, 205, 751–758. [Google Scholar] [CrossRef]
- Yokota, S.; Nakao, M.; Horiike, S.; Seriu, T.; Iwai, T.; Kaneko, H.; Azuma, H.; Oka, T.; Takeda, T.; Watanabe, A.; Kikuta, A.; Asami, K.; Sekine, I.; Matsushita, T.; Tsuhciya, T.; Mimaya, J.; Koizumi, S.; Miyake, M.; Nishikawa, K.; Takaue, Y.; Kawano, Y.; Iwai, A.; Ishida, Y.; Matsumoto, K.; Fujimoto, T. Mutational analysis of the N-ras gene in acute lymphoblastic leukemia: A study of 125 Japanese pediatric cases. Int. J. Hematol. 1998, 67, 379–387. [Google Scholar] [CrossRef]
- Paietta, E.; Ferrando, A.A.; Neuberg, D.; Bennett, J.M.; Racevskis, J.; Lazarus, H.; Dewald, G.; Rowe, J.M.; Wiernik, P.H.; Tallman, M.S.; Look, A.T. Activating FLT3 mutations in CD117/KIT+ T-cell acute lymphoblastic leukemias. Blood 2004, 104, 558–560. [Google Scholar] [CrossRef]
- Graux, C.; Cools, J.; Melotte, C.; Quentmeier, H.; Ferrando, A.; Levine, R.; Vermeesch, J.R.; Stul, M.; Dutta, B.; Boeckx, N.; Bosly, A.; Heimann, P.; Uyttebroeck, A.; Mentens, N.; Somers, R.; MacLeod, R.A.F.; Drexler, H.G.; Look, A.T.; Gilliland, D.G.; Michaux, L.; Vandenberghe, P.; Wlodarska, I.; Marynen, P.; Hagemeijer, A. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat. Genet. 2004, 36, 1084–1089. [Google Scholar] [CrossRef]
- Clappier, E.; Cuccuini, W.; Kalota, A.; Crinquette, A.; Cayuela, J.; Dik, W.A.; Langerak, A.W.; Montpellier, B.; Nadel, B.; Walrafen, P.; Delattre, O.; Aurias, A.; Leblanc, T.; Dombret, H.; Gewirtz, A.M.; Baruchel, A.; Sigaux, F.; Soulier, J. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 2007, 110, 1251–1261. [Google Scholar] [CrossRef]
- Lahortiga, I.; De Keersmaecker, K.; Van Vlierberghe, P.; Graux, C.; Cauwelier, B.; Lambert, F.; Mentens, N.; Beverloo, H.B.; Pieters, R.; Speleman, F.; Odero, M.D.; Bauters, M.; Froyen, G.; Marynen, P.; Vandenberghe, P.; Wlodarska, I.; Meijerink, J.P.P.; Cools, J. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat. Genet. 2007, 39, 593–595. [Google Scholar] [CrossRef]
- O'Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; Draetta, G.; Sears, R.; Clurman, B.E.; Look, A.T. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Invest. 2008, 118, 3762–3774. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; Bhagat, G.; Agarwal, A.M.; Basso, G.; Castillo, M.; Nagase, S.; Cordon-Cardo, C.; Parsons, R.; Zúñiga-Pflücker, J.C.; Dominguez, M.; Ferrando, A.A. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef]
- Gutierrez, A.; Tschumper, R.C.; Wu, X.; Shanafelt, T.D.; Eckel-Passow, J.; Huddleston, P.M.; Slager, S.L.; Kay, N.E.; Jelinek, D.F. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B cell lymphocytosis. Blood 2010, 115, 2845–2851. [Google Scholar] [CrossRef]
- Kleppe, M.; Lahortiga, I.; El Chaar, T.; De Keersmaecker, K.; Mentens, N.; Graux, C.; Van Roosbroeck, K.; Ferrando, A.A.; Langerak, A.W.; Meijerink, J.P.P.; Sigaux, F.; Haferlach, T.; Wlodarska, I.; Vandenberghe, P.; Soulier, J.; Cools, J. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 530–535. [Google Scholar] [CrossRef]
- Van Vlierberghe, P.; Palomero, T.; Khiabanian, H.; Van der Meulen, J.; Castillo, M.; Van Roy, N.; De Moerloose, B.; Philippé, J.; González-García, S.; Toribio, M.L.; Taghon, T.; Zuurbier, L.; Cauwelier, B.; Harrison, C.J.; Schwab, C.; Pisecker, M.; Strehl, S.; Langerak, A.W.; Gecz, J.; Sonneveld, E.; Pieters, R.; Paietta, E.; Rowe, J. M.; Wiernik, P.H.; Benoit, Y.; Soulier, J.; Poppe, B.; Yao, X.; Cordon-Cardo, C.; Meijerink, J.; Rabadan, R.; Speleman, F.; Ferrando, A. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 338–342. [Google Scholar] [CrossRef]
- Cardoso, B.A.; Gírio, A.; Henriques, C.; Martins, L.R.; Santos, C.; Silva, A.; Barata, J.T. Aberrant signaling in T-cell acute lymphoblastic leukemia: Biological and therapeutic implications. Braz. J. Med. Biol. Res. 2008, 41, 344–350. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Langerak, A.W. Signaling pathways involved in the development of T-cell acute lymphoblastic leukemia. Haematologica 2008, 93, 493–497. [Google Scholar] [CrossRef]
- Kordes, U.; Krappmann, D.; Heissmeyer, V.; Ludwig, W.D.; Scheidereit, C. Transcription factor NF-κB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia 2000, 14, 399–402. [Google Scholar] [CrossRef]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.; Kee, B.L.; Ferrando, A.; Miele, L.; Aifantis, I. Targeting the NF-κB signaling pathway in Notch1-induced T-cell leukemia. Nat. Med. 2007, 13, 70–77. [Google Scholar] [CrossRef]
- Bellavia, D.; Campese, A.F.; Alesse, E.; Vacca, A.; Felli, M.P.; Balestri, A.; Stoppacciaro, A.; Tiveron, C.; Tatangelo, L.; Giovarelli, M.; Gaetano, C.; Ruco, L.; Hoffman, E.S.; Hayday, A.C.; Lendahl, U.; Frati, L.; Gulino, A.; Screpanti, I. Constitutive activation of NF-κB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO J. 2000, 19, 3337–3348. [Google Scholar] [CrossRef]
- dos Santos, N.R.; Williame, M.; Gachet, S.; Cormier, F.; Janin, A.; Weih, D.; Weih, F.; Ghysdael, J. RelB-dependent stromal cells promote T-cell leukemogenesis. PLoS ONE 2008, 3, e2555. [Google Scholar]
- O'Neil, J.; Ventura, J.; Cusson, N.; Kelliher, M. NF-κB activation in premalignant mouse tal-1/scl thymocytes and tumors. Blood 2003, 102, 2593–2596. [Google Scholar] [CrossRef]
- Espinosa, L.; Cathelin, S.; D'Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Inglés-Esteve, J.; Nomdedeu, J.; Bellosillo, B.; Besses, C.; Abdel-Wahab, O.; Kucine, N.; Sun, S.; Song, G.; Mullighan, C.C.; Levine, R.L.; Rajewsky, K.; Aifantis, I.; Bigas, A. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell 2010, 18, 268–281. [Google Scholar] [CrossRef]
- García, M.G.; Alaniz, L.; Lopes, E.C.; Blanco, G.; Hajos, S.E.; Alvarez, E. Inhibition of NF-κB activity by BAY 11-7082 increases apoptosis in multidrug resistant leukemic T-cell lines. Leuk. Res. 2005, 29, 1425–1434. [Google Scholar] [CrossRef]
- Karin, M. The IκB kinase - a bridge between inflammation and cancer. Cell Res. 2008, 18, 334–342. [Google Scholar] [CrossRef]
- Neumann, M.; Naumann, M. Beyond IκBs: Alternative regulation of NF-κB activity. FASEB J. 2007, 21, 2642–2654. [Google Scholar] [CrossRef]
- Jamieson, C.; McCaffrey, P.G.; Rao, A.; Sen, R. Physiologic activation of T cells via the T cell receptor induces NF-κB. J. Immunol. 1991, 147, 416–420. [Google Scholar]
- Aifantis, I.; Gounari, F.; Scorrano, L.; Borowski, C.; von Boehmer, H. Constitutive pre-TCR signaling promotes differentiation through Ca2+ mobilization and activation of NF-κB and NFAT. Nat. Immunol. 2001, 2, 403–409. [Google Scholar]
- Voll, R.E.; Jimi, E.; Phillips, R.J.; Barber, D.F.; Rincon, M.; Hayday, A.C.; Flavell, R.A.; Ghosh, S. NF-κB activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity 2000, 13, 677–689. [Google Scholar] [CrossRef]
- Rawlings, D.J.; Sommer, K.; Moreno-García, M.E. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat. Rev. Immunol. 2006, 6, 799–812. [Google Scholar] [CrossRef]
- Akagi, T.; Motegi, M.; Tamura, A.; Suzuki, R.; Hosokawa, Y.; Suzuki, H.; Ota, H.; Nakamura, S.; Morishima, Y.; Taniwaki, M.; Seto, M. A novel gene, MALT1 at 18q21, is involved in t(11;18) (q21;q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene 1999, 18, 5785–5794. [Google Scholar] [CrossRef]
- Dierlamm, J.; Baens, M.; Wlodarska, I.; Stefanova-Ouzounova, M.; Hernandez, J.M.; Hossfeld, D.K.; De Wolf-Peeters, C.; Hagemeijer, A.; Van den Berghe, H.; Marynen, P. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood 1999, 93, 3601–3609. [Google Scholar]
- Strzadala, L.; Miazek, A.; Matuszyk, J.; Kisielow, P. Role of thymic selection in the development of thymic lymphomas in TCR transgenic mice. Int. Immunol. 1997, 9, 127–138. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; Xu, W.; Shaffer, A.L.; Wright, G.; Xiao, W.; Powell, J.; Jiang, J.; Thomas, C.J.; Rosenwald, A.; Ott, G.; Muller-Hermelink, H.K.; Gascoyne, R.D.; Connors, J.M.; Johnson, N.A.; Rimsza, L.M.; Campo, E.; Jaffe, E.S.; Wilson, W.H.; Delabie, J.; Smeland, E.B.; Fisher, R.I.; Braziel, R.M.; Tubbs, R.R.; Cook, J.R.; Weisenburger, D.D.; Chan, W.C.; Pierce, S.K.; Staudt, L.M. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Asnafi, V.; Beldjord, K.; Boulanger, E.; Comba, B.; Le Tutour, P.; Estienne, M.; Davi, F.; Landman-Parker, J.; Quartier, P.; Buzyn, A.; Delabesse, E.; Valensi, F.; Macintyre, E. Analysis of TCR, pTα, and RAG-1 in T-acute lymphoblastic leukemias improves understanding of early human T-lymphoid lineage commitment. Blood 2003, 101, 2693–2703. [Google Scholar] [CrossRef]
- Burger, R.; Hansen-Hagge, T.E.; Drexler, H.G.; Gramatzki, M. Heterogeneity of T-acute lymphoblastic leukemia (T-ALL) cell lines: Suggestion for classification by immunophenotype and T-cell receptor studies. Leuk. Res. 1999, 23, 19–27. [Google Scholar] [CrossRef]
- Vacca, A.; Felli, M.P.; Palermo, R.; Di Mario, G.; Calce, A.; Di Giovine, M.; Frati, L.; Gulino, A.; Screpanti, I. Notch3 and pre-TCR interaction unveils distinct Dierlamm, J.; Baens, M pathways in T-cell development and leukemia. EMBO J. 2006, 25, 1000–1008. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O'Neil, J.; Neuberg, D.; Weng, A.P.; Aster, J.C.; Sigaux, F.; Soulier, J.; Look, A.T.; Young, R.A.; Califano, A.; Ferrando, A.A. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef]
- Osipo, C.; Golde, T.E.; Osborne, B.A.; Miele, L.A. Off the beaten pathway: The complex cross talk between Notch and NF-κB. Lab. Invest. 2008, 88, 11–17. [Google Scholar] [CrossRef]
- Oswald, F.; Liptay, S.; Adler, G.; Schmid, R.M. NF-κB2 is a putative target gene of activated Notch-1 via RBP-Jκ. Mol. Cell. Biol. 1998, 18, 2077–2088. [Google Scholar]
- Shin, H.M.; Minter, L.M.; Cho, O.H.; Gottipati, S.; Fauq, A.H.; Golde, T.E.; Sonenshein, G.E.; Osborne, B.A. Notch1 augments NF-κB activity by facilitating its nuclear retention. EMBO J. 2006, 25, 129–138. [Google Scholar] [CrossRef]
- Song, L.L.; Peng, Y.; Yun, J.; Rizzo, P.; Chaturvedi, V.; Weijzen, S.; Kast, W.M.; Stone, P.J.B.; Santos, L.; Loredo, A.; Lendahl, U.; Sonenshein, G.; Osborne, B.; Qin, J.; Pannuti, A.; Nickoloff, B.J.; Miele, L. Notch-1 associates with IKKα and regulates IKK activity in cervical cancer cells. Oncogene 2008, 27, 5833–5844. [Google Scholar] [CrossRef]
- Chang, P.; Draheim, K.; Kelliher, M.A.; Miyamoto, S. NFKB1 is a direct target of the TAL1 oncoprotein in human T leukemia cells. Cancer Res. 2006, 66, 6008–6013. [Google Scholar] [CrossRef]
- Carron, C.; Cormier, F.; Janin, A.; Lacronique, V.; Giovannini, M.; Daniel, M.T.; Bernard, O.; Ghysdael, J. TEL-JAK2 transgenic mice develop T-cell leukemia. Blood 2000, 95, 3891–3899. [Google Scholar]
- dos Santos, N.R.; Ghysdael, J. A transgenic mouse model for TEL-JAK2-induced B-cell lymphoma/leukemia. Leukemia 2006, 20, 182–185. [Google Scholar] [CrossRef]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffé, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; Bernard, O.A. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef]
- Peeters, P.; Raynaud, S.D.; Cools, J.; Wlodarska, I.; Grosgeorge, J.; Philip, P.; Monpoux, F.; Van Rompaey, L.; Baens, M.; Van den Berghe, H.; Marynen, P. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood 1997, 90, 2535–2540. [Google Scholar]
- Schwaller, J.; Frantsve, J.; Aster, J.; Williams, I.R.; Tomasson, M.H.; Ross, T.S.; Peeters, P.; Van Rompaey, L.; Van Etten, R.A.; Ilaria, R.; Marynen, P.; Gilliland, D.G. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J. 1998, 17, 5321–5333. [Google Scholar] [CrossRef]
- Malinge, S.; Monni, R.; Bernard, O.; Penard-Lacronique, V. Activation of the NF-κB pathway by the leukemogenic TEL-Jak2 and TEL-Abl fusion proteins leads to the accumulation of antiapoptotic IAP proteins and involves IKKα. Oncogene 2006, 25, 3589–3597. [Google Scholar] [CrossRef]
- Santos, S.C.; Monni, R.; Bouchaert, I.; Bernard, O.; Gisselbrecht, S.; Gouilleux, F.; Penard-Lacronique, V. Involvement of the NF-κB pathway in the transforming properties of the TEL-Jak2 leukemogenic fusion protein. FEBS Lett. 2001, 497, 148–152. [Google Scholar] [CrossRef]
- Burnett, R.C.; Thirman, M.J.; Rowley, J.D.; Diaz, M.O. Molecular analysis of the T-cell acute lymphoblastic leukemia-associated t(1;7)(p34;q34) that fuses LCK and TCRB. Blood 1994, 84, 1232–1236. [Google Scholar]
- Livolsi, A.; Busuttil, V.; Imbert, V.; Abraham, R.T.; Peyron, J.F. Tyrosine phosphorylation-dependent activation of NF-κB. Requirement for p56 LCK and ZAP-70 protein tyrosine kinases. Eur. J. Biochem. 2001, 268, 1508–1515. [Google Scholar] [CrossRef]
- Hamdane, M.; David-Cordonnier, M.H.; D'Halluin, J.C. Activation of p65 NF-κB protein by p210BCR-ABL in a myeloid cell line (P210BCR-ABL activates p65 NF-κB). Oncogene 1997, 15, 2267–2275. [Google Scholar]
- Reuther, J.Y.; Reuther, G.W.; Cortez, D.; Pendergast, A.M.; Baldwin, A.S. A requirement for NF-κB activation in Bcr-Abl-mediated transformation. Genes Dev. 1998, 12, 968–981. [Google Scholar] [CrossRef]
- Narayan, P.; Holt, B.; Tosti, R.; Kane, L.P. CARMA1 is required for Akt-mediated NF-κB activation in T cells. Mol. Cell. Biol 2006, 26, 2327–2336. [Google Scholar] [CrossRef]
- Garaude, J.; Cherni, S.; Kaminski, S.; Delepine, E.; Chable-Bessia, C.; Benkirane, M.; Borges, J.; Pandiella, A.; Iñiguez, M.A.; Fresno, M.; Hipskind, R.A.; Villalba, M. ERK5 activates NF-κB in leukemic T cells and is essential for their growth in vivo. J.Immunol. 2006, 177, 7607–7617. [Google Scholar]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef]
- Herreros, B.; Sanchez-Aguilera, A.; Piris, M.A. Lymphoma microenvironment: Culprit or innocent? Leukemia 2008, 22, 49–58. [Google Scholar] [CrossRef]
- dos Santos, N.R.; Rickman, D.S.; de Reynies, A.; Cormier, F.; Williame, M.; Blanchard, C.; Stern, M.H.; Ghysdael, J. Pre-TCR expression cooperates with TEL-JAK2 to transform immature thymocytes and induce T-cell leukemia. Blood 2007, 109, 3972–3981. [Google Scholar] [CrossRef]
- Burkly, L.; Hession, C.; Ogata, L.; Reilly, C.; Marconi, L.A.; Olson, D.; Tizard, R.; Cate, R.; Lo, D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature 1995, 373, 531–536. [Google Scholar] [CrossRef]
- Gray, D.H.D.; Seach, N.; Ueno, T.; Milton, M.K.; Liston, A.; Lew, A.M.; Goodnow, C.C.; Boyd, R.L. Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells. Blood 2006, 108, 3777–3785. [Google Scholar] [CrossRef]
- Weih, F.; Carrasco, D.; Durham, S.K.; Barton, D.S.; Rizzo, C.A.; Ryseck, R.P.; Lira, S.A.; Bravo, R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell 1995, 80, 331–340. [Google Scholar] [CrossRef]
- Wu, L.; D'Amico, A.; Winkel, K.D.; Suter, M.; Lo, D.; Shortman, K. RelB is essential for the development of myeloid-related CD8α- dendritic cells but not of lymphoid-related CD8α+ dendritic cells. Immunity 1998, 9, 839–847. [Google Scholar] [CrossRef]
- Bonizzi, G.; Bebien, M.; Otero, D.C.; Johnson-Vroom, K.E.; Cao, Y.; Vu, D.; Jegga, A.G.; Aronow, B.J.; Ghosh, G.; Rickert, R.C.; Karin, M. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. EMBO J. 2004, 23, 4202–4210. [Google Scholar] [CrossRef]
- Buonamici, S.; Trimarchi, T.; Ruocco, M.G.; Reavie, L.; Cathelin, S.; Mar, B.G.; Klinakis, A.; Lukyanov, Y.; Tseng, J.; Sen, F.; Gehrie, E.; Li, M.; Newcomb, E.; Zavadil, J.; Meruelo, D.; Lipp, M.; Ibrahim, S.; Efstratiadis, A.; Zagzag, D.; Bromberg, J.S.; Dustin, M.L.; Aifantis, I. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 2009, 459, 1000–1004. [Google Scholar] [CrossRef]
- Scupoli, M.T.; Vinante, F.; Krampera, M.; Vincenzi, C.; Nadali, G.; Zampieri, F.; Ritter, M.A.; Eren, E.; Santini, F.; Pizzolo, G. Thymic epithelial cells promote survival of human T-cell acute lymphoblastic leukemia blasts: The role of interleukin-7. Haematologica 2003, 88, 1229–1237. [Google Scholar]
- Scupoli, M.T.; Perbellini, O.; Krampera, M.; Vinante, F.; Cioffi, F.; Pizzolo, G. Interleukin 7 requirement for survival of T-cell acute lymphoblastic leukemia and human thymocytes on bone marrow stroma. Haematologica 2007, 92, 264–266. [Google Scholar] [CrossRef]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; Di Mario, G.; Screpanti, I.; Ponzoni, M.; Doglioni, C.; Amadori, A. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar] [CrossRef]
- Yan, X.Q.; Sarmiento, U.; Sun, Y.; Huang, G.; Guo, J.; Juan, T.; Van, G.; Qi, M.Y.; Scully, S.; Senaldi, G.; Fletcher, F.A. A novel Notch ligand, Dll4, induces T-cell leukemia/lymphoma when overexpressed in mice by retroviral-mediated gene transfer. Blood 2001, 98, 3793–3799. [Google Scholar] [CrossRef]
- Pui, C.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Hannun, Y.A. Apoptosis and the dilemma of cancer chemotherapy. Blood 1997, 89, 1845–1853. [Google Scholar]
- Das, K.C.; White, C.W. Activation of NF-κB by antineoplastic agents. Role of protein kinase C. J. Biol. Chem. 1997, 272, 14914–14920. [Google Scholar] [CrossRef]
- Nakanishi, C.; Toi, M. Nuclear factor-κB inhibitors as sensitizers to anticancer drugs. Nat. Rev. Cancer 2005, 5, 297–309. [Google Scholar] [CrossRef]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J. Clin. Invest. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- Wuerzberger-Davis, S.M.; Chang, P.; Berchtold, C.; Miyamoto, S. Enhanced G2-M arrest by nuclear factor-κB-dependent p21waf1/cip1 induction. Mol. Cancer Res. 2005, 3, 345–353. [Google Scholar] [CrossRef]
- Chiarini, F.; Falà, F.; Tazzari, P.L.; Ricci, F.; Astolfi, A.; Pession, A.; Pagliaro, P.; McCubrey, J.A.; Martelli, A.M. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009, 69, 3520–3528. [Google Scholar] [CrossRef]
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181. [Google Scholar] [CrossRef]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [Google Scholar] [CrossRef]
- Qiuping, Z.; Jei, X.; Youxin, J.; Wei, J.; Chun, L.; Jin, W.; Qun, W.; Yan, L.; Chunsong, H.; Mingzhen, Y.; Qingping, G.; Kejian, Z.; Zhimin, S.; Qun, L.; Junyan, L.; Jinquan, T. CC chemokine ligand 25 enhances resistance to apoptosis in CD4+ T cells from patients with T-cell lineage acute and chronic lymphocytic leukemia by means of livin activation. Cancer Res. 2004, 64, 7579–7587. [Google Scholar] [CrossRef]
- Crazzolara, R.; Kreczy, A.; Mann, G.; Heitger, A.; Eibl, G.; Fink, F.M.; Möhle, R.; Meister, B. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2001, 115, 545–553. [Google Scholar] [CrossRef]
- Winter, S.S.; Sweatman, J.J.; Lawrence, M.B.; Rhoades, T.H.; Hart, A.L.; Larson, R.S. Enhanced T-lineage acute lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA-1 and ICAM-1. Br. J. Haematol. 2001, 115, 862–871. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dos Santos, N.R.; Ghezzo, M.N.; Da Silva, R.C.; Fernandes, M.T. NF-κB in T-cell Acute Lymphoblastic Leukemia: Oncogenic Functions in Leukemic and in Microenvironmental Cells. Cancers 2010, 2, 1838-1860. https://doi.org/10.3390/cancers2041838
Dos Santos NR, Ghezzo MN, Da Silva RC, Fernandes MT. NF-κB in T-cell Acute Lymphoblastic Leukemia: Oncogenic Functions in Leukemic and in Microenvironmental Cells. Cancers. 2010; 2(4):1838-1860. https://doi.org/10.3390/cancers2041838
Chicago/Turabian StyleDos Santos, Nuno R., Marinella N. Ghezzo, Ricardo C. Da Silva, and Mónica T. Fernandes. 2010. "NF-κB in T-cell Acute Lymphoblastic Leukemia: Oncogenic Functions in Leukemic and in Microenvironmental Cells" Cancers 2, no. 4: 1838-1860. https://doi.org/10.3390/cancers2041838
APA StyleDos Santos, N. R., Ghezzo, M. N., Da Silva, R. C., & Fernandes, M. T. (2010). NF-κB in T-cell Acute Lymphoblastic Leukemia: Oncogenic Functions in Leukemic and in Microenvironmental Cells. Cancers, 2(4), 1838-1860. https://doi.org/10.3390/cancers2041838