2.1. DLEC1 Expression Is Altered by Epigenetic Regulations in HCC Cell Lines
DLEC1 has been found to be silenced in human cancers including HCC. Consistently, its overexpression in cell lines led to a decrease in colony formation, cell growth and cell size, and an induction of G1 arrest in the cell cycle [
6]. The silencing of DLEC1 is associated with DNA methylation [
6,
10], as well as histone acetylation [
11,
16,
21]. However, the reported epigenetic studies of DLEC1 have been focused on clinical observations in human cancers without revealing underlying mechanisms.
In the case of HCC associated with HBV infection, epigenetic modifications may well implicate viral proteins including HBx, which has been linked to the silencing of tumor suppressor genes and development of HCC [
3]. Therefore in this study, we focused on DLEC1 expression in HCC cell lines, and explored the role of HBV replication and HBx protein in the regulation of DLEC1 expression.
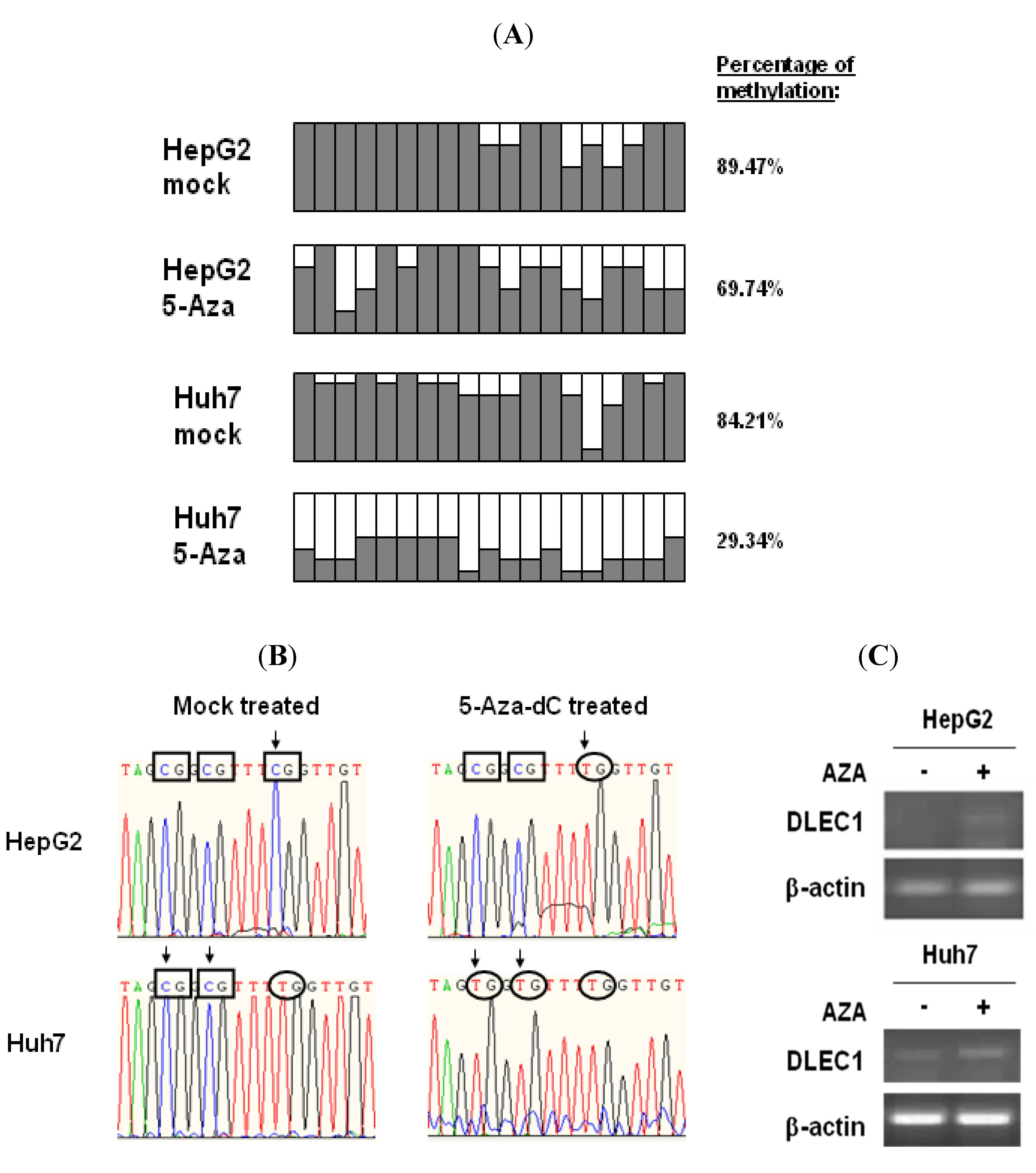
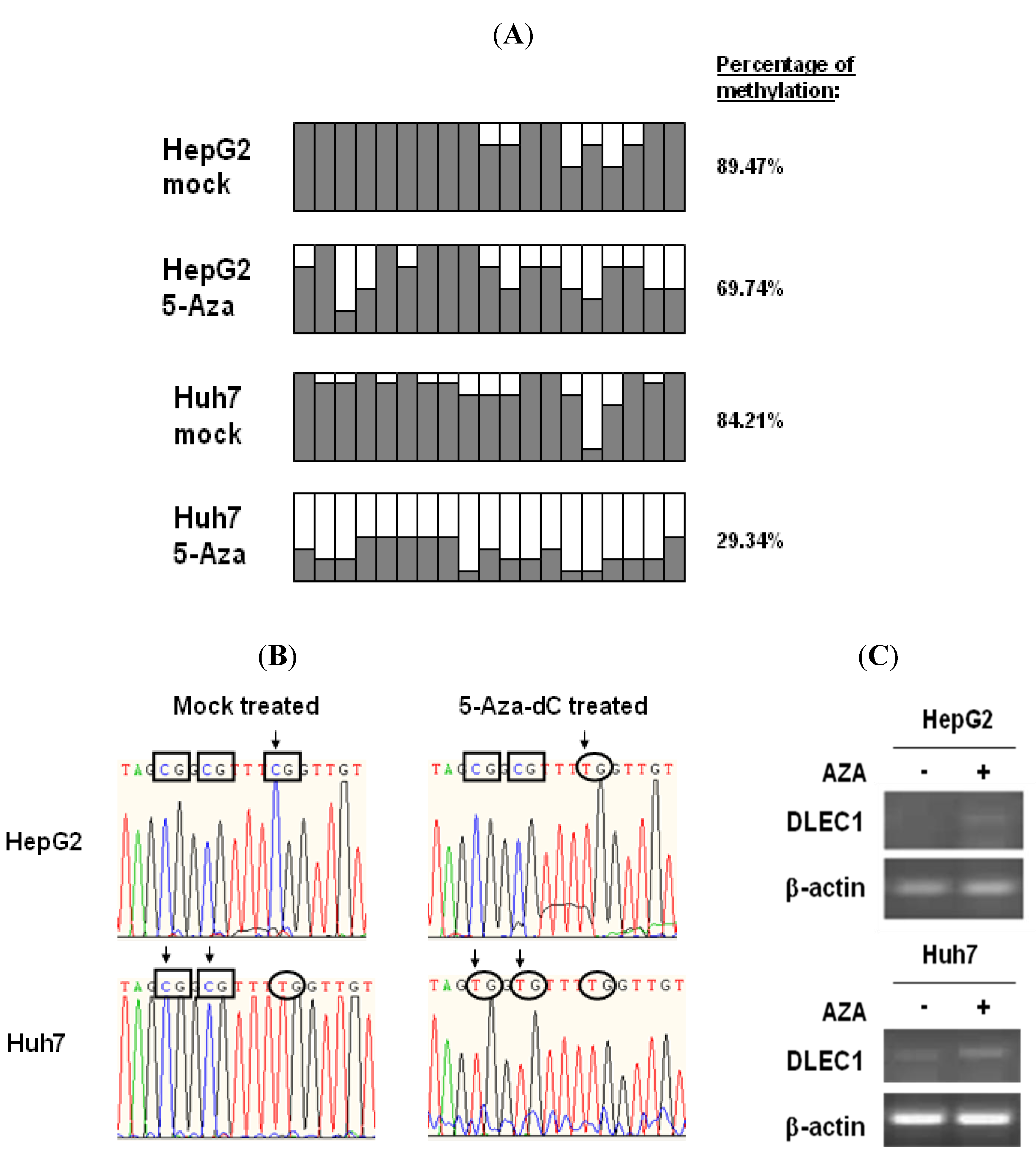
To assess the status of DNA methylation in the DLEC1 promoter, we performed bisulfite genome sequencing (BGS) of the DLEC1 promoter in two of HCC cell lines, HepG2 and Huh7.
The DLEC1 promoters of the above cell lines were analyzed after the treatment of DNMT inhibitor 5-Aza-dC or mock treatment. Results shown in
Figure 1A indicate that the DLEC1 promoter was highly methylated in both Huh7 (84.21%,
Figure 1A row 1) and HepG2 cells (89.47%,
Figure 1A row 3). On the other hand, methylation in the DLEC1 promoter was decreased after the cells were incubated with the DNMT inhibitor 5-Aza-dC. Specifically, the methylation was found to be at 29.34% in Huh7 (
Figure 1A row 2), 69.74% in HepG2 (
Figure 1A row 4). Our findings in HepG2 were consistent with the reported investigation on the methylation of the DLEC1 promoter in HepG2 [
6]. Combined with our findings in Huh7 cells, our results indicated that DLEC1 was significantly methylated in HCC-derived cell lines.
Representative BGS results of CpG methylation status changed before and after incubation of the cells with 5-Aza-dC are shown in
Figure 1B. In HepG2 cells, the three presented CpG sites in DLEC1 promoter were methylated before incubation with 5-Aza-dC (
Figure 1B, upper left panel); while one of them (rectangle in left panel with arrow) was demethylated after incubation (
Figure 1B, upper right panel). The methylation status of the same three CpG sites was different in Huh7 cells. Before the incubation of the cells with 5-Aza-dC, two of the CpG sites were methylated (lower left panel,
Figure 1B), whereas these two CpG sites were demethylated after incubation (lower right panel,
Figure 1B).
To correlate the methylation of DLEC1 promoter with its expression, RT-PCR was performed in both HepG2 and Huh7 cells. As shown in
Figure 1C, DLEC1 expression was upregulated in both types of cells incubated with 5-Aza-dC compared with those without any incubation with 5-Aza-dC. Our results therefore suggest that DLEC1 expression was inhibited in HepG2 and Huh7 cells and the DNA methylation was directly involved in this suppression.
Figure 1.
DLEC1 promoter hypermethylation and its expression in HCC cell lines. (A) Promoter methylation analysis by bisulfite genome sequencing of DLEC1 5’ CpG sites in both of HepG2 and Huh7 with or without treatment of 5-Aza-dC. 19 CpG dinucleotides were analyzed in the 5’ promoter of DLEC1 (represented as 19 columns). 6–8 clones of each sample were sequenced and the results were presented as follow: Open area indicates the percentage of unmethylated CpG sites, while dark area indicates percentage of methylated CpG sites. The quantitative result in percentage of methylated CpG sites was listed on the right of the column groups. Three independent experiments were carried out and representative result from one of them is listed here. (B) Representative bisulfite sequencing results of the DLEC1 promoter region showing the CpG methylation status of bisulfite treated DNA from two cell lines mock treated or 5-Aza-dC treated. Rectangles indicate the methylated CpG sites, while ellipses for unmethylated CpG sites. Arrows mark the CpG sites whose methylation status was changed by 5-Aza-dC treatment. (C) RT-PCR analysis of DLEC1 expression in two cell lines mock treated or 5-Aza-dC treated.
Figure 1.
DLEC1 promoter hypermethylation and its expression in HCC cell lines. (A) Promoter methylation analysis by bisulfite genome sequencing of DLEC1 5’ CpG sites in both of HepG2 and Huh7 with or without treatment of 5-Aza-dC. 19 CpG dinucleotides were analyzed in the 5’ promoter of DLEC1 (represented as 19 columns). 6–8 clones of each sample were sequenced and the results were presented as follow: Open area indicates the percentage of unmethylated CpG sites, while dark area indicates percentage of methylated CpG sites. The quantitative result in percentage of methylated CpG sites was listed on the right of the column groups. Three independent experiments were carried out and representative result from one of them is listed here. (B) Representative bisulfite sequencing results of the DLEC1 promoter region showing the CpG methylation status of bisulfite treated DNA from two cell lines mock treated or 5-Aza-dC treated. Rectangles indicate the methylated CpG sites, while ellipses for unmethylated CpG sites. Arrows mark the CpG sites whose methylation status was changed by 5-Aza-dC treatment. (C) RT-PCR analysis of DLEC1 expression in two cell lines mock treated or 5-Aza-dC treated.
![Cancers 02 01689 g001]()
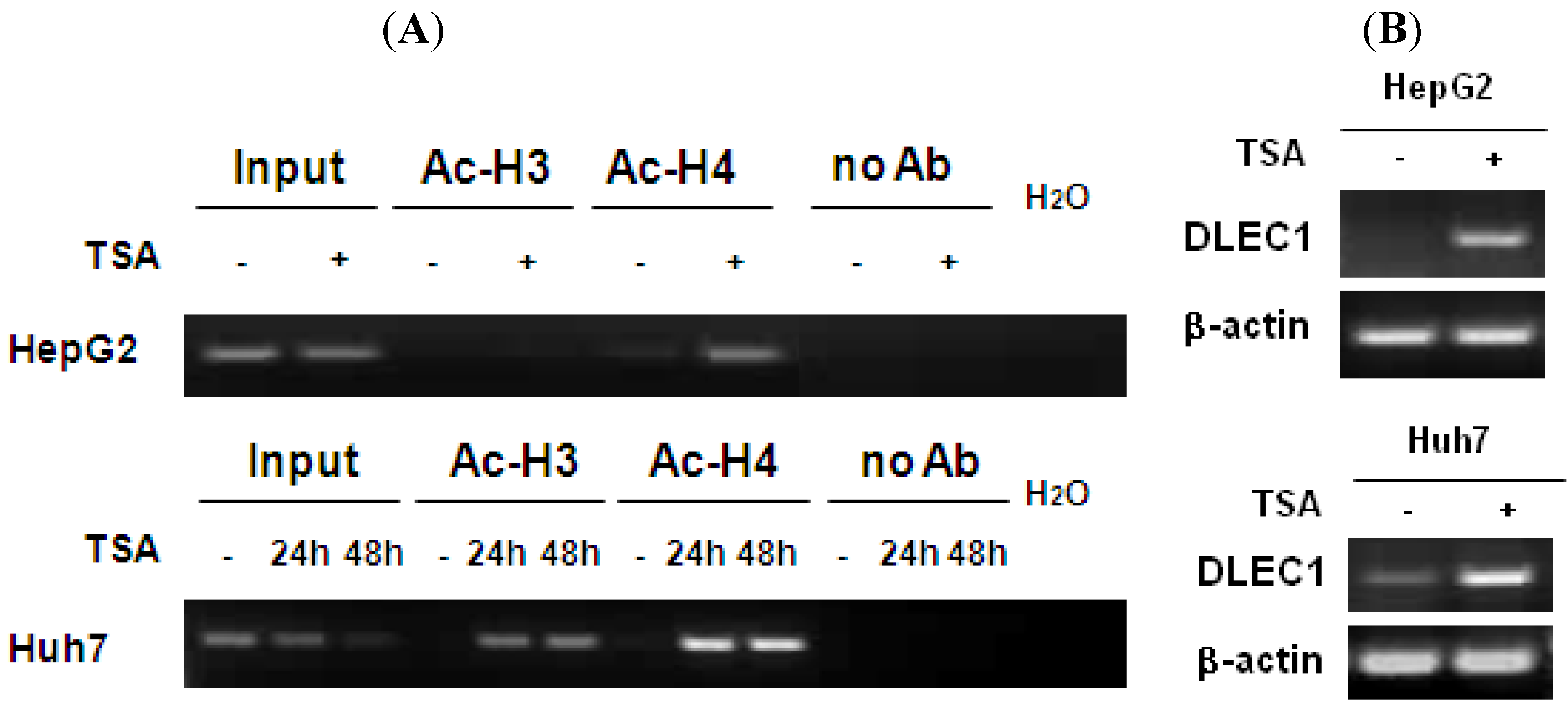
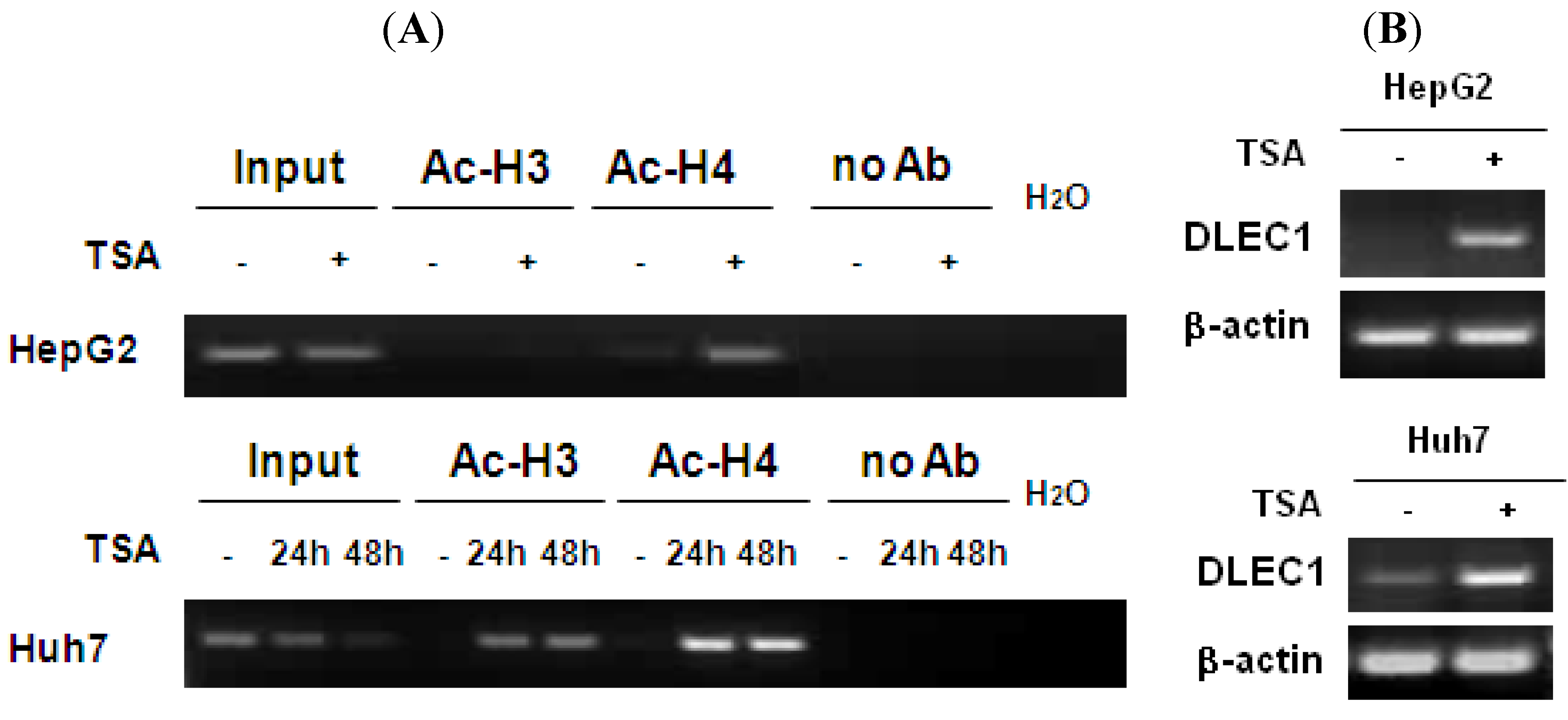
In addition to DNA methylation, the involvement of histone acetylation in regulating DLEC1 expression was analyzed by chromatin immunoprecipitation (ChIP) assay. The results shown in
Figure 2A indicate that low level of histone H4 acetylation was detected in both HepG2 (lane 5, upper panel) and Huh7 (lane 7, lower panel). In addition, the acetylation of histone H3 was not detected in HepG2 (lane 3, upper panel) and Huh7 (lane 4, lower panel). The acetylation levels for both histones 3 and 4 increased in Huh7 cells (lanes 5, 6, 8 and 9, lower panel) incubated with the histone deacetylase inhibitor TSA, for HepG2, the acetylated histone H4 was significantly increased (lanes 6, upper panel), while the increase of acetylation of histone H3 was less significant (lanes 4, upper panel).
Figure 2.
Histone H3 and H4 hypoacetylation in DLEC1 5’ region and its expression in HCC cell lines. (A) ChIP analysis of the 5’ region of DLEC1 in HepG2 and Huh7 mock treated or TSA treated. DNA sequences cross-linked to acetylated histone H3 and H4 were co-immunoprecipitated as part of the chromatin complex with antibodies specific to acetylated histone H3 and H4. The reaction without antibody immunoprecipitation (no Ab) serves as negative control. Input indicates 1% of total DNA prior to immunoprecipitation. (B) RT-PCR analysis of DLEC1 expression in two cell lines mock treated or TSA treated.
Figure 2.
Histone H3 and H4 hypoacetylation in DLEC1 5’ region and its expression in HCC cell lines. (A) ChIP analysis of the 5’ region of DLEC1 in HepG2 and Huh7 mock treated or TSA treated. DNA sequences cross-linked to acetylated histone H3 and H4 were co-immunoprecipitated as part of the chromatin complex with antibodies specific to acetylated histone H3 and H4. The reaction without antibody immunoprecipitation (no Ab) serves as negative control. Input indicates 1% of total DNA prior to immunoprecipitation. (B) RT-PCR analysis of DLEC1 expression in two cell lines mock treated or TSA treated.
To correlate the histone acetylation with DLEC1 expression, RT-PCR was performed. Results shown in
Figure 2B indicate that the expression of DLEC1 was reactivated in both HepG2 cells (upper panel) and Huh7 cells (lower panel) after their incubation with TSA. Hence, DLEC1 expression was significantly correlated with their histone acetylation status (
Figure 2B). Interestingly, the increase of DLEC1 expression after the incubation of cells with TSA (
Figure 2B) was more significant than that in cells incubated with 5-Aza-dC (
Figure 1C). These results suggest that both DNA hypermethylation and histone hypoacetylation are involved in inhibiting DLEC1 expression, with histone acetylation having a more active role.
2.2. DLEC1 Is Inhibited in HBV-Producing HepG2.2.15 Cells
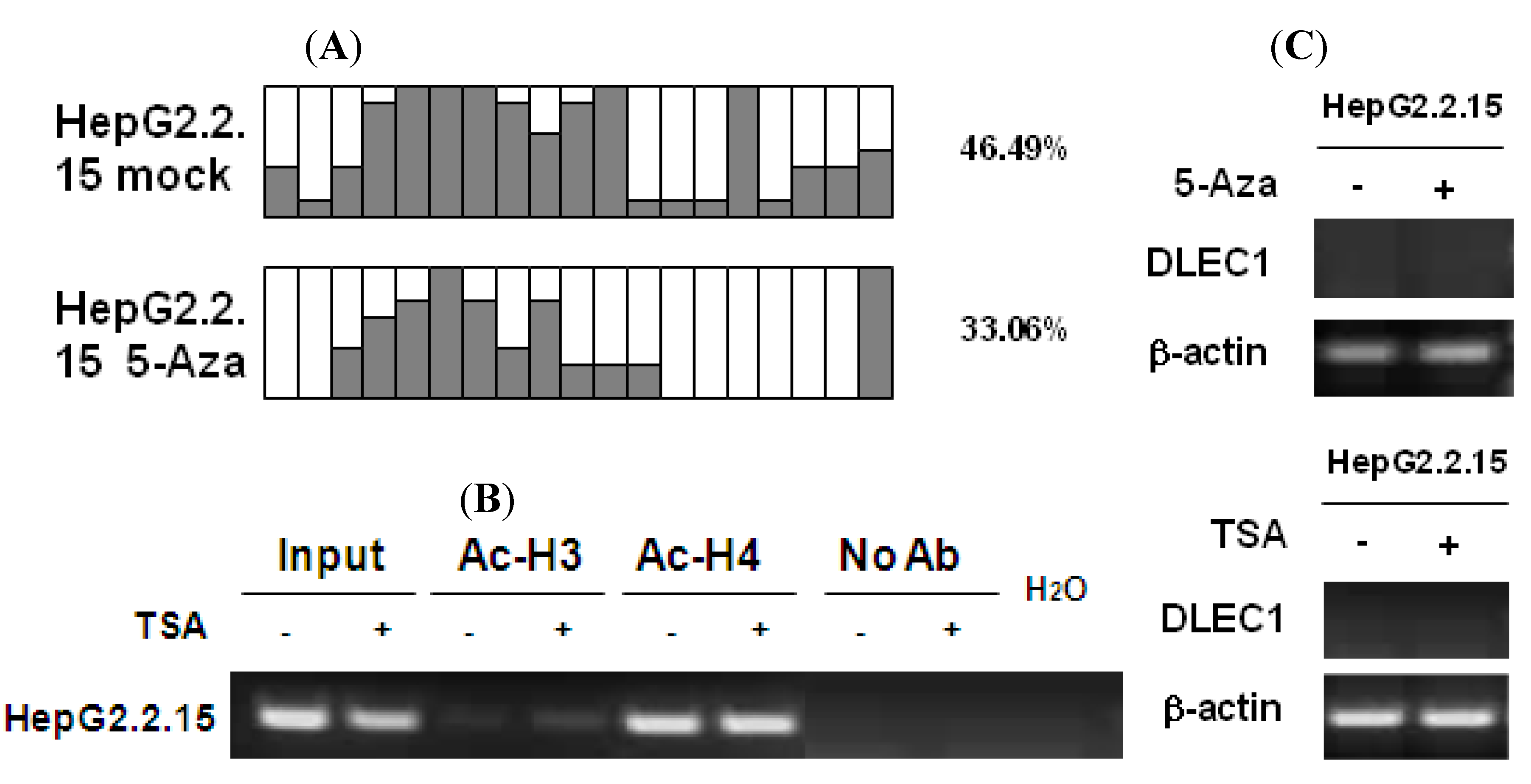
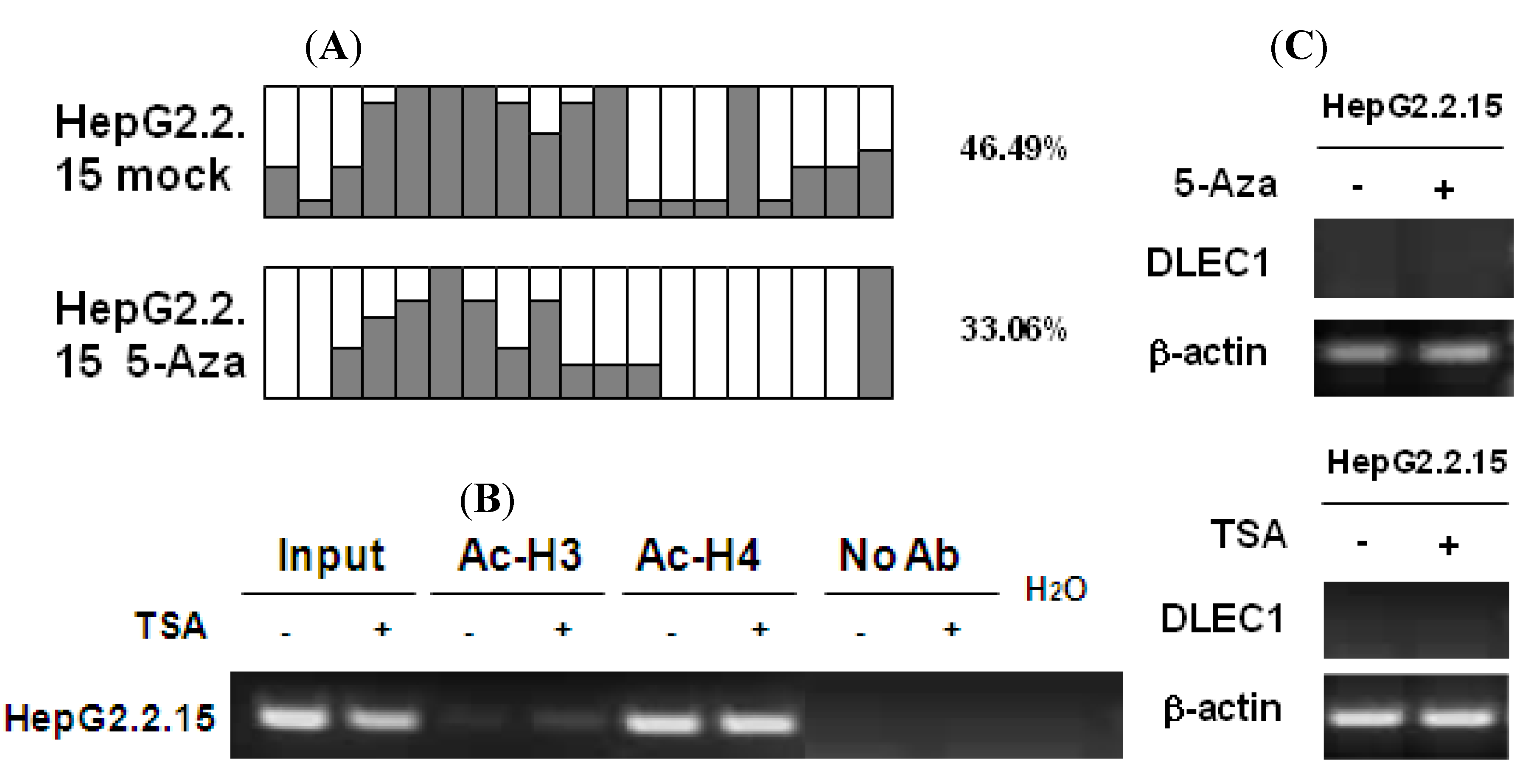
To investigate the effects of HBV replication on epigenetic modifications in the context of DLEC1 expression, analyses on DNA methylation and histone acetylation were carried out in HepG2.2.15 cells which constitutively produce HBV particles. HepG2.2.15 is derived from HepG2 cells with integration of a replicative HBV genome (genotype D), and it supports both assembly and secretion of replicative intermediates of HBV DNA and Dane particles [
22]. Results of methylation analysis by BGS shown in
Figure 3A indicated that the methylation of DLEC1 promoter was lower in HepG2.2.15 (46.49%) than that in HepG2 (89.47%,
Figure 1A). It was further decreased to 33.06% after the incubation of the cells with 5-Aza-dC (
Figure 3A). In addition, the acetylation level of histones H3 and H4 was found to be higher in HepG2.2.15 (lanes 3 and 5,
Figure 3B) compared with that in HepG2 (lanes 3 and 5,
Figure 2A). The level of acetylation of histone 3 was further increased after HepG2.2.15 cells were incubated with TSA (lane 4,
Figure 3B).
In contrast to HepG2 and Huh7 cells, the expression of DLEC1 in HepG2.2.15 was not detectable by RT-PCR (
Figure 3C). DLEC1 expression was not reactivated in HepG2.2.15 cells incubated with either 5-Aza-dC or TSA (
Figure 3C)
Our results therefore suggest that HBV replication is involved in the suppression of DLEC1 in HepG2.2.15, and likely to be independent of the epigenetic mechanisms (including both DNA methylation and histone acetylation). However, the exact underlying mechanism remains unclear. On the other hand, since the HBV genotype expressed in HepG2.2.15 was genotype D, the association between HBV genotype and DLEC1 regulation was further investigated.
Figure 3.
DNA methylation and histone acetylation status of DLEC1 and its expression in HepG2.2.15 cells. (A) Promoter methylation analysis by bisulfite genome sequencing of DLEC1 5’ CpG sites in HepG2.2.15 with or without treatment of 5-Aza-dC. Open area indicates the percentage of unmethylated CpG sites, while dark area indicates percentage of methylated CpG sites. The quantitative result in percentage of methylated CpG sites is listed on the right of the column groups. Three independent experiments were carried out and representative result from one of them was listed here. (B) ChIP analysis of 5’ region of DLEC1 in HepG2.2.15 mock treated or TSA treated. Input indicated 1% of total DNA prior to immunoprecipitation. (C) RT-PCR analysis of DLEC1 expression in HepG2.2.15 treated by either 5-Aza-dC or TSA.
Figure 3.
DNA methylation and histone acetylation status of DLEC1 and its expression in HepG2.2.15 cells. (A) Promoter methylation analysis by bisulfite genome sequencing of DLEC1 5’ CpG sites in HepG2.2.15 with or without treatment of 5-Aza-dC. Open area indicates the percentage of unmethylated CpG sites, while dark area indicates percentage of methylated CpG sites. The quantitative result in percentage of methylated CpG sites is listed on the right of the column groups. Three independent experiments were carried out and representative result from one of them was listed here. (B) ChIP analysis of 5’ region of DLEC1 in HepG2.2.15 mock treated or TSA treated. Input indicated 1% of total DNA prior to immunoprecipitation. (C) RT-PCR analysis of DLEC1 expression in HepG2.2.15 treated by either 5-Aza-dC or TSA.
2.3. DLEC1 Expression Is Modulated by HBx Genotypes
Among the HBV proteins, HBx has been suggested in epigenetic regulation of gene expression [
3,
18] and recent evidence has revealed genotype specific effects of HBx in host cells [
15,
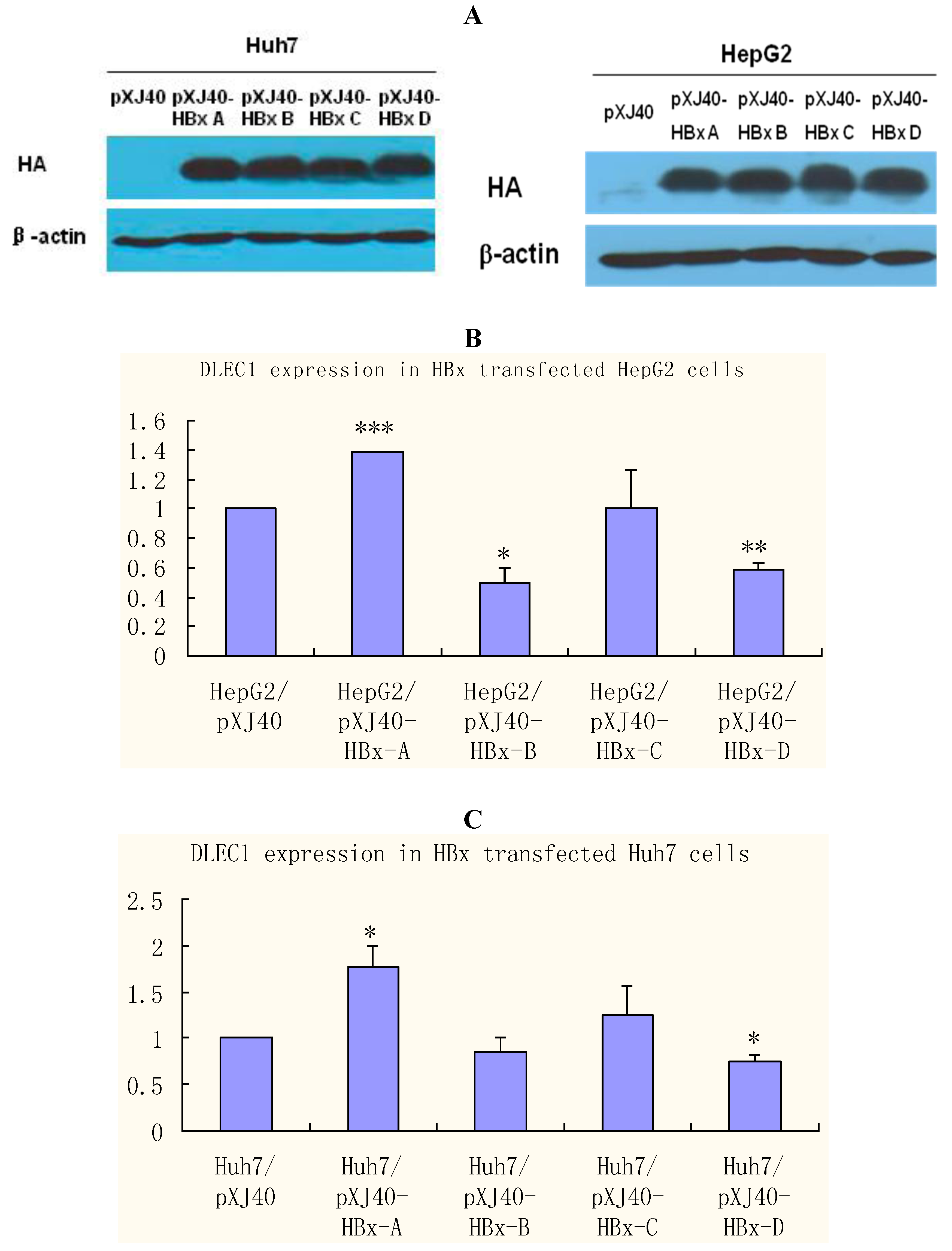
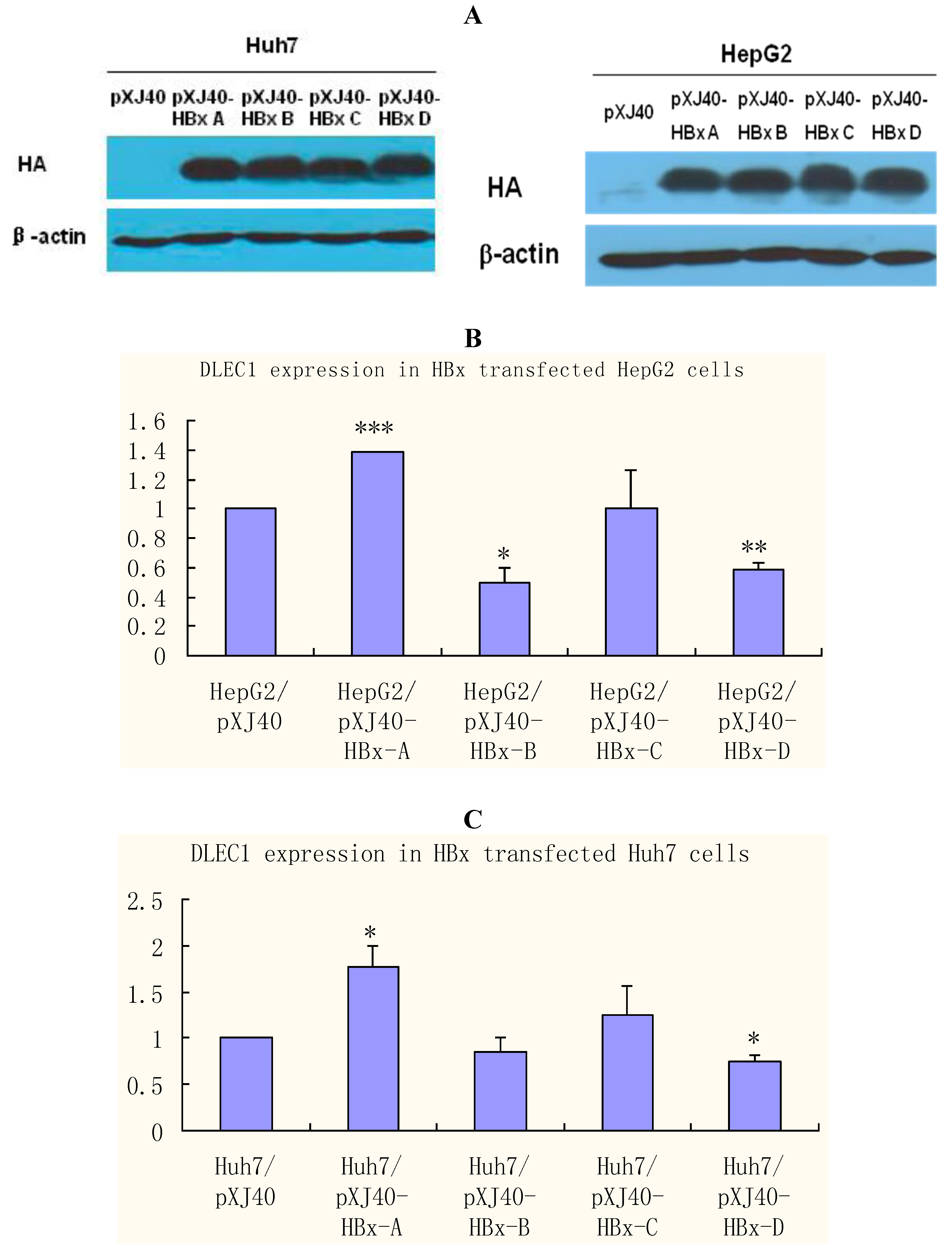
23]. HBx was therefore selected to investigate further the role of HBV in epigenetic regulations of DLEC1. HepG2 and Huh7 cells were transfected separately with plasmids containing HBx of different genotypes (A, B, C and D). The results of Western blot analysis shown in
Figure 4A indicate that the expression of HBx was comparable among different genotypes, whereas no HBx was detected in control cells transfected with the empty plasmid pXJ40.
DLEC1 expression was then examined in cells transfected with HBx of various genotypes by real-time RT-PCR (
Figure 4B and C). The results in HepG2 cells shown in
Figure 4B indicate that DLEC1 expression was modulated in relation to HBx genotypes. In comparison with the level in cells transfected with the empty plasmid pXJ40 (column 1), DLEC1 expression was significantly increased in cells transfected with HBx genotype A (column 2), and significantly decreased in cells transfected with either HBx genotype B or D (columns 3 and 5, respectively). In contrast, there was no significant effect of HBx C on expression of DLEC1 compared with that in cells transfected with pXJ40 (column 4). A similar pattern was observed in Huh7 cells (
Figure 4C).
Taken together, our results suggest that DLEC1 expression is modulated differently in cells transfected with different HBx genotypes.
The biological significance of demethylation/acetylation of DLEC1 and its up-regulation HBx may be of particular interest. HBx has been generally considered as an onco-protein that promotes tumorigenesis. Our results appeared to suggest the opposite, in that HBx enhanced the expression of the tumor suppressor gene, DLEC1 leading to the suppression of tumor formation.
HBx has been shown to be a multifunctional viral protein involved in a number of cellular activities with contrasting effects. Therefore, it is not unexpected to see some contradictory phenotypes related to HBx functions, such as the role of HBx in apoptosis [
24], both pro-apoptotic [
15,
25] and anti-apoptotic [
26,
27]. Our findings on the upregulation of TSGs by HBx were consistent with reports on other TSGs. For example, for SOCS-1, which is silenced by hypermethylation in HCC and shows growth-suppression activity [
28], its CpG island hypermethylation has been found to be inversely associated with HBV infection status [
29]. Similarly, IGFBP3, a potential tumor suppressor previously reported to be repressed in HCC, has been found to be both downregulated [
3] and upregulated [
30] by HBx. This contradiction suggests a role of HBx genotypes.
Therefore, the effects of HBx genotypes on the regulation of DLEC1 expression as well as epigenetic alterations of DLEC1 promoter were further investigated in this study.
2.4. DLEC1 Is Modulated by HBx-Modulated Epigenetic Modifications
Inspired by the genotype specific effects of HBx on DLEC1 expression, correlation of such effects to epigenetic alterations was next examined. Both BGS and ChIP assay were performed to evaluate the DNA methylation and histone acetylation status in Huh7 cells transfected with HBx of different genotypes (
Figure 5).
Figure 4.
DLEC1 expression in HBx genotype A-D expressing HepG2 and Huh7. (A) Western analysis of HBx expression in HepG2 and Huh7 cells. HBx expression was detected by HA tag contained in the vector pXJ40. β-actin was detected as the internal control. (B) Analysis of DLEC1 expression in HBx genotype A-D expressing HepG2 by real-time RT-PCR. (C) Analysis of DLEC1 expression in HBx genotype A-D expressing Huh7 by real-time RT-PCR. Experiment was done three times. pXJ40 was included as an empty vector control. Experimental differences were tested for statistical significance using t-test. * p < 0.05; ** p < 0.01; ***p < 0.001.
Figure 4.
DLEC1 expression in HBx genotype A-D expressing HepG2 and Huh7. (A) Western analysis of HBx expression in HepG2 and Huh7 cells. HBx expression was detected by HA tag contained in the vector pXJ40. β-actin was detected as the internal control. (B) Analysis of DLEC1 expression in HBx genotype A-D expressing HepG2 by real-time RT-PCR. (C) Analysis of DLEC1 expression in HBx genotype A-D expressing Huh7 by real-time RT-PCR. Experiment was done three times. pXJ40 was included as an empty vector control. Experimental differences were tested for statistical significance using t-test. * p < 0.05; ** p < 0.01; ***p < 0.001.
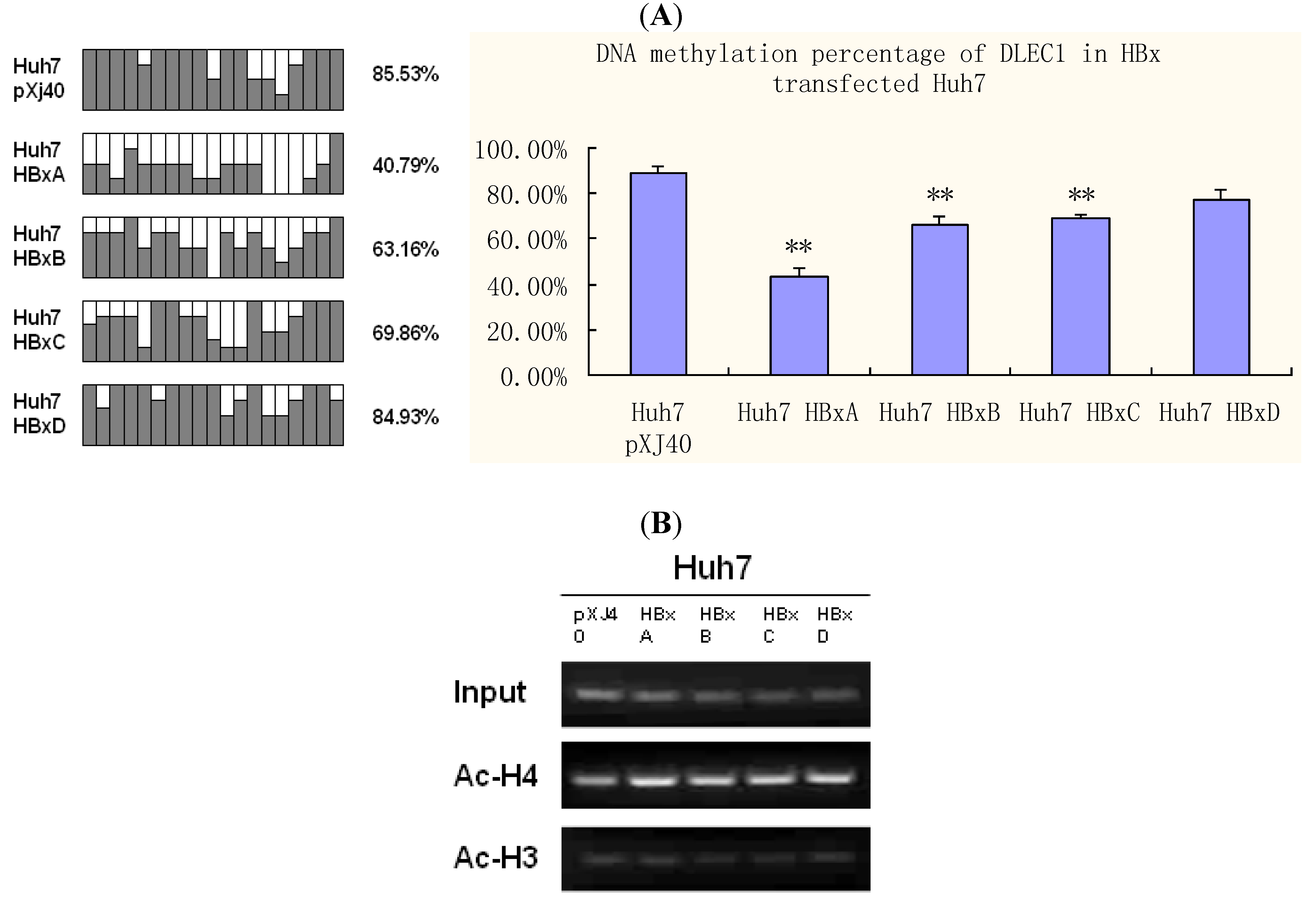
Figure 5.
DNA methylation and histone acetylation status of DLEC1 in Huh7 expressing HBx genotypes A-D. (A) Promoter methylation analysis by bisulfite genome sequencing of DLEC1 5’ CpG sites in Huh7 transfected by HBx genotypes A-D. Open area indicates the percentage of unmethylated CpG sites, while dark area indicates percentage of methylated CpG sites. The quantitative result in percentage of methylated CpG sites is listed on the right of the column groups. Three independent experiments were carried out and representative result from one of them was listed here as the upper panel; the lower panel summarized the results from all three experiments. (B) ChIP analysis of the 5’ region of DLEC1 in Huh7 expressing HBx. Input indicates 1% of total DNA prior to immunoprecipitation. pXJ40 was included as an empty vector control. Experiments were done three times. Experimental differences were tested for statistical significance using t-test. * p < 0.05; **p < 0.01.
Figure 5.
DNA methylation and histone acetylation status of DLEC1 in Huh7 expressing HBx genotypes A-D. (A) Promoter methylation analysis by bisulfite genome sequencing of DLEC1 5’ CpG sites in Huh7 transfected by HBx genotypes A-D. Open area indicates the percentage of unmethylated CpG sites, while dark area indicates percentage of methylated CpG sites. The quantitative result in percentage of methylated CpG sites is listed on the right of the column groups. Three independent experiments were carried out and representative result from one of them was listed here as the upper panel; the lower panel summarized the results from all three experiments. (B) ChIP analysis of the 5’ region of DLEC1 in Huh7 expressing HBx. Input indicates 1% of total DNA prior to immunoprecipitation. pXJ40 was included as an empty vector control. Experiments were done three times. Experimental differences were tested for statistical significance using t-test. * p < 0.05; **p < 0.01.
The results shown in
Figure 5A indicate that DNA methylation of the DLEC1 promoter in Huh7 transfected by HBx A-D varied from each other. The lowest percentage of methylation was in Huh7 transfected by HBx A (40.79%), which was half of that observed in cells transfected by vector pXJ40 (85.53%). HBx genotype B (63.16%) and C (69.86%) also led to lower percentage of methylation than vector, but HBx D (84.93%) had no obvious effect on DNA methylation compared with the vector control.
On the other hand, histone acetylation status was explored by ChIP as indicated in
Figure 5B. One percent of the total DNA before the immunoprecipitation was used as the input control. Compared with the input control, the acetylated histone H4 (
Figure 5B, panel 2) was increased to a different extent in Huh7 transfected by HBx of various genotypes, especially in HBx A (
Figure 5B, panel 2, lane 2), compared with that in cells transfected with pXJ40 (lane 1). No significant changes in the acetylation of histone 3 were observed in cells transfected with pXJ40 or individual HBx genotypes (
Figure 5B, panel 3).
Combined with DNA methylation alterations, our results suggest a significant role of HBx genotype A in inducing epigenetic modifications, which in turn modulates the expression of DLEC1. Our results therefore indicate that the DLEC1 expression is modulated by epigenetic modifications, which are influenced by HBx in a genotype specific manner.
Among the four genotypes of HBx, HBx A showed significant correlation between epigenetic analyses and DLEC1 expression. Interestingly, HBx D had no significant effect on DNA methylation (
Figure 5A) nor on the histone acetylation. However, its expression led to a decrease in DLEC1 expression. Our findings indicate that the HBV genotype D repressed DLEC1 in HepG2.2.15 independently of the epigenetic alterations in its promoter (part 2.2). However, the underlying mechanism of repression of DLEC1 by HBV/HBx genotype D remains to be explored.
The mechanism of genotype-specific effects of HBx on epigenetic alterations of DLEC1 needs further characterization. One possible focus for such investigations may be the proline-rich region identified in our lab [
23]. This proline-rich region is hitherto the only noticeable variable region among various HBx genotypes. In addition to its role in interacting with cytoskeletal proteins [
31,
32], further investigation should shed new light on the role of this proline-rich region in HBx-mediated epigenetic alterations. Another possible mechanism could be involved here is the direct interaction of HBx with
de novo DNA methyltransferase DNMT3A [
30]. It has been reported that HBx recruits DNMT3A to the promoter of some genes and subsequently silenced their transcription via de novo DNA methylation; HBx can also trigger the transcription of some genes via deprivation of DNMT3A from their promoters [
30]. In our case, transcription of DLEC1 could be affected by HBx A via deprivation of DNMT3A from its promoter and thus upregulated. The sequence variations among different genotypes of HBx may as well affect the interaction of HBx with DNMT3A and lead to genotype specific effects on DLEC1 expression. The detailed mechanism remains to be explored.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}