The Evolution of Biomarkers in Thyroid Cancer—From Mass Screening to a Personalized Biosignature

Abstract
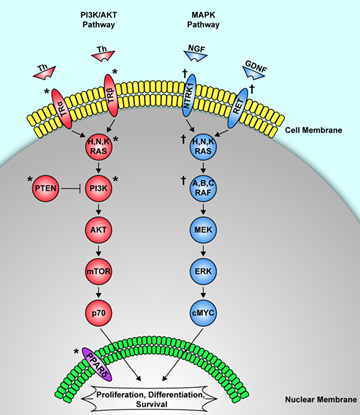
:
1. Introduction
2. Serum-Based Biomarkers
2.1. Calcitonin
2.2. Thyroglobulin
3. Mutation Based Biomarkers

{kind=link}
{kind=link}
| Mutation | Adenoma | PTC | FTC | Anaplastic | Ref. |
|---|---|---|---|---|---|
| BRAF | 0% | 44% | <1% | 24% | [39] |
| RET/PTC | Controversial | 35% | 0% | 0% | [40] |
| RAS | 13% | 10% | 40% | 22% | [41] |
| PAX8/PPARδ | 11% | 0% | 36% | 0% | [42] |
| P53 | 0% | 1% | 1% | 55% | [43] |
| NTRK1 | Unknown | 12% | Unknown | Unknown | [44] |
| PTC: papillary thyroid cancer, FTC: follicular thyroid cancer | |||||
3.1. BRAF
3.2. RET/PTC Rearrangements (Sporadic Papillary Thyroid Cancer)
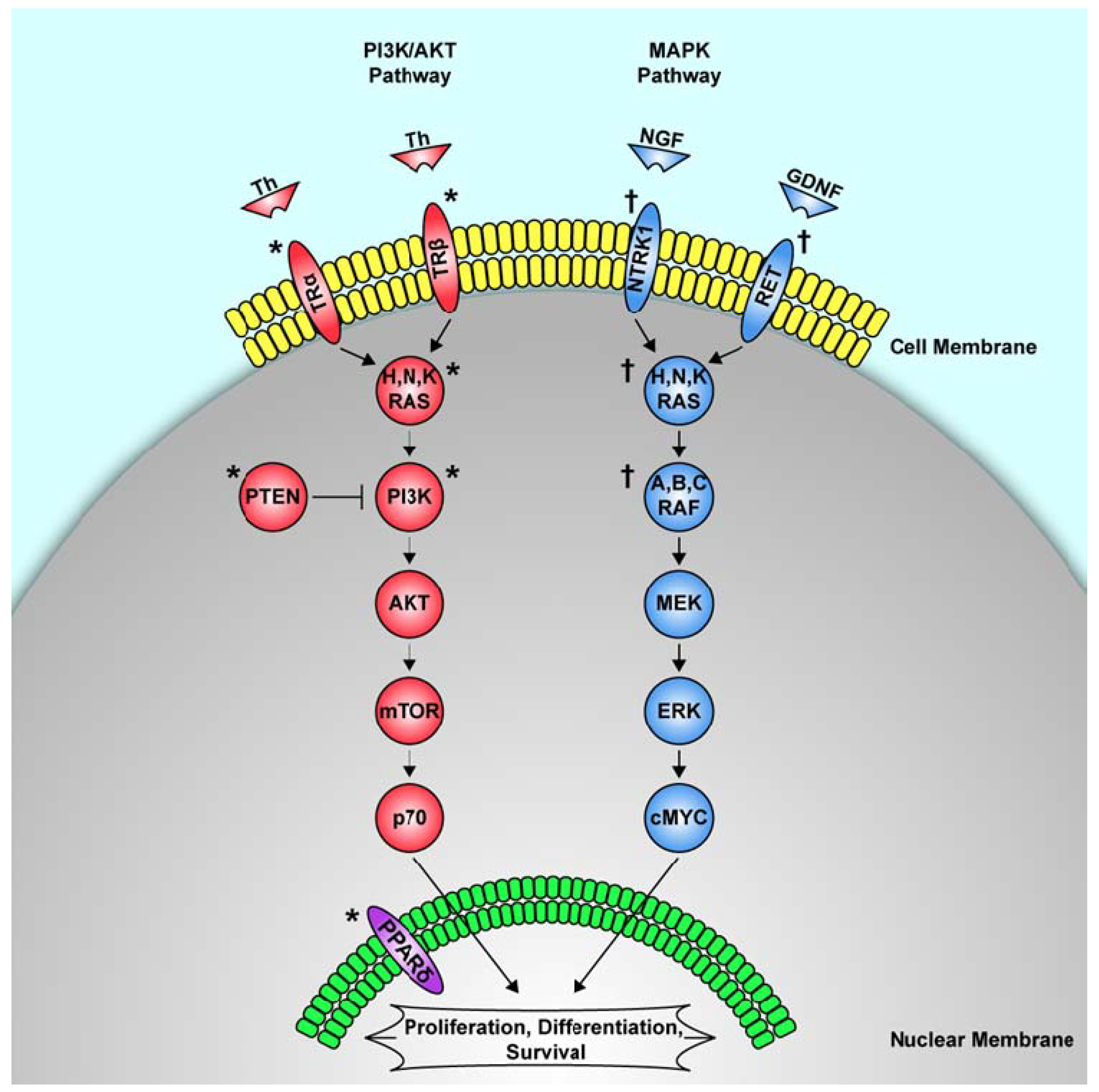
3.3. RET Point Mutations (Familial Medullary Thyroid Cancer)
3.4. RAS
3.5. PAX8/PPARgamma
3.6. P53 Tumor Suppressor
3.7. NTRK1
3.8. DNA Mutation Panels
3.9. Microsatellite Instability
4. Epigenetic Biomarkers
4.1. Hypermethylation
4.2. MicroRNA
5. Genomics
6. Proteomics
6.1. Immunohistochemistry
| Marker | Expression in thyroid cancer | Marker | Expression in thyroid cancer |
|---|---|---|---|
| Galectin-3 | Upregulated | HER4 | Upregulated |
| CK19 | Upregulated | TG | Downregulated |
| VEGF | Downregulated | MIB-1 | Upregulated |
| Aurora-A | Upregulated | Caveolin | Upregulated |
| p16 | Upregulated | Aurora-C | Upregulated |
| AR | Upregulated | S100 | Upregulated |
| HBME-1 | Upregulated | MRAS | Downregulated |
| Bcl-2 | Downregulated | c-kit | Downregulated |
| Cyclin-D1 | Upregulated | HER3 | Upregulated |
| CAV-1 | Upregulated | RET | Upregulated |
| Cyclin-E | Upregulated | AMF-R | Upregulated |
| E-CAD | Downregulated | MLH1 | Downregulated |
| Clusterin | Upregulated | AAT | Upregulated |
| CR | Upregulated | TTF-1 | Upregulated |
| IGFBP5 | Upregulated | PGI | Upregulated |
| P21 | Upregulated | HSP-27 | Downregulated |
| IGFBP2 | Upregulated | Syntrophin | Upregulated |
| CTNNB1 | Upregulated |
6.2. High-Throughput Proteomics
7. Conclusions
Acknowledgements
References
- Milhaud, G.; Calmette, C.; Taboulet, J.; Julienne, A.; Moukhtar, M.S. Letter: Hypersecretion of calcitonin in neoplastic conditions. Lancet 1974, 1, 462–463. [Google Scholar]
- Melvin, K.E.; Miller, H.H.; Tashjian, A.H. Early diagnosis of medullary carcinoma of the thyroid gland by means of calcitonin assay. N. Engl. J. Med. 1971, 285, 1115–1120. [Google Scholar] [CrossRef]
- Srinivas, P.R.; Kramer, B.S.; Srivastava, S. Trends in biomarker research for cancer detection. Lancet Oncol. 2001, 2, 698–704. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar] [CrossRef]
- Davies, L.; Welch, H.G. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA 2006, 295, 2164–2167. [Google Scholar] [CrossRef]
- National Cancer Institute. Age-Adjusted SEER Incidence Rates By Cancer Site Ages < 50, All Races, Female 2000 – 2006 (SEER 17). Surveillance, Epidemiology, and End Results (SEER) Program. Available online: http://www.seer.cancer.gov/ (accessed on 24 February 2010).
- Shibru, D.; Chung, K.W.; Kebebew, E. Recent developments in the clinical application of thyroid cancer biomarkers. Curr. Opin. Oncol. 2008, 20, 13–18. [Google Scholar] [CrossRef]
- Van Herle, A.J.; Uller, R.P.; Matthews, N.I.; Brown, J. Radioimmunoassay for measurement of thyroglobulin in human serum. J. Clin. Invest. 1973, 52, 1320–1327. [Google Scholar] [CrossRef]
- Tashijan, A.H.; Howland, B.G.; Melvin, K.E.; Hill, C.S. Immunoassay of human calcitonin. N. Engl. J. Med. 1970, 283, 890–895. [Google Scholar] [CrossRef]
- Jensen, M.B.; Chacon, M.R.; Sattin, J.A.; Aleu, A.; Lyden, P.D. The promise and potential pitfalls of serum biomarkers for ischemic stroke and transient ischemic attack. Neurologist 2008, 14, 243–246. [Google Scholar] [CrossRef]
- Herberman, R.B. Immunologic tests in diagnosis of cancer. Am. J. Clin. Pathol. 1977, 68, 688–698. [Google Scholar]
- Holyoke, E.D.; Block, G.E.; Jensen, E.; Sizemore, G.W.; Heath, H.; Chu, T.M.; Murphy, G.P.; Mittelman, A.; Ruddon, R.W.; Arnott, M.S. Biologic markers in cancer diagnosis and treatment. Curr. Probl. Cancer 1981, 6, 1–68. [Google Scholar]
- Bieglmayer, C.; Vierhapper, H.; Dudczak, R.; Niederle, B. Measurement of calcitonin by immunoassay analyzers. Clin. Chem. Lab. Med. 2007, 45, 662–666. [Google Scholar]
- Guilloteau, D.; Perdrisot, R.; Calmettes, C.; Baulieu, J.L.; Lecomte, P.; Kaphan, G.; Milhaud, G.; Besnard, J.C.; Jallet, P.; Bigorgne, J.C. Diagnosis of medullary carcinoma of the thyroid (MCT) by calcitonin assay using monoclonal antibodies: criteria for the pentagastrin stimulation test in hereditary MCT. J. Clin. Endocrinol. Metab. 1990, 71, 1064–1067. [Google Scholar] [CrossRef]
- d'Herbomez, M.; Caron, P.; Bauters, C.; Cao, C.D.; Schlienger, J.L.; Sapin, R.; Baldet, L.; Carnaille, B.; Wémeau, J.L. French Group GTE (Groupe des Tumeurs Endocrines) Reference range of serum calcitonin levels in humans: influence of calcitonin assays, sex, age, and cigarette smoking. Eur. J. Endocrinol. 2007, 157, 749–755. [Google Scholar] [CrossRef]
- Toledo, S.P.; Lourenço, D.M.; Santos, M.A.; Tavares, M.R.; Toledo, R.A.; Correia-Deur, J.E. Hypercalcitoninemia is not pathognomonic of medullary thyroid carcinoma. Clinics (Sao Paulo) 2009, 64, 699–706. [Google Scholar]
- van Veelen, W.; de Groot, J.W.; Acton, D.S.; Hofstra, R.M.; Höppener, J.W.; Links, T.P.; Lips, C.J. Medullary thyroid carcinoma and biomarkers: past, present and future. J. Intern. Med. 2009, 266, 126–140. [Google Scholar] [CrossRef]
- Cohen, R.; Campos, J.M.; Salaün, C.; Heshmati, H.M.; Kraimps, J.L.; Proye, C.; Sarfati, E.; Henry, J.F.; Niccoli–Sire, P.; Modigliani, E. Preoperative calcitonin levels are predictive of tumor size and postoperative calcitonin normalization in medullary thyroid carcinoma. Groupe d'Etudes des Tumeurs a Calcitonine (GETC). J. Clin. Endocrinol. Metab. 2000, 85, 919–922. [Google Scholar] [CrossRef]
- Laure Giraudet, A.; Al Ghulzan, A.; Aupérin, A.; Leboulleux, S.; Chehboun, A.; Troalen, F.; Dromain, C.; Lumbroso, J.; Baudin, E.; Schlumberger, M. Progression of medullary thyroid carcinoma: assessment with calcitonin and carcinoembryonic antigen doubling times. Eur. J. Endocrinol. 2008, 158, 239–246. [Google Scholar] [CrossRef]
- Lippman, S.M.; Mendelsohn, G.; Trump, D.L.; Wells, S.A.; Baylin, S.B. The prognostic and biological significance of cellular heterogeneity in medullary thyroid carcinoma: a study of calcitonin, L-dopa decarboxylase, and histaminase. J. Clin. Endocrinol. Metab. 1982, 54, 233–240. [Google Scholar] [CrossRef]
- Wells, S.A.; Chi, D.D.; Toshima, K.; Dehner, L.P.; Coffin, C.M.; Dowton, S.B.; Ivanovich, J.L.; DeBenedetti, M.K.; Dilley, W.G.; Moley, J.F. Predictive DNA testing and prophylactic thyroidectomy in patients at risk for multiple endocrine neoplasia type 2A. Ann. Surg. 1994, 220, 237–250. [Google Scholar] [CrossRef]
- Eng, C.; Smith, D.P.; Mulligan, L.M.; Healey, C.S.; Zvelebil, M.J.; Stonehouse, T.J.; Ponder, M.A.; Jackson, C.E.; Waterfield, M.D.; Ponder, B.A. A novel point mutation in the tyrosine kinase domain of the RET proto-oncogene in sporadic medullary thyroid carcinoma and in a family with FMTC. Oncogene 1995, 10, 509–513. [Google Scholar]
- Wong, J.; Lu, Z.; Doery, J.; Fuller, P. Lessons from a review of thyroglobulin assays in the management of thyroid cancer. Intern. Med. J. 2008, 38, 441–444. [Google Scholar] [CrossRef]
- Whitley, R.J.; Ain, K.B. Thyroglobulin: a specific serum marker for the management of thyroid carcinoma. Clin. Lab. Med. 2004, 24, 29–47. [Google Scholar] [CrossRef]
- Gupta, M.; Chia, S.Y. Circulating thyroid cancer markers. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 383–388. [Google Scholar] [CrossRef]
- Spencer, C.A.; Bergoglio, L.M.; Kazarosyan, M.; Fatemi, S.; LoPresti, J.S. Clinical impact of thyroglobulin (Tg) and Tg autoantibody method differences on the management of patients with differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2005, 90, 5566–5575. [Google Scholar] [CrossRef]
- Haugen, B.R.; Pacini, F.; Reiners, C.; Schlumberger, M.; Ladenson, P.W.; Sherman, S.I.; Cooper, D.S.; Graham, K.E.; Braverman, L.E.; Skarulis, M.C.; et al. A comparison of recombinant human thyrotropin and thyroid hormone withdrawal for the detection of thyroid remnant or cancer. J. Clin. Endocrinol. Metab. 1999, 84, 3877–3885. [Google Scholar] [CrossRef]
- Refetoff, S.; Lever, E.G. The value of serum thyroglobulin measurement in clinical practice. JAMA 1983, 250, 2352–2357. [Google Scholar] [CrossRef]
- Ishikawa, T.; Miwa, M.; Uchida, K. Quantitation of thyroid peroxidase mRNA in peripheral blood for early detection of thyroid papillary carcinoma. Thyroid 2006, 16, 435–442. [Google Scholar] [CrossRef]
- Mercken, L.; Simons, M.J.; Brocas, H.; Vassart, G. Alternative splicing may be responsible for heterogeneity of thyroglobulin structure. Biochimie 1989, 71, 223–226. [Google Scholar] [CrossRef]
- Chinnappa, P.; Taguba, L.; Arciaga, R.; Faiman, C.; Siperstein, A.; Mehta, A.E.; Reddy, S.K.; Nasr, C.; Gupta, M.K. Detection of thyrotropin-receptor messenger ribonucleic acid (mRNA) and thyroglobulin mRNA transcripts in peripheral blood of patients with thyroid disease: sensitive and specific markers for thyroid cancer. J. Clin. Endocrinol. Metab. 2004, 89, 3705–3709. [Google Scholar] [CrossRef]
- Mulligan, L.M.; Kwok, J.B.; Healey, C.S.; Elsdon, M.J.; Eng, C.; Gardner, E.; Love, D.R.; Mole, S.E.; Moore, J.K.; Papi, L. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363, 458–460. [Google Scholar] [CrossRef]
- Knyazev, P.G.; Fedorov, S.N.; Serova, O.M.; Pluzhnikova, G.F.; Novikov, L.B.; Kalinovsky, V.P.; Seitz, J.F. Molecular-genetic analysis of myc and c-Ha-ras proto-oncogene alterations in human carcinoma. Haematol. Blood Transfus. 1987, 31, 469–473. [Google Scholar]
- Grieco, M.; Santoro, M.; Berlingieri, M.T.; Melillo, R.M.; Donghi, R.; Bongarzone, I.; Pierotti, M.A.; Della Porta, G.; Fusco, A.; Vecchio, G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 1990, 60, 557–563. [Google Scholar] [CrossRef]
- Ito, T.; Seyama, T.; Mizuno, T.; Tsuyama, N.; Hayashi, T.; Hayashi, Y.; Dohi, K.; Nakamura, N.; Akiyama, M. Unique association of p53 mutations with undifferentiated but not with differentiated carcinomas of the thyroid gland. Cancer Res. 1992, 52, 1369–1371. [Google Scholar]
- Greco, A.; Pierotti, M.A.; Bongarzone, I.; Pagliardini, S.; Lanzi, C.; Della Porta, G. TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene 1992, 7, 237–242. [Google Scholar]
- Kroll, T.G.; Sarraf, P.; Pecciarini, L.; Chen, C.J.; Mueller, E.; Spiegelman, B.M.; Fletcher, J.A. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science 2000, 289, 1357–1360. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef]
- Nikiforov, Y.E. RET/PTC rearrangement in thyroid tumors. Endocr. Pathol. 2002, 13, 3–16. [Google Scholar] [CrossRef]
- Ezzat, S.; Zheng, L.; Kolenda, J.; Safarian, A.; Freeman, J.L.; Asa, S.L. Prevalence of activating ras mutations in morphologically characterized thyroid nodules. Thyroid 1996, 6, 409–416. [Google Scholar]
- Placzkowski, K.A.; Reddi, H.V.; Grebe, S.K.; Eberhardt, N.L.; McIver, B. The Role of the PAX8/PPARgamma Fusion Oncogene in Thyroid Cancer. PPAR Res. 2008, 2008, 672829. [Google Scholar]
- Sobrinho-Simões, M.; Máximo, V.; Rocha, A.S.; Trovisco, V.; Castro, P.; Preto, A.; Lima, J.; Soares, P. Intragenic mutations in thyroid cancer. Endocrinol. Metab. Clin. North Am. 2008, 37, 333–362. [Google Scholar] [CrossRef]
- Greco, A.; Miranda, C.; Pierotti, M.A. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol. Cell Endocrinol. 2009, 321, 44–49. [Google Scholar]
- Kebebew, E.; Weng, J.; Bauer, J.; Ranvier, G.; Clark, O.H.; Duh, Q.Y.; Shibru, D.; Bastian, B.; Griffin, A. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann. Surg. 2007, 246, 466–471. [Google Scholar] [CrossRef]
- Soares, P.; Trovisco, V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Máximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simões, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580. [Google Scholar] [CrossRef]
- Trovisco, V.; Vieira de Castro, I.; Soares, P.; Máximo, V.; Silva, P.; Magalhães, J.; Abrosimov, A.; Guiu, X.M.; Sobrinho-Simões, M. BRAF mutations are associated with some histological types of papillary thyroid carcinoma. J. Pathol. 2004, 202, 247–251. [Google Scholar] [CrossRef]
- Xing, M.; Westra, W.H.; Tufano, R.P.; Cohen, Y.; Rosenbaum, E.; Rhoden, K.J.; Carson, K.A.; Vasko, V.; Larin, A.; Tallini, G.; et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2005, 90, 6373–6379. [Google Scholar] [CrossRef]
- Kim, T.Y.; Kim, W.B.; Song, J.Y.; Rhee, Y.S.; Gong, G.; Cho, Y.M.; Kim, S.Y.; Kim, S.C.; Hong, S.J.; Shong, Y.K. The BRAF mutation is not associated with poor prognostic factors in Korean patients with conventional papillary thyroid microcarcinoma. Clin. Endocrinol. (Oxf.) 2005, 63, 588–593. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, E.S.; Kim, Y.S. Clinicopathologic significance of BRAF V600E mutation in papillary carcinomas of the thyroid: a meta-analysis. Cancer 2007, 110, 38–46. [Google Scholar] [CrossRef]
- Takahashi, M.; Ritz, J.; Cooper, G.M. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985, 42, 581–588. [Google Scholar] [CrossRef]
- Simpson, N.E.; Kidd, K.K.; Goodfellow, P.J.; McDermid, H.; Myers, S.; Kidd, J.R.; Jackson, C.E.; Duncan, A.M.; Farrer, L.A.; Brasch, K. Assignment of multiple endocrine neoplasia type 2A to chromosome 10 by linkage. Nature 1987, 328, 528–530. [Google Scholar] [CrossRef]
- Takahashi, M.; Buma, Y.; Iwamoto, T.; Inaguma, Y.; Ikeda, H.; Hiai, H. Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene 1988, 3, 571–578. [Google Scholar]
- Ishizaka, Y.; Itoh, F.; Tahira, T.; Ikeda, I.; Sugimura, T.; Tucker, J.; Fertitta, A.; Carrano, A.V.; Nagao, M. Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene 1989, 4, 1519–1521. [Google Scholar]
- Yamamoto, M.; Miki, T.; Tanaka, N.; Miya, A.; Shin, E.; Karakawa, K.; Kobayashi, T.; Tahira, T.; Ishizaka, Y.; Itoh, F. Tight linkage of the ret proto-oncogene with the multiple endocrine neoplasia type 2A locus. Jpn. J. Clin. Oncol. 1991, 21, 149–152. [Google Scholar]
- Mole, S.E.; Mulligan, L.M.; Healey, C.S.; Ponder, B.A.; Tunnacliffe, A. Localisation of the gene for multiple endocrine neoplasia type 2A to a 480 kb region in chromosome band 10q11.2. Hum. Mol. Genet. 1993, 2, 247–252. [Google Scholar] [CrossRef]
- Donis-Keller, H.; Dou, S.; Chi, D.; Carlson, K.M.; Toshima, K.; Lairmore, T.C.; Howe, J.R.; Moley, J.F.; Goodfellow, P.; Wells, S.A. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet. 1993, 2, 851–856. [Google Scholar] [CrossRef]
- Skinner, M.A.; Moley, J.A.; Dilley, W.G.; Owzar, K.; Debenedetti, M.K.; Wells, S.A. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N. Engl. J. Med. 2005, 353, 1105–1113. [Google Scholar] [CrossRef]
- Machens, A.; Niccoli-Sire, P.; Hoegel, J.; Frank-Raue, K.; van Vroonhoven, T.J.; Roeher, H.D.; Wahl, R.A.; Lamesch, P.; Raue, F.; Conte-Devolx, B.; et al. Early malignant progression of hereditary medullary thyroid cancer. N. Engl. J. Med. 2003, 349, 1517–1525. [Google Scholar] [CrossRef]
- American Thyroid Association Guidelines Task Force; Kloos, R.T.; Eng, C.; Evans, D.B.; Francis, G.L.; Gagel, R.F.; Gharib, H.; Moley, J.F.; Pacini, F.; Ringel, M.D.; et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009, 19, 565–612. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W.; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef]
- Morita, N.; Ikeda, Y.; Takami, H. Clinical significance of p53 protein expression in papillary thyroid carcinoma. World J. Surg. 2008, 32, 2617–2622. [Google Scholar] [CrossRef]
- Jossart, G.H.; Epstein, H.D.; Shaver, J.K.; Weier, H.U.; Greulich, K.M.; Tezelman, S.; Grossman, R.F.; Siperstein, A.E.; Duh, Q.Y.; Clark, O.H. Immunocytochemical detection of p53 in human thyroid carcinomas is associated with mutation and immortalization of cell lines. J. Clin. Endocrinol. Metab. 1996, 81, 3498–3504. [Google Scholar] [CrossRef]
- Park, K.Y.; Koh, J.M.; Kim, Y.I.; Park, H.J.; Gong, G.; Hong, S.J.; Ahn, I.M. Prevalences of Gs alpha, ras, p53 mutations and ret/PTC rearrangement in differentiated thyroid tumours in a Korean population. Clin. Endocrinol. (Oxf) 1998, 49, 317–323. [Google Scholar] [CrossRef]
- Pollina, L.; Pacini, F.; Fontanini, G.; Vignati, S.; Bevilacqua, G.; Basolo, F. bcl-2, p53 and proliferating cell nuclear antigen expression is related to the degree of differentiation in thyroid carcinomas. Br. J. Cancer 1996, 73, 139–143. [Google Scholar] [CrossRef]
- Musholt, T.J.; Musholt, P.B.; Khaladj, N.; Schulz, D.; Scheumann, G.F.W.; Klempnauer, J. Prognostic significance of RET and NTRK 1 rearrangements in sporadic papillary thyoid carcinoma. Br. J. Surg. 2000, 87, 1256–1278. [Google Scholar]
- Nikiforov, Y.E.; Steward, D.L.; Robinson-Smith, T.M.; Haugen, B.R.; Klopper, J.P.; Zhu, Z.; Fagin, J.A.; Falciglia, M.; Weber, K.; Nikiforova, M.N. Molecular testing for mutations in improving the fine-needle aspiration diagnosis of thyroid nodules. J. Clin. Endocrinol. Metab. 2009, 94, 2092–2098. [Google Scholar] [CrossRef]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef]
- Soares, P.; dos Santos, N.R.; Seruca, R.; Lothe, R.A.; Sobrinho-Simões, M. Benign and malignant thyroid lesions show instability at microsatellite loci. Eur. J. Cancer 1997, 33, 293–296. [Google Scholar]
- Onda, M.; Nakamura, I.; Suzuki, S.; Takenoshita, S.; Brogren, C.H.; Stampanoni, S.; Li, D.; Rampino, N. Microsatellite instability in thyroid cancer: hot spots, clinicopathological implications, and prognostic significance. Clin. Cancer Res. 2001, 7, 3444–3449. [Google Scholar]
- Mitmaker, E.; Alvarado, C.; Bégin, L.R.; Trifiro, M. Microsatellite instability in benign and malignant thyroid neoplasms. J. Surg. Res. 2008, 150, 40–48. [Google Scholar] [CrossRef]
- Vermiglio, F.; Schlumberger, M.; Lazar, V.; Lefrere, I.; Bressac, B. Absence of microsatellite instability in thyroid carcinomas. Eur. J. Cancer 1995, 31, 128–128. [Google Scholar]
- Xing, M. Gene methylation in thyroid tumorigenesis. Endocrinology 2007, 148, 948–953. [Google Scholar] [CrossRef]
- Alvarez-Nuñez, F.; Bussaglia, E.; Mauricio, D.; Ybarra, J.; Vilar, M.; Lerma, E.; de Leiva, A.; Matias-Guiu, X. Thyroid Neoplasia Study Group PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid 2006, 16, 17–23. [Google Scholar] [CrossRef]
- Hoque, M.O.; Rosenbaum, E.; Westra, W.H.; Xing, M.; Ladenson, P.; Zeiger, M.A.; Sidransky, D.; Umbricht, C.B. Quantitative assessment of promoter methylation profiles in thyroid neoplasms. J. Clin. Endocrinol. Metab. 2005, 90, 4011–4018. [Google Scholar] [CrossRef]
- Hu, S.; Liu, D.; Tufano, R.P.; Carson, K.A.; Rosenbaum, E.; Cohen, Y.; Holt, E.H.; Kiseljak-Vassiliades, K.; Rhoden, K.J.; Tolaney, S.; et al. Association of aberrant methylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int. J. Cancer 2006, 119, 2322–2329. [Google Scholar] [CrossRef]
- Kondo, Y.; Issa, J.P.J. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 29–39. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Drakaki, A.; Iliopoulos, D. MicroRNA Gene Networks in Oncogenesis. Curr. Genomics 2009, 10, 35–41. [Google Scholar] [CrossRef]
- Medina, P.P.; Slack, F.J. microRNAs and cancer: an overview. Cell Cycle 2008, 7, 2485–2492. [Google Scholar] [CrossRef]
- He, H.; Jazdzewski, K.; Li, W.; Liyanarachchi, S.; Nagy, R.; Volinia, S.; Calin, G.A.; Liu, C.G.; Franssila, K.; Suster, S.; et al. The role of microRNA genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. U S A 2005, 102, 19075–19080. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Chiosea, S.I.; Nikiforov, Y.E. MicroRNA expression profiles in thyroid tumors. Endocr. Pathol. 2009, 20, 85–91. [Google Scholar] [CrossRef]
- Chen, Y.T.; Kitabayashi, N.; Zhou, X.K.; Fahey, T.J.; Scognamiglio, T. MicroRNA analysis as a potential diagnostic tool for papillary thyroid carcinoma. Mod. Pathol. 2008, 21, 1139–1146. [Google Scholar] [CrossRef]
- McKusick, V.A.; Ruddle, F.H. A new discipline, a new name, a new journal. Genomics 1987, 1, 1–2. [Google Scholar] [CrossRef]
- Mandruzzato, S. Technological platforms for microarray gene expression profiling. Adv. Exp. Med. Biol. 2007, 593, 12–18. [Google Scholar] [CrossRef]
- Lee, N.H.; Saeed, A.I. Microarrays: an overview. Methods Mol. Biol. 2007, 353, 265–300. [Google Scholar]
- Hoheisel, J.D. Microarray technology: beyond transcript profiling and genotype analysis. Nat. Rev. Genet. 2006, 7, 200–210. [Google Scholar] [CrossRef]
- Huang, Y.; Prasad, M.; Lemon, W.J.; Hampel, H.; Wright, F.A.; Kornacker, K.; LiVolsi, V.; Frankel, W.; Kloos, R.T.; Eng, C.; et al. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc. Natl. Acad. Sci. USA 2001, 98, 15044–15049. [Google Scholar] [CrossRef]
- Giordano, T.J.; Kuick, R.; Thomas, D.G.; Misek, D.E.; Vinco, M.; Sanders, D.; Zhu, Z.; Ciampi, R.; Roh, M.; Shedden, K.; et al. Molecular classification of papillary thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene 2005, 24, 6646–6656. [Google Scholar] [CrossRef]
- Lui, W.O.; Foukakis, T.; Lidén, J.; Thoppe, S.R.; Dwight, T.; Höög, A.; Zedenius, J.; Wallin, G.; Reimers, M.; Larsson, C. Expression profiling reveals a distinct transcription signature in follicular thyroid carcinomas with a PAX8-PPAR(gamma) fusion oncogene. Oncogene 2005, 24, 1467–1476. [Google Scholar] [CrossRef]
- Vasko, V.; Espinosa, A.V.; Scouten, W.; He, H.; Auer, H.; Liyanarachchi, S.; Larin, A.; Savchenko, V.; Francis, G.L.; de la Chapelle, A.; et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc. Natl. Acad. Sci. USA 2007, 104, 2803–2808. [Google Scholar] [CrossRef]
- Detours, V.; Delys, L.; Libert, F.; Weiss Solís, D.; Bogdanova, T.; Dumont, J.E.; Franc, B.; Thomas, G.; Maenhaut, C. Genome-wide gene expression profiling suggests distinct radiation susceptibilities in sporadic and post-Chernobyl papillary thyroid cancers. Br. J. Cancer 2007, 97, 818–825. [Google Scholar] [CrossRef]
- Griffith, O.L.; Melck, A.; Jones, S.J.; Wiseman, S.M. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J. Clin. Oncol. 2006, 24, 5043–5051. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Pasquali, C.; Appel, R.D.; Ou, K.; Golaz, O.; Sanchez, J.C.; Yan, J.X.; Gooley, A.A.; Hughes, G.; Humphery-Smith, I.; et al. From Proteins to Proteomes: Large Scale Protein Identification by Two-Dimensional Electrophoresis and Arnino Acid Analysis. Nat. Biotech. 1996, 14, 61–65. [Google Scholar] [CrossRef]
- Fischer, S.; Asa, S.L. Application of immunohistochemistry to thyroid neoplasms. Arch. Pathol. Lab. Med. 2008, 132, 359–372. [Google Scholar]
- Volante, M.; Bozzalla-Cassione, F.; Orlandi, F.; Papotti, M. Diagnostic role of galectin-3 in follicular thyroid tumors. Virchows Arch. 2004, 444, 309–312. [Google Scholar]
- Torregrossa, L.; Faviana, P.; Camacci, T.; Materazzi, G.; Berti, P.; Minuto, M.; Elisei, R.; Vitti, P.; Miccoli, P.; Basolo, F. Galectin-3 is highly expressed in nonencapsulated papillary thyroid carcinoma but weakly expressed in encapsulated type; comparison with Hector Battifora mesothelial cell 1 immunoreactivity. Hum. Pathol. 2007, 38, 1482–1488. [Google Scholar]
- Türköz, H.K.; Oksüz, H.; Yurdakul, Z.; Ozcan, D. Galectin-3 expression in tumor progression and metastasis of papillary thyroid carcinoma. Endocr. Pathol. 2008, 19, 92–96. [Google Scholar] [CrossRef]
- Saussez, S.; Glinoer, D.; Chantrain, G.; Pattou, F.; Carnaille, B.; André, S.; Gabius, H.J.; Laurent, G. Serum galectin-1 and galectin-3 levels in benign and malignant nodular thyroid disease. Thyroid 2008, 18, 705–712. [Google Scholar] [CrossRef]
- Rossi, E.D.; Raffaelli, M.; Mule', A.; Miraglia, A.; Lombardi, C.P.; Vecchio, F.M.; Fadda, G. Simultaneous immunohistochemical expression of HBME-1 and galectin-3 differentiates papillary carcinomas from hyperfunctioning lesions of the thyroid. Histopathology 2006, 48, 795–800. [Google Scholar] [CrossRef]
- Saggiorato, E.; De Pompa, R.; Volante, M.; Cappia, S.; Arecco, F.; Dei Tos, A.P.; Orlandi, F.; Papotti, M. Characterization of thyroid 'follicular neoplasms' in fine-needle aspiration cytological specimens using a panel of immunohistochemical markers: a proposal for clinical application. Endocr. Relat. Cancer 2005, 12, 305–317. [Google Scholar] [CrossRef]
- Scognamiglio, T.; Hyjek, E.; Kao, J.; Chen, Y.T. Diagnostic usefulness of HBME1, galectin-3, CK19, and CITED1 and evaluation of their expression in encapsulated lesions with questionable features of papillary thyroid carcinoma. Am. J. Clin. Pathol. 2006, 126, 700–708. [Google Scholar] [CrossRef]
- Wiseman, S.M.; Melck, A.; Masoudi, H.; Ghaidi, F.; Goldstein, L.; Gown, A.; Jones, S.J.; Griffith, O.L. Molecular phenotyping of thyroid tumors identifies a marker panel for differentiated thyroid cancer diagnosis. Ann. Surg. Oncol. 2008, 15, 2811–2826. [Google Scholar] [CrossRef]
- Krause, K.; Jessnitzer, B.; Fuhrer, D. Proteomics in thyroid tumor research. J. Clin. Endocrinol. Metab. 2009, 94, 2717–2724. [Google Scholar] [CrossRef]
- Manne, U.; Srivastava, R.G.; Srivastava, S. Recent advances in biomarkers for cancer diagnosis and treatment. Drug Discov. Today 2005, 10, 965–976. [Google Scholar] [CrossRef]
- Wang, J.X.; Yu, J.K.; Wang, L.; Liu, Q.L.; Zhang, J.; Zheng, S. Application of serum protein fingerprint in diagnosis of papillary thyroid carcinoma. Proteomics 2006, 6, 5344–5349. [Google Scholar] [CrossRef]
- Wulfkuhle, J.D.; Liotta, L.A.; Petricoin, E.F. Proteomic applications for the early detection of cancer. Nat. Rev. Cancer 2003, 3, 267–275. [Google Scholar] [CrossRef]
- Gillette, M.A.; Mani, D.R.; Carr, S.A. Place of pattern in proteomic biomarker discovery. J. Proteome Res. 2005, 4, 1143–1154. [Google Scholar] [CrossRef]
- Shen, Y.; Jacobs, J.M.; Camp, D.G.; Fang, R.; Moore, R.J.; Smith, R.D.; Xiao, W.; Davis, R.W.; Tompkins, R.G. Ultra-High-Efficiency Strong Cation Exchange LC/RPLC/MS/MS for High Dynamic Range Characterization of the Human Plasma Proteome. Anal. Chem. 2004, 76, 1134–1144. [Google Scholar] [CrossRef]
- Srisomsap, C.; Subhasitanont, P.; Otto, A.; Mueller, E.C.; Punyarit, P.; Wittmann-Liebold, B.; Svasti, J. Detection of cathepsin B up-regulation in neoplastic thyroid tissues by proteomic analysis. Proteomics 2002, 2, 706–712. [Google Scholar] [CrossRef]
- Brown, L.M.; Helmke, S.M.; Hunsucker, S.W.; Netea-Maier, R.T.; Chiang, S.A.; Heinz, D.E.; Shroyer, K.R.; Duncan, M.W.; Haugen, B.R. Quantitative and qualitative differences in protein expression between papillary thyroid carcinoma and normal thyroid tissue. Mol. Carcinog. 2006, 45, 613–626. [Google Scholar] [CrossRef]
- Netea-Maier, R.T.; Hunsucker, S.W.; Hoevenaars, B.M.; Helmke, S.M.; Slootweg, P.J.; Hermus, A.R.; Haugen, B.R.; Duncan, M.W. Discovery and validation of protein abundance differences between follicular thyroid neoplasms. Cancer Res. 2008, 68, 1572–1580. [Google Scholar] [CrossRef]
- Nair, S.; Wearsch, P.A.; Mitchell, D.A.; Wassenberg, J.J.; Gilboa, E.; Nicchitta, C.V. Calreticulin displays in vivo peptide-binding activity and can elicit CTL responses against bound peptides. J. Immunol. 1999, 162, 6426–6432. [Google Scholar]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Giusti, L.; Iacconi, P.; Ciregia, F.; Giannaccini, G.; Basolo, F.; Donatini, G.; Miccoli, P.; Lucacchini, A. Proteomic analysis of human thyroid fine needle aspiration fluid. J. Endocrinol. Invest. 2007, 30, 865–869. [Google Scholar]
- Giusti, L.; Iacconi, P.; Ciregia, F.; Giannaccini, G.; Donatini, G.L.; Basolo, F.; Miccoli, P.; Pinchera, A.; Lucacchini, A. Fine-needle aspiration of thyroid nodules: proteomic analysis to identify cancer biomarkers. J. Proteome Res. 2008, 7, 4079–4088. [Google Scholar] [CrossRef]
- Torres-Cabala, C.; Bibbo, M.; Panizo-Santos, A.; Barazi, H.; Krutzsch, H.; Roberts, D.D.; Merino, M.J. Proteomic identification of new biomarkers and application in thyroid cytology. Acta Cytol. 2006, 50, 518–528. [Google Scholar] [CrossRef]
- Krause, K.; Karger, S.; Schierhorn, A.; Poncin, S.; Many, M.C.; Fuhrer, D. Proteomic profiling of cold thyroid nodules. Endocrinology 2007, 148, 1754–1763. [Google Scholar]
- Moretz, W.H.; Gourin, C.G.; Terris, D.J.; Xia, Z.S.; Liu, Z.; Weinberger, P.M.; Chin, E.; Adam, B.L. Detection of papillary thyroid carcinoma with serum protein profile analysis. Arch. Otolaryngol. Head Neck Surg. 2008, 134, 198–202. [Google Scholar] [CrossRef]
- Fan, Y.; Shi, L.; Liu, Q.; Dong, R.; Zhang, Q.; Yang, S.; Fan, Y.; Yang, H.; Wu, P.; Yu, J.; et al. iscovery and identification of potential biomarkers of papillary thyroid carcinoma. Mol. Cancer. 2009, 8, 79. [Google Scholar] [CrossRef]
- Suriano, R.; Lin, Y.; Ashok, B.T.; Schaefer, S.D.; Schantz, S.P.; Geliebter, J.; Tiwari, R.K. Pilot study using SELDI-TOF-MS based proteomic profile for the identification of diagnostic biomarkers of thyroid proliferative diseases. J. Proteome Res. 2006, 5, 856–861. [Google Scholar] [CrossRef]
- Iannetti, A.; Pacifico, F.; Acquaviva, R.; Lavorgna, A.; Crescenzi, E.; Vascotto, C.; Tell, G.; Salzano, A.M.; Scaloni, A.; Vuttariello, E.; et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc. Natl. Acad. Sci. USA 2008, 105, 14058–14063. [Google Scholar] [CrossRef]
- Dunn, L.L.; Rahmanto, Y.S.; Richardson, D.R. Iron uptake and metabolism in the new millennium. Trends Cell Biol. 2007, 17, 93–100. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef]
- Arcinas, A.; Yen, T.Y.; Kebebew, E.; Macher, B.A. Cell surface and secreted protein profiles of human thyroid cancer cell lines reveal distinct glycoprotein patterns. J. Proteome Res. 2009, 8, 3958–3968. [Google Scholar] [CrossRef]
- Gorla, L.; Mondellini, P.; Cuccuru, G.; Miccichè, F.; Cassinelli, G.; Cremona, M.; Pierotti, M.A.; Lanzi, C.; Bongarzone, I. Proteomics study of medullary thyroid carcinomas expressing RET germ-line mutations: identification of new signaling elements. Mol. Carcinog. 2009, 48, 220–231. [Google Scholar] [CrossRef]
- Dietel, M.; Sers, C. Personalized medicine and development of targeted therapies: The upcoming challenge for diagnostic molecular pathology. A review. Virchows Arch. 2006, 448, 744–755. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grogan, R.H.; Mitmaker, E.J.; Clark, O.H. The Evolution of Biomarkers in Thyroid Cancer—From Mass Screening to a Personalized Biosignature. Cancers 2010, 2, 885-912. https://doi.org/10.3390/cancers2020885
Grogan RH, Mitmaker EJ, Clark OH. The Evolution of Biomarkers in Thyroid Cancer—From Mass Screening to a Personalized Biosignature. Cancers. 2010; 2(2):885-912. https://doi.org/10.3390/cancers2020885
Chicago/Turabian StyleGrogan, Raymon H., Elliot J. Mitmaker, and Orlo H. Clark. 2010. "The Evolution of Biomarkers in Thyroid Cancer—From Mass Screening to a Personalized Biosignature" Cancers 2, no. 2: 885-912. https://doi.org/10.3390/cancers2020885
APA StyleGrogan, R. H., Mitmaker, E. J., & Clark, O. H. (2010). The Evolution of Biomarkers in Thyroid Cancer—From Mass Screening to a Personalized Biosignature. Cancers, 2(2), 885-912. https://doi.org/10.3390/cancers2020885