Oxidative Stress Induced Mechanisms in the Progression of Periodontal Diseases and Cancer: A Common Approach to Redox Homeostasis?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Association between Periodontal Disease and Cancer; Case Definition of Periodontal Disease
1.2. Inflammatory Loading Imposed by Chronic Periodontitis and Predisposition to Carcinogenesis

1.3. Oral Lesions and Immunosurveillance
2. Mechanisms Involved
2.1. Implications of Helicobacter pylori (H. pylori) in Periodontal Disease, Oral and Gastric Cancer

2.2. Oncogene c-Src and Cancer Invasion
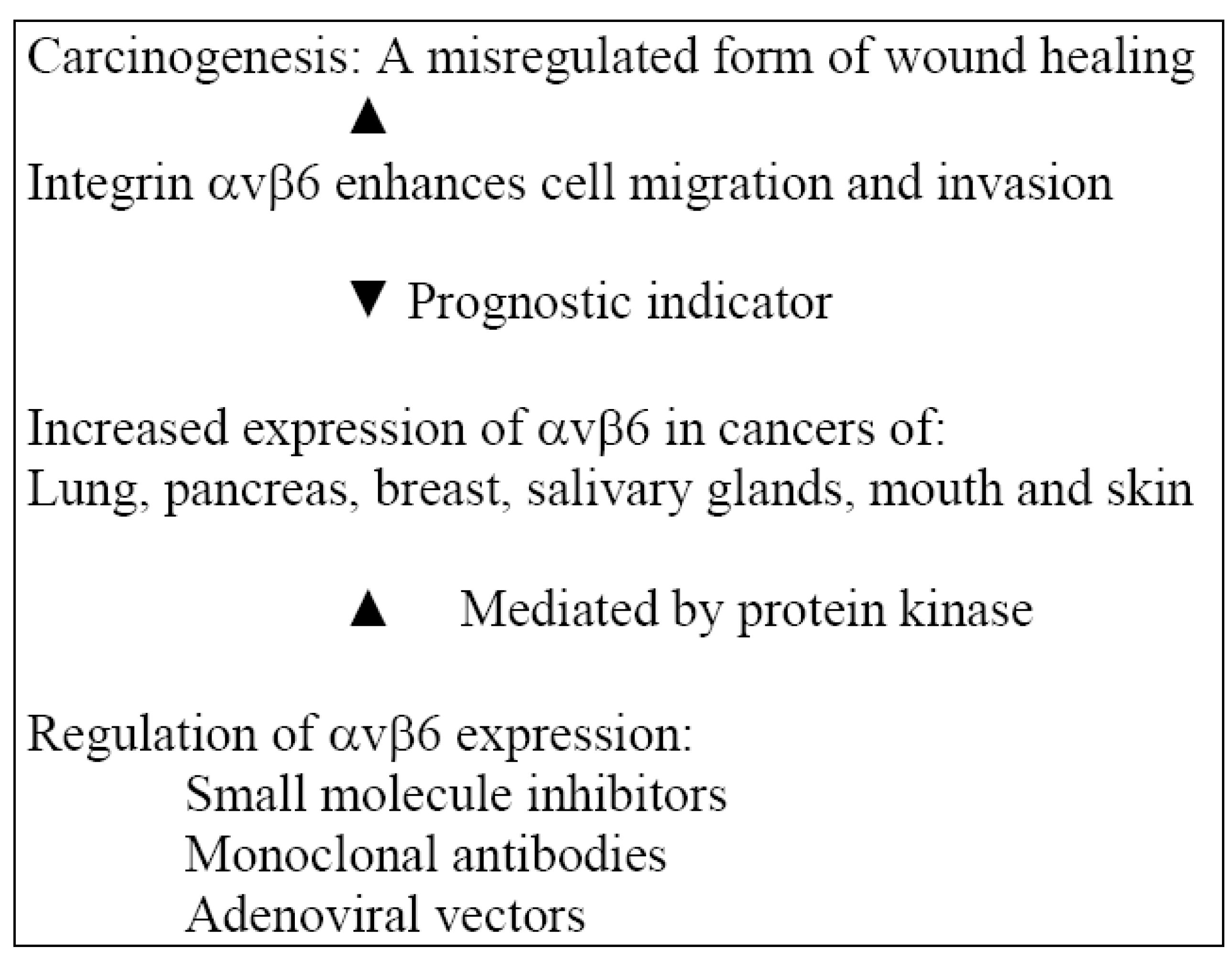
2.3. Actions of Integrin αvβ6 in an Inflammatory Model
2.4. Regulation of αvβ6 Expression
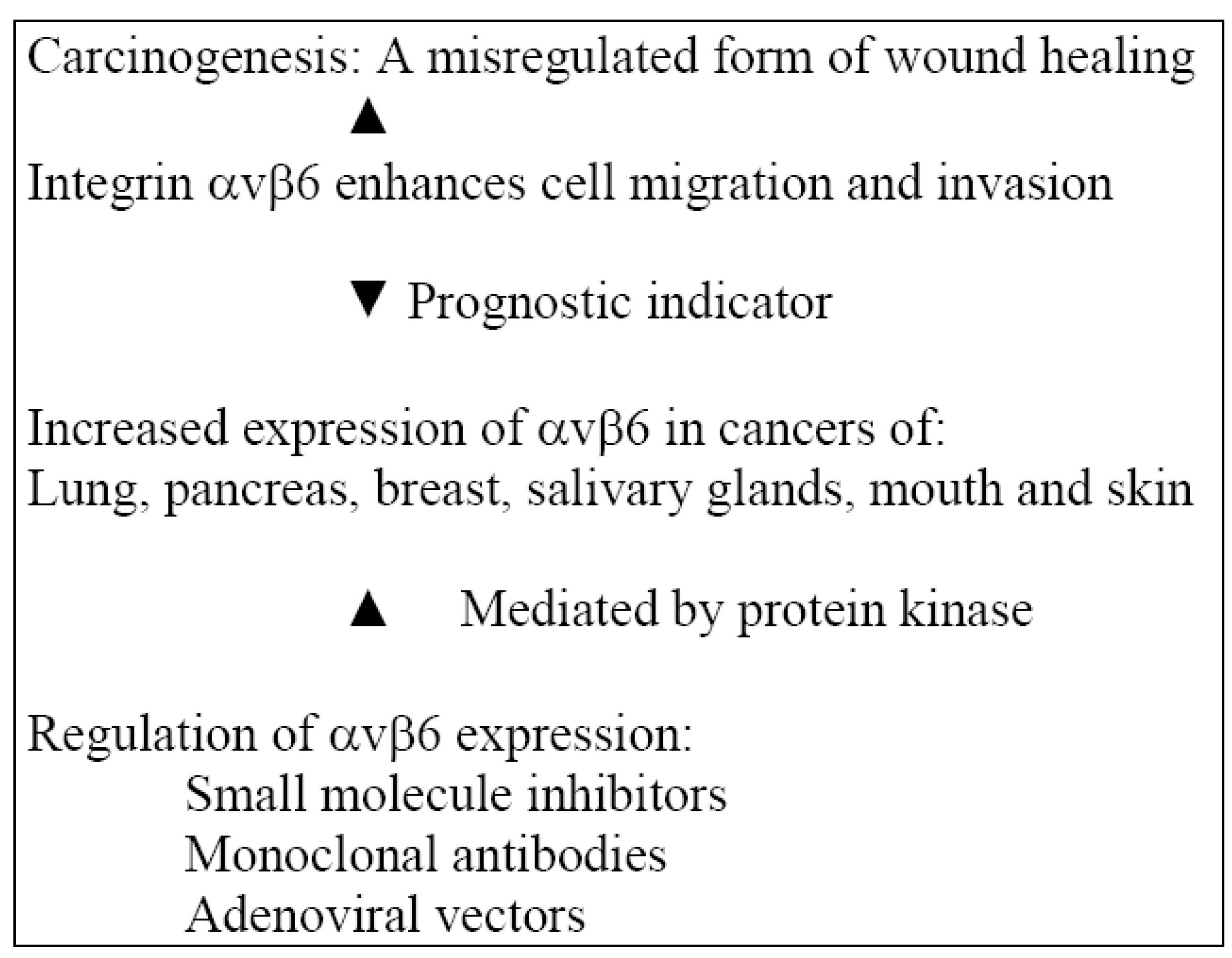
3. Adjunctive Measures for Management and Mechanisms of Action in an Inflammatory Model
3.1. Treatment with αvβ6 Inhibition
3.2. Resolution of Inflammation with Lipoxins, Resolvins and Protectins
3.3. Lipoxins
3.4. Resolvins and Protectins
3.5. Efficacy of Polyphenols in Preventing Cancer
3.6. Treatment with Green Tea Polyphenols
4. Summary and Conclusions
References
- Haffajee, A.D.; Socransky, S.S. Microbial aetiological agents and destructive periodontal diseases. Periodontology 2000 1994, 5, 78–111. [Google Scholar] [CrossRef]
- Page, R.C.; Offenbacher, S.; Schroeder, H.E.; Seymour, G.J.; Kornman, K.S. Advances in the pathogenesis of periodontitis: Summary of developments, clinical implications and future directions. Periodontology 2000 1997, 14, 216–248. [Google Scholar] [CrossRef]
- Page, R.C.; Eke, P.I. Case definitions for use in population-based surveillance of periodontitis. J. Periodontol. 2007, 78, 1387–1399. [Google Scholar] [CrossRef]
- Vargas, C.M.; Arevalo, O. How dental care can preserve and improve oral health. Dent. Clin. North Am. 2009, 53, 399–420. [Google Scholar] [CrossRef]
- Hujoel, P.; Drangsholt, M.; Spiekerman, C.; Weiss, N. An exploration of the periodontitis-cancer association. Ann. Epidemiol. 2003, 13, 312–316. [Google Scholar] [CrossRef]
- Pihlstrom, B.; Michalowicz, B.; Johnson, N. Periodontal diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef]
- Soder, B.; Jin, L.J.; Klinge, B.; Soder, P-O. Periodontitis and premature death: a 16-year longitudinal study in a Swedish urban population. J. Periodont. Res. 2007, 42, 361–366. [Google Scholar] [CrossRef]
- Michaud, D.S.; Liu, Y.; Meyer, M.; Giovannucci, E.; Joshipura, K. Periodontal disease, tooth loss, and cancer risk in male health professionals: a prospective cohort study. Lancet Oncol. 2008, 9, 550–558. [Google Scholar] [CrossRef]
- Michaud, D.; Joshipura, K.; Giovannucci, E.; Fuchs, C. A prospective study of periodontal disease and pancreatic cancer in US male health professionals. J. Natl. Cancer Inst. 2007, 99, 171–175. [Google Scholar] [CrossRef]
- Tezal, M.; Sullivan, M.; Reid, M.; Marshall, J.R.; Hyland, A.; Loree, T.; Lillis, C.; Linda Hauck, L.; Wactawski-Wende, J.; Scannapieco, F.A. Chronic periodontitis and the risk of tongue cancer. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 450–454. [Google Scholar] [CrossRef]
- Migliorati, C.A. Periodontal diseases and cancer. Lancet Oncol. 2008, 9, 510–512. [Google Scholar] [CrossRef]
- Migliorati, C.A.; Madrid, C. The interface between oral and systemic health: the need for more collaboration. Clin. Microbiol. Infect. 2007, 13 Suppl. 4, 11–16. [Google Scholar] [CrossRef]
- Gandolfo, S.; Castellani, R.; Pentenero, M. Proliferative verrucous leukoplakia: a potentially malignant disorder involving periodontal sites. J. Periodontol. 2009, 80, 274–281. [Google Scholar] [CrossRef]
- Asqah, M.; Al Hamoudi, N.; Anil, S.; Al Jebreen, A.; Al-Hamoudi, W.K. (2009). Is the presence of Helicobacter pylori in dental plaque of patients with chronic periodontitis a risk factor for gastric infection? Can. J. Gastroenterol. 2009, 23, 177–179. [Google Scholar]
- Kilmartin, C.M. Dental implications of Helicobacter pylori. J. Can. Dent. Assoc. 2002, 68, 489–493. [Google Scholar]
- Dixon, M.F. Helicobacter pylori and peptic ulceration. Histopathological aspects. J. Gastroenterol. Hepatol. 1991, 6, 125–130. [Google Scholar] [CrossRef]
- Genta, R.M.; Hammer, H.W.; Graham, D.Y. Gastric lymphoid follicles in Helicobacter pylori infection: Frequency, distribution, and response to triple therapy. Hum. Pathol. 1993, 24, 577–583. [Google Scholar] [CrossRef]
- Graham, D.Y. Helicobacter pylori: Its epidemiology and its role in duodenal ulcer disease. J. Gastroenterol. Hepatol. 1991, 6, 105–113. [Google Scholar] [CrossRef]
- Tytgat, G.N.; Noach, L.A.; Rauws, E.A. Helicobacter pylori infection and duodenal ulcer disease. Gastroenterol. Clin. North Am. 1993, 22, 127–139. [Google Scholar]
- Wotherspoon, A.C.; Ortiz-Hidalgo, C.; Falzon, M.R.; Isaacson, P.G. Helicobacter pylori–associated gastritis and primary B-cell gastric lymphoma. Lancet 1991, 338, 1175–1176. [Google Scholar] [CrossRef]
- Dye, B.A.; Kruszon-Moran, D.; McQuillan, D. The relationship between periodontal disease attributes and Helicobacter pylori infection among adults in the United States. Am. J. Public Health 2002, 92, 1809–1815. [Google Scholar] [CrossRef]
- Gebara, E.C.E.; Pannuti, C.; Faria, C.M.; Chehter, L.; Mayer, M.P.A.; Lima, L.A.P.A. Prevalence of Helicobacter pylori detected by polymerase chain reaction in the oral cavity of periodontitis patients. Oral Microbiol. Immunol. 2004, 19, 277–280. [Google Scholar] [CrossRef]
- Nguyen, A.M.; Engstrand, L.; Genta, R.M.; Graham, D.Y.; El-Zaatari, F.A.K. Detection of Helicobacter pylori in dental plaque by reverse transcription polymerase chain reaction. J. Clin. Microbiol. 1993, 31, 783–787. [Google Scholar]
- Riggio, M.P.; Lennon, A. Identification by PCR of Helicobacter pylori in subgingival plaque of adult periodontitis patients. J. Med. Microbiol. 1999, 48, 317–322. [Google Scholar] [CrossRef]
- Gebara, E.C.E.; Faria, C.M.; Pannuti, C.; Chehter, L.; Mayer, M.P.A.; Lima, L.A.P.A. Persistence of Helicobacter pylori in the oral cavity after systemic eradication therapy. J. Clin. Periodontol. 2006, 33, 329–333. [Google Scholar] [CrossRef]
- Umeda, M.; Kobayashi, H.; Takeuchi, Y.; Hayashi, J.; Mototome-Hayashi, Y.; Yano, K. High prevalence of Helicobacter pylori detected by PCR in the oral cavities of periodontitis patients. J. Periodontol. 2003, 74, 129–134. [Google Scholar] [CrossRef]
- Nguyen, A.M.; El-Zaatari, F.A.K.; Graham, D.Y. Helicobacter pylori in the oral cavity. A critical review of the literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1995, 79, 705–709. [Google Scholar] [CrossRef]
- Avcu, N.; Avcu, F.; Beyan, C.; Ural, A.; Kaptan, K.; Ozyurt, M. The relationship between gastric-oral Helicobacter pylori and oral hygiene in patients with vitamin B-12 deficiency anemia. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2001, 92, 166–169. [Google Scholar] [CrossRef]
- Ferguson, D.A.; Chuanfu, L.I, Jr.; Patel, N.R.; Mayberry, W.R.; Chi, D.S.; Thomas, E. Isolation of Helicobacter pylori from saliva. J. Clin. Microbiol. 1993, 31, 2802–2804. [Google Scholar]
- Mapstone, N.P.; Lynch, D.A.F.; Lewis, F.A.; Axon, A.T.R.; Tompkins, D.S.; Dixon, M.F. Identification of Helicobacter pylori DNA in the mouths and stomachs of patients with gastritis using PCR. J. Clin. Pathol. 1993, 46, 540–543. [Google Scholar] [CrossRef]
- Oshowo, A.; Gillam, D.; Botha, A.; Turnio, M.; Holton, J.; Boulos, P. Helicobacter pylori: The mouth, stomach and gut axis. Ann. Periodontol. 1998, 3, 276–280. [Google Scholar] [CrossRef]
- Song, Q.; Lange, T.A.; Spahr, A.; Adler, G.; Bode, G. Characteristic distribution pattern of Helicobacter pylori in dental plaque and saliva detected with nested PCR. J. Med. Microbiol. 2000, 49, 349–353. [Google Scholar]
- Miyabayashi, H.; Furihata, K.; Shimizu, T.; Ueno, I.; Akamatsu, T. Influence of oral Helicobacter pylori on the success of eradication therapy against gastric Helicobacter pylori. Helicobacter 2000, 5, 30–37. [Google Scholar] [CrossRef]
- Souto, R.; Colombo, A.P. Detection of Helicobacter pylori by polymerase chain reaction in the subgingival biofilm and saliva of non-dyspeptic periodontal patients. J. Periodontol. 2008, 79, 97–103. [Google Scholar] [CrossRef]
- Andersen, R.N.; Ganeshkumar, N.; Kolenbrander, P.E. Helicobacter pylori adheres selectively to Fusobacterium spp. Oral Microbiol. Immunol. 1998, 13, 51–54. [Google Scholar] [CrossRef]
- Paster, B.J.; Gibbons, R.J. Chemotactic response to formate by Campylobacter concisus and its potential role in gingival colonization. Infect. Immun. 1986, 52, 378–383. [Google Scholar]
- Okuda, K.; Kimizuka, R.; Katakura, A.; Nakagawa, T.; Ishihara, K. Ecological and immunopathological implications of oral bacteria in Helicobacter pylori-infected disease. J. Periodontol. 2003, 74, 123–128. [Google Scholar] [CrossRef]
- Albandar, J.M. Epidemiology and risk factors of periodontal diseases. Dent. Clin. North Am. 2005, 49, 517–532. [Google Scholar] [CrossRef]
- Goodman, K.J.; Correa, P. The transmission of Helycobacter pylori. A critical review of the evidence. Int. J. Epidemiol. 1995, 24, 875–877. [Google Scholar] [CrossRef]
- Fernando, N.; Jayakumar, G.; Perera, N.; Amarasingha, I.; Meedin, F.; Holton, J. Presence of Helicobacter pylori in betel chewers and non-betel chewers with and without oral cancers. BMC Oral Health 2009, 9, 23. [Google Scholar] [CrossRef]
- Farinati, F.; Cardin, R.; Cassaro, M.; Bortolami, M.; Nitti, D.; Tieppo, C.; Zaninotto, G.; Rugge, M. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: a morphological, biological and molecular pathway. Eur. J. Cancer Prev. 2008, 17, 195–200. [Google Scholar] [CrossRef]
- Rautelin, H.I.; Oksanen, A.M.; Veijola, L.I.; Sipponen, P.I.; Tervaharrtiala, T.I.; Sorsa, T.A.; Lauhio, A. Enhanced systemic matrix metalloproteinase response in Helicobacter pylori gastritis. Ann. Med. 2009, 41, 208–215. [Google Scholar] [CrossRef]
- Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis 2008, 29, 2400–2405. [Google Scholar] [CrossRef]
- Calvino-Fernandez, M.; Benito-Martinez, S.; Parra-Cid, T. Oxidative stress by Helicobacter pylori causes apoptosis through mitochondrial pathway in gastric epithelial cells. Apoptosis 2008, 13, 1267–1280. [Google Scholar] [CrossRef]
- Rucci, N.; Susa, M.; Teti, A. Inhibition of protein kinase c-Src as a therapeutic approach for cancer and bone metastases. Anti-Cancer Agents Med. Chem. 2008, 8, 342–349. [Google Scholar] [CrossRef]
- Guarino, M. Src signalling in cancer invasion. J. Cell. Physiol. 2010, 223, 14–26. [Google Scholar]
- Cudic, M.; Fields, G.B. Extracellular proteases as targets for drug development. Curr. Protein Pept. Sci. 2009, 10, 297–307. [Google Scholar] [CrossRef]
- Jedinak, A.; Maliar, T. Inhibitors of proteases as anti-cancer drugs. Neoplasma 2005, 52, 85–92. [Google Scholar]
- Thomas, G.J.; Nystrom, M.L.; Marshall, J.F. v6 Integrin in wound healing and cancer of the oral cavity. J. Oral Pathol. Med. 2006, 35, 1–10. [Google Scholar] [CrossRef]
- Huang, X.; Wu, J.; Spong, S.; Sheppard, D. The integrin alpha v beta 6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. J. Cell Sci. 1998, 111 (Pt 15), 2189–2195. [Google Scholar]
- Thomas, G.J.; Poomsawat, S.; Lewis, M.P.; Hart, I.R.; Speight, P.M.; Marshall, J.F. alpha v beta 6 Integrin upregulates matrix metalloproteinase 9 and promotes migration of normal oral keratinocytes. J. Invest. Dermatol. 2001, 116, 898–904. [Google Scholar] [CrossRef]
- Janes, S.M.; Watt, F.M. Switch from alphavbeta5 to alphavbeta6 integrin expression protects squamous cell carcinomas from anoikis. J. Cell Biol. 2004, 166, 419–431. [Google Scholar] [CrossRef]
- Breuss, J.M.; Gallo, J.; DeLisser, H.M.; Klimanskaya, I.V.; Folkesson, H.G.; Pittet, J.F.; Nishimura, S.L.; Aldape, K.; Lander, D.V.; Carpenter, W.; Gillett, N.; Sheppard, D.; Matthay, M.A.; Albelda, S.M.; Kramer, R.H.; Pytela, R. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J. Cell Sci. 1995, 108, 2241–2251. [Google Scholar]
- Clark, R.A.; Ashcroft, G.S.; Spencer, M.J.; Larjava, H.; Ferguson, M.W. Re-epithelialization of normal human excisional wounds is associated with a switch from alpha v beta 5 to alpha v beta 6 integrins. Br. J. Dermatol. 1996, 135, 46–51. [Google Scholar]
- Haapasalmi, K.; Zhang, K.; Tonnesen, M.; Olerud, J.; Sheppard, D.; Salo, T.; Kramer, R.; Clark, R.A.; Uitto, VJ.; Larjava, H. Keratinocytes in human wounds express alpha v beta 6 integrin. J. Invest. Dermatol. 1996, 106, 42–48. [Google Scholar]
- Ghannad, F.; Nica, D.; Garcia Fulle, M.I.; Grenier, D.; Putnins, E.E.; Johnston, S.; Eslami, A.; Koivisto, L.; Jiang, G.; McKee, M.D.; Häkkinen, L.; Larjava, H. Absence of αvβ6 Integrin Is Linked to Initiation and Progression of Periodontal Disease. Am. J. Pathol. 2008, 172, 1271–1286. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Jones, J.; Watt, F.M.; Speight, P.M. Changes in the expression of alpha v integrins in oral squamous cell carcinomas. J. Oral Pathol. Med. 1997, 26, 63–68. [Google Scholar] [CrossRef]
- Ahmed, N.; Riley, C.; Rice, G.E.; Quinn, M.A.; Baker, M.S. Alpha (v) beta (6) integrin – A marker for the malignant potential of epithelial ovarian cancer. J. Histochem. Cytochem. 2002, 50, 1371–1380. [Google Scholar] [CrossRef]
- Arihiro, K.; Kaneko, M.; Fujii, S.; Inai, K.; Yokosaki, Y. Significance of alpha 9 beta 1 and alpha v beta 6 integrin expression in breast carcinoma. Breast Cancer 2000, 7, 19–26. [Google Scholar] [CrossRef]
- Bates, R.C.; Bellovin, D.I.; Brown, C.; Maynard, E.; Wu, B.; Kawakatsu, H.; Sheppard, D.; Oettgen, P.; Mercurio, A.M. Transcriptional activation of integrin beta 6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J. Clin. Invest. 2005, 115, 339–347. [Google Scholar]
- Kawashima, A.; Tsugawa, S.; Boku, A.; Kobayashi, M.; Minamoto, T.; Nakanishi, I.; Oda, Y. Expression of alpha v integrin family in gastric carcinomas: increased alphavbeta 6 is associated with lymph node metastasis. Pathol. Res. Pract. 2003, 199, 57–64. [Google Scholar] [CrossRef]
- Sipos, B.; Hahn, D.; Carceller, A.; Piulats, J.; Hedderich, J.; Kalthoff, H.; Goodman, S.L.; Kosmahl, M.; Klöppel, G. Immunohistochemical screening for beta 6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology 2004, 45, 226–236. [Google Scholar] [CrossRef]
- Westernoff, T.H.; Jordan, R.C.; Regezi, J.A.; Ramos, D.M.; Schmidt, B.L. Beta-6 Integrin, tenascin-C, and MMP-1 expression in salivary gland neoplasms. Oral Oncol. 2005, 41, 170–174. [Google Scholar]
- Hamidi, S.; Salo, T.; Kainulainen, T.; Epstein, J.; Lerner, K.; Larjava, H. Expression of alpha(v)beta6 integrin in oral leukoplakia. Br. J. Cancer 2000, 82, 1433–1440. [Google Scholar] [CrossRef]
- Regezi, J.A.; Ramos, D.M.; Pytela, R.; Dekker, N.P.; Jordan, R.C. Tenascin and beta 6 integrin are overexpressed in floor of mouth in situ carcinomas and invasive squamous cell carcinomas. Oral Oncol. 2002, 38, 332–336. [Google Scholar] [CrossRef]
- Niu, J.; Dorahy, D.J.; Gu, X.; Scott, R.J.; Draganic, B.; Ahmed, N.; Agrez, M.V. Integrin expression in colon cancer cells is regulated by the cytoplasmic domain of the beta 6 integrin subunit. Int. J. Cancer 2002, 99, 529–537. [Google Scholar] [CrossRef]
- Niu, J.; Gu, X.; Ahmed, N.; Andrews, S.; Turton, J.; Bates, R.; Agrez, M. The alphaVbeta6 integrin regulates its own expression with cell crowding: implications for tumour progression. Int. J. Cancer 2001, 92, 40–48. [Google Scholar] [CrossRef]
- Scott, K.A.; Arnott, C.H.; Robinson, S.C.; Moore, R.J.; Thompson, R.G.; Marshall, J.F.; Balkwill, F.R. TNF-alpha regulates epithelial expression of MMP-9 and integrin alphavbeta6 during tumour promotion. A role for TNF- alpha in keratinocyte migration? Oncogene 2004, 23, 6954–6966. [Google Scholar] [CrossRef]
- Banno, T.; Gazel, A.; Blumenberg, M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 2004, 279, 32633–32642. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Raghavan, S. Defining the role of integrin alphavbeta 6 in cancer. Curr Drug Targets 2009, 10, 645–652. [Google Scholar] [CrossRef]
- Müller-Hermelink, N.; Braumüller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A.; Mocikat, R.; Schwaiger, M.; Förster, I.; Huss, R.; Weber, W.A.; Kneilling, M.; Röcken, M. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008, 13, 507–518. [Google Scholar] [CrossRef]
- Goodman, S.L.; Holzemann, G.; Sulyok, G.A.; Kessler, H. Nanomolar small molecule inhibitors for alpha v(beta) 6, alpha v (beta)5, and alpha v(beta)3 integrins. J. Med. Chem. 2002, 45, 1045–1051. [Google Scholar] [CrossRef]
- Weinreb, P.H.; Simon, K.J.; Rayhorn, P.; Yang, W.J.; Leone, D.R.; Dolinski, B.M.; Pearse, B.R.; Yokota, Y.; Kawakatsu, H.; Atakilit, A.; Sheppard, D.; Violette, S.M. Functionblocking integrin alpha v beta 6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J. Biol. Chem. 2004, 279, 17875–17887. [Google Scholar] [CrossRef]
- Xue, H.; Atakilit, A.; Zhu, W.; Li, X.; Ramos, D.M.; Pytela, R. Role of the alpha (v) beta 6 integrin in human oral squamous cell carcinoma growth in vivo and in vitro. Biochem. Biophys. Res. Commun. 2001, 288, 610–618. [Google Scholar] [CrossRef]
- Liu, M.; Acres, B.; Balloul, J.M.; Bizouarne, N.; Paul, S.; Slos, P.; Squiban, P. Gene-based vaccines and immunotherapeutics. Proc. Natl. Acad. Sci.USA 2004, 101 (Suppl. 2), 14567–14571. [Google Scholar]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Cirielli, C.; Serino, F.; Straino, S.; Toietta, G.; Abeni, D.; Ventoruzzo, G.; Orlando, G.; Mazzanti, P.; Melillo, G.; Whickham, T.J.; Kovesdi, I.; Biglioli, P.; Gaetano, C.; Capogrossi, M.C. Adenovirus vectors targeting alphaV integrin or heparan sulfate receptors display different distribution of transgene activity after intramuscular injection. J. Gene Med. 2004, 6, 309–316. [Google Scholar] [CrossRef]
- Serhan, C.N. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef]
- Serhan, C.N. Controlling the resolution of acute inflammation: a new genus of dual anti-inflammatory and proresolving mediators. J. Periodontol. 2008, 79, 1520–1526. [Google Scholar] [CrossRef]
- Janakiram, N.B.; Rao, C.V. Role of lipoxins and resolvins as anti-inflammatory and proresolving mediators in colon cancer. Curr. Mol. Med. 2009, 9, 565–579. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S200–S215. [Google Scholar]
- Majino, G.; Joris, I. Cells, Tissues and Disease: Principles of General Pathology; Oxford University Press: New York, NY, USA, 2004. [Google Scholar]
- Godson, C.; Mitchell, S.; Harvey, K.; Petasis, NA; Hogg, N; Brady, HR. Cutting edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte derived macrophages. J. Immunol. 2000, 164, 1663–1667. [Google Scholar]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Nicos A. Petasis, N.A.; Serhan, C.N. Sterochemical assignment, anti-inflammatory properties and receptor for the omega-3 lipid mediator resolving E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Campbell, E.L.; Louis, N.A.; Tomassetti, S.E.; Canny, G.O.; Arita, M.; Serhan, C.N.; Colgan, S.P. Resolvin E1 promotes mucosal surface clearance of neutrophils: A new paradigm for inflammatory resolution. FASEB J. 2007, 21, 3162–3170. [Google Scholar] [CrossRef]
- Serhan, C.N.; Gotlinger, K.; Hong, S.; Lu, Y.; Siegelman, J.; Baer, T.; Yang, R.; Colgan, S.P.; Petasis, N.A. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: Assignments of dihydroxy- containing docosatrienes. J. Immunol. 2006, 176, 1848–1859. [Google Scholar]
- Canny, G.; Levy, O.; Furuta, G.T.; Narravula-Alipati, S.; Sisson, R.B; Serhan, C.N.; Colgan, S.P. Lipid mediator- induced expression of bactericidal/permeability- increasing protein (BPI) in human mucosal epithelia. Proc. Natl. Acad. Sci. USA 2002, 99, 3902–3907. [Google Scholar]
- Chiang, N.; Serhan, C.N.; Dahlen, S.E.; Drazen, J.M.; Hay, D.W.P.; Govati, G.E.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor ALX: Potent ligand specific and stereoselective actions in vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef]
- Claria, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eocosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef]
- Mitchell, S.; Thomas, G.; Harvey, K.; Cottell, D.; Reville, K.; Berlasconi, G.; Petasis, N.A.; Erwig, L.; Rees, A.J.; Savill, J.; Brady, H.R.; Godson, C. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: Stimulation of of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 2002, 13, 2497–2507. [Google Scholar] [CrossRef]
- McMahon, B.; Godson, C. Lipoxins: Endogenous regulators of inflammation. Am. J. Physiol. Renal Physiol. 2004, 286, F189–F201. [Google Scholar] [CrossRef]
- Guilford, W.J.; Parkinson, J.F. Second-generation beta oxidation resistant 3-oxa-lipoxin A4 analogs. Prostaglandins Leukot Essent Fatty Acids 2005, 73, 245–250. [Google Scholar] [CrossRef]
- Petasis, N.A.; Keledijan, R.; Sun, Y-P.; Nagulapalli, K.C.; Tjonahen, E.; Yang, R.; Serhan, C.N. Design and synthesis of benzo-lipoxin A4 analogs with enhanced stability and potent anti-inflammatory properties. Bioorg. Med. Chem. Lett. 2008, 18, 1382–1387. [Google Scholar] [CrossRef]
- Hong, S.; Gronert, K.; Devchand, P.R.; Moussignac, R.L.; Serhan, C.N. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood and glial cells. Autacoids in anti-inflammation. J. Biol. Chem. 278, 14677–14687. [Google Scholar]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid derived mediators with anti-inflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-non-steroidal anti-inflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef]
- Sun, Y.P.; Oh, S.F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S.P.; Petasis, N.A.; Serhan, C.N. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties and enzymatic inactivation. J. Biol. Chem. 2007, 282, 9323–9334. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Marcheselli, V.L.; Serhan, C.N.; Bazan, N.G. Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci.USA 2004, 101, 8491–8496. [Google Scholar]
- Gronert, K.; Maheshwari, N.; Khan, N.; hassan, I.R.; Dunn, M.; laniado Schwartzman, M. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defence. J. Biol. Chem. 2005, 280, 15267–15278. [Google Scholar]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation. The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Maddox, J.F.; Serhan, C.N. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: Selective inactivation by dehydrogenation and reduction. J. Exp. Med. 1996, 183, 137–146. [Google Scholar] [CrossRef]
- Blumberg, R.S.; Strober, W. Prospects for research in inflammatory bowel disease. JAMA 2001, 285, 643–647. [Google Scholar] [CrossRef]
- Fiorucci, S.; Wallace, J.L.; Mencarelli, A; Distrutti, J.L.; Rizzo, G.; Farneti, S.; Morelli, A.; Tseng, J.L.; Suramanyam, B.; Guilford, W.J.; Parkinson, J.F. A beta-oxidation-resistant lipoxin A4 analog treats hapten induced colitis by attenuating inflammation and immune dysfunction. Proc. Natl. Acad. Sci. USA 2004, 101, 15736–15741. [Google Scholar] [CrossRef]
- Soory, M. A role for non-antimicrobial actions of tetracyclines in combating oxidative stress in periodontal and metabolic diseases. Open Dent. J. 2008, 2, 5–12. [Google Scholar] [CrossRef]
- Rahman, Z.A.; Soory, M. Antioxidant effects of glutathione and IGF in a hyperglycaemic cell culture model of fibroblasts: some actions of advanced glycaemic end products (AGE) and nicotine. Endocr. Metab. Immune Disorders-Drug Targets 2006, 6, 279–286. [Google Scholar] [CrossRef]
- Figuero-Ruiz, E.; Soory, M.; Cerero, R.; Bascones, A. Oxidant/antioxidant interactions of nicotine, Coenzyme Q10, Pycnogenol and phytoestrogens in oral periosteal fibroblasts and MG63 osteoblasts. Steroids 2006, 71, 1062–1072. [Google Scholar] [CrossRef]
- Soory, M. Redox status in periodontal and systemic inflammatory conditions including associated neoplasias: Antioxidants as adjunctive therapy? Infect. Disord. Drug Targets 2009, 9, 415–427. [Google Scholar] [CrossRef]
- Soory, M. Relevance of nutritional antioxidants in metabolic syndrome, ageing and cancer: Potential for therapeutic targeting. Infect. Disord. Drug Targets 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Soory, M. Periodontal diseases and rheumatoid arthritis: A coincident model for therapeutic intervention. Curr. Drug Metab. 2007, 8, 750–757. [Google Scholar] [CrossRef]
- Soory, M. Relevance of dyslipidaemia and its consequences in periodontal patients with co-existing cardiovascular disease and diabetes mellitus: Therapeutic targets. Rec. Patents Endocr. Metab. Imm. Drug Discovery 2009, 3, 214–224. [Google Scholar] [CrossRef]
- Eley, B.M.; Soory, M.; Manson, D. Periodontics, 6th ed.; Elsevier: New York, NY, USA, 2010; pp. 107–125. [Google Scholar]
- Petti, S.; Scully, C. Polyphenols, oral health and disease: A review. J. Dent. 2009, 37, 413–423. [Google Scholar] [CrossRef]
- Williams, R.J.; Spencer, J.P.E.; Rice-Evans, C. Flavonoids: antioxidants or signaling molecules? Free Radical Biol. Med. 2004, 36, 838–849. [Google Scholar] [CrossRef]
- Galati, G.; O’Brien, P.J. Potential toxicity of flavonoids and other dietary phenolics: significance for their chemopreventive and anti-cancer properties. Free Radical Biol. Med. 2004, 37, 287–303. [Google Scholar] [CrossRef]
- Decker, E.A. phenolics: prooxidants or antioxidants? Nutr. Rev. 1997, 55, 396–407. [Google Scholar] [CrossRef]
- Hirvonen, T.; Virtamo, J.; Korhonen, P.; Albanes, D.; Poetinen, P. Flavonol and flavone intake and the risk of cancer in male smokers (Finland). Cancer Causes Contr. 2001, 12, 789–796. [Google Scholar]
- Ho, Y.C.; Yang, S.F.; Peng, C.Y.; Chou, M.Y.; Chang, Y.C. Epigallocatechin-3-gallate inhibits the invasion of human oral cancer cells and decreases the production of matrix metalloproteinases and urokinase-plasminogen activator. J. Oral Pathol. Med. 2007, 36, 588–593. [Google Scholar] [CrossRef]
- Hsu, S.; Lewis, J.B.; Borke, J.L.; Singh, B.; Dickinson, D.P.; Caughman, G.B. Chemoprevention effects of green tea polyphenols correlate with reversible induction of p57 expression. Anticancer Res. 2001, 21, 3743–3748. [Google Scholar]
- Masuda, M.; Suzui, M.; Weinstein, I.B. effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin. Cancer Res. 2001, 7, 4220–4229. [Google Scholar]
- Volate, S.R.; Muga, S.J.; Issa, A.Y.; Nitcheva, D.; Smith, T.; Wargovich, M.J. Epigenetic modulation of the retinoid X receptor alpha by green tea in the azoxymethane-Apc Min/+ mouse model of intestinal cancer. Mol. Carcinog. 2009, 48, 920–933. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Soory, M. Oxidative Stress Induced Mechanisms in the Progression of Periodontal Diseases and Cancer: A Common Approach to Redox Homeostasis? Cancers 2010, 2, 670-692. https://doi.org/10.3390/cancers2020670
Soory M. Oxidative Stress Induced Mechanisms in the Progression of Periodontal Diseases and Cancer: A Common Approach to Redox Homeostasis? Cancers. 2010; 2(2):670-692. https://doi.org/10.3390/cancers2020670
Chicago/Turabian StyleSoory, Mena. 2010. "Oxidative Stress Induced Mechanisms in the Progression of Periodontal Diseases and Cancer: A Common Approach to Redox Homeostasis?" Cancers 2, no. 2: 670-692. https://doi.org/10.3390/cancers2020670
APA StyleSoory, M. (2010). Oxidative Stress Induced Mechanisms in the Progression of Periodontal Diseases and Cancer: A Common Approach to Redox Homeostasis? Cancers, 2(2), 670-692. https://doi.org/10.3390/cancers2020670