Mini-Review: Novel Therapeutic Strategies to Blunt Actions of Pneumolysin in the Lungs


{kind=link}
{kind=link}
Abstract
:1. Severe Pneumonia: A Huge Medical Problem
2. Importance of Pulmonary Barrier Integrity
3. Pneumolysin: Structure and Activities
4. PLY Interactions with the Immune System
5. PLY Compromises the Barrier Function of the Lung
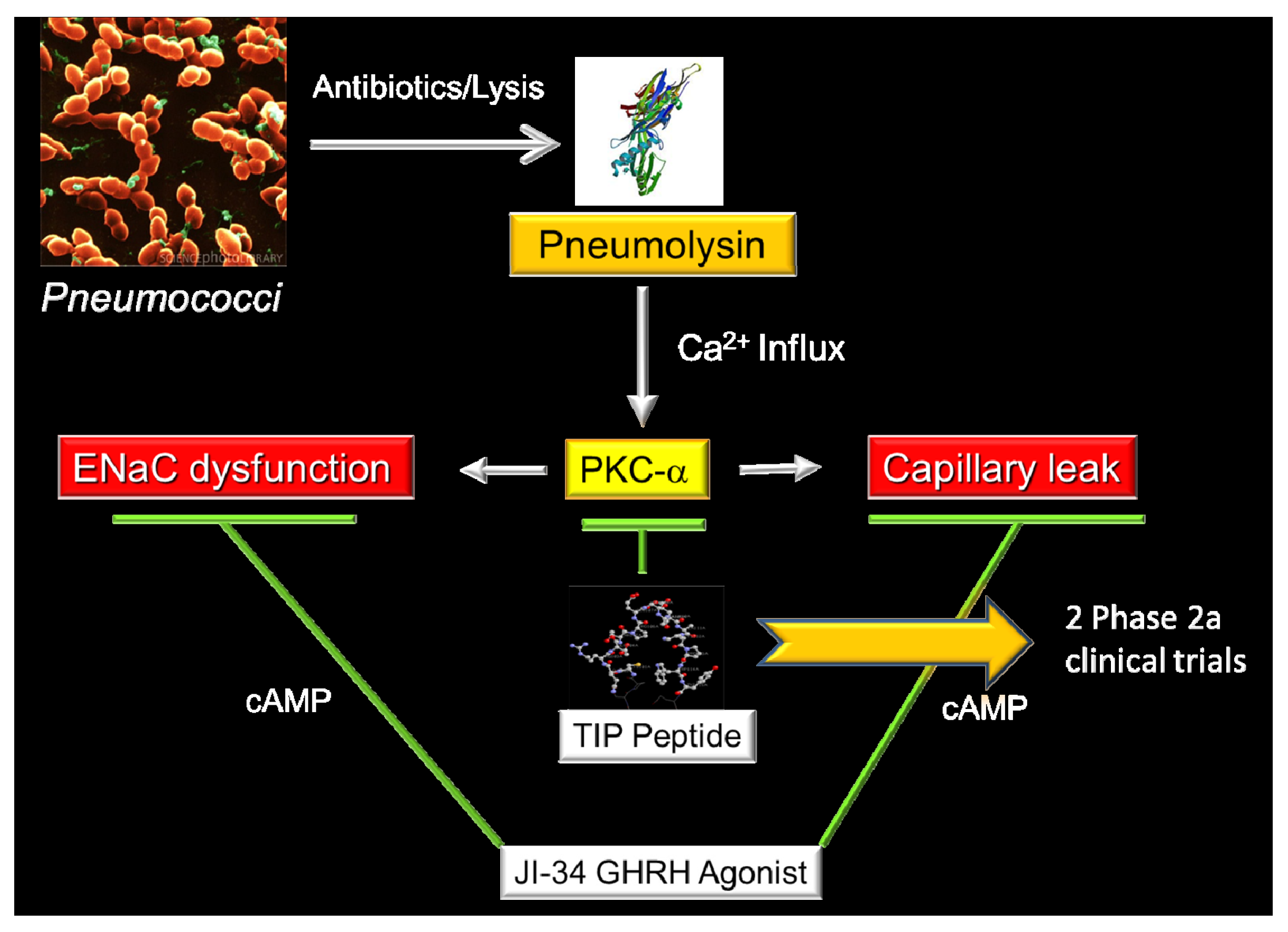
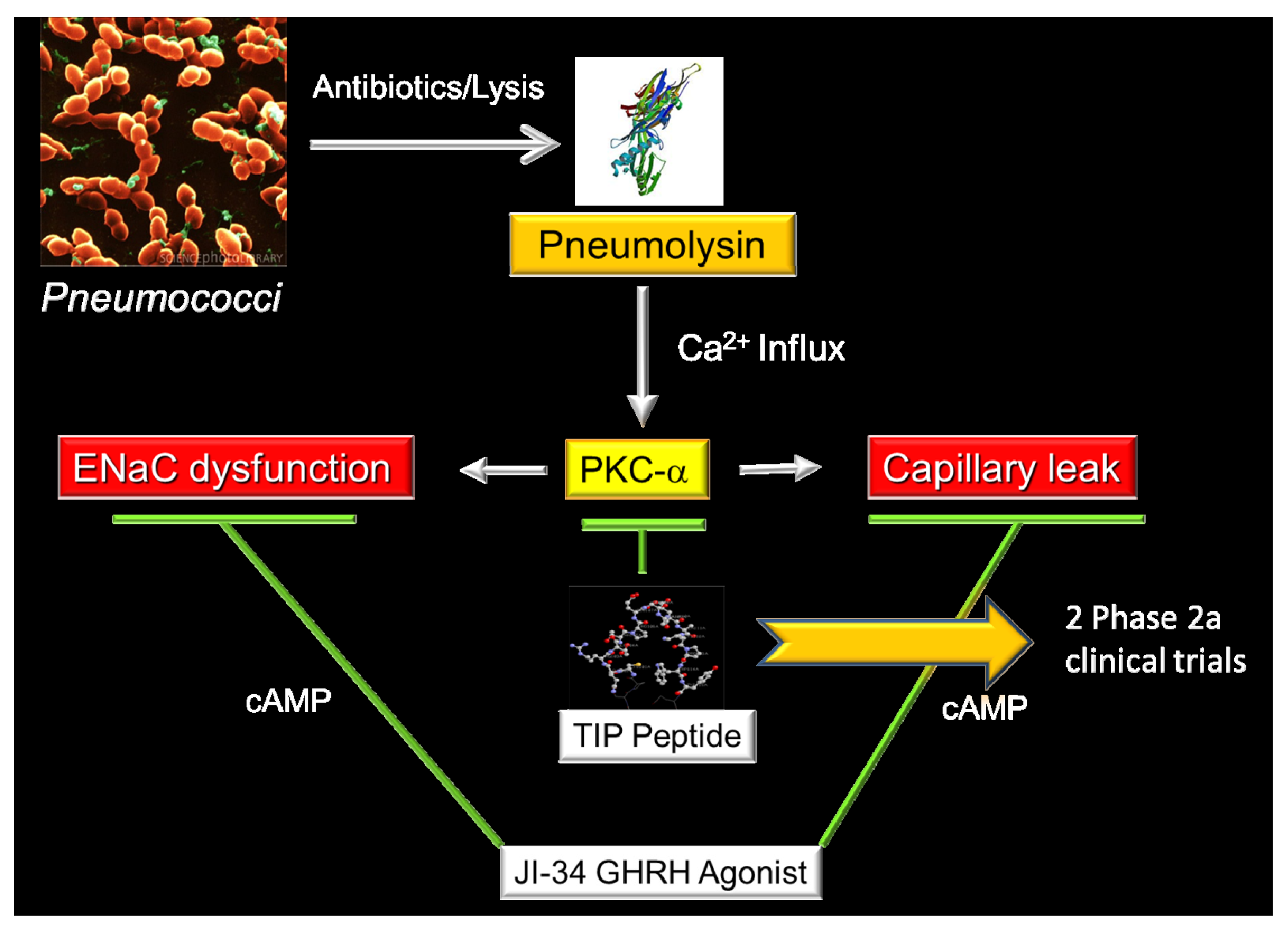
6. Does the Brain Protect Our Lungs During Pneumonia? The Unexpected Story of Growth Hormone-Releasing Hormone
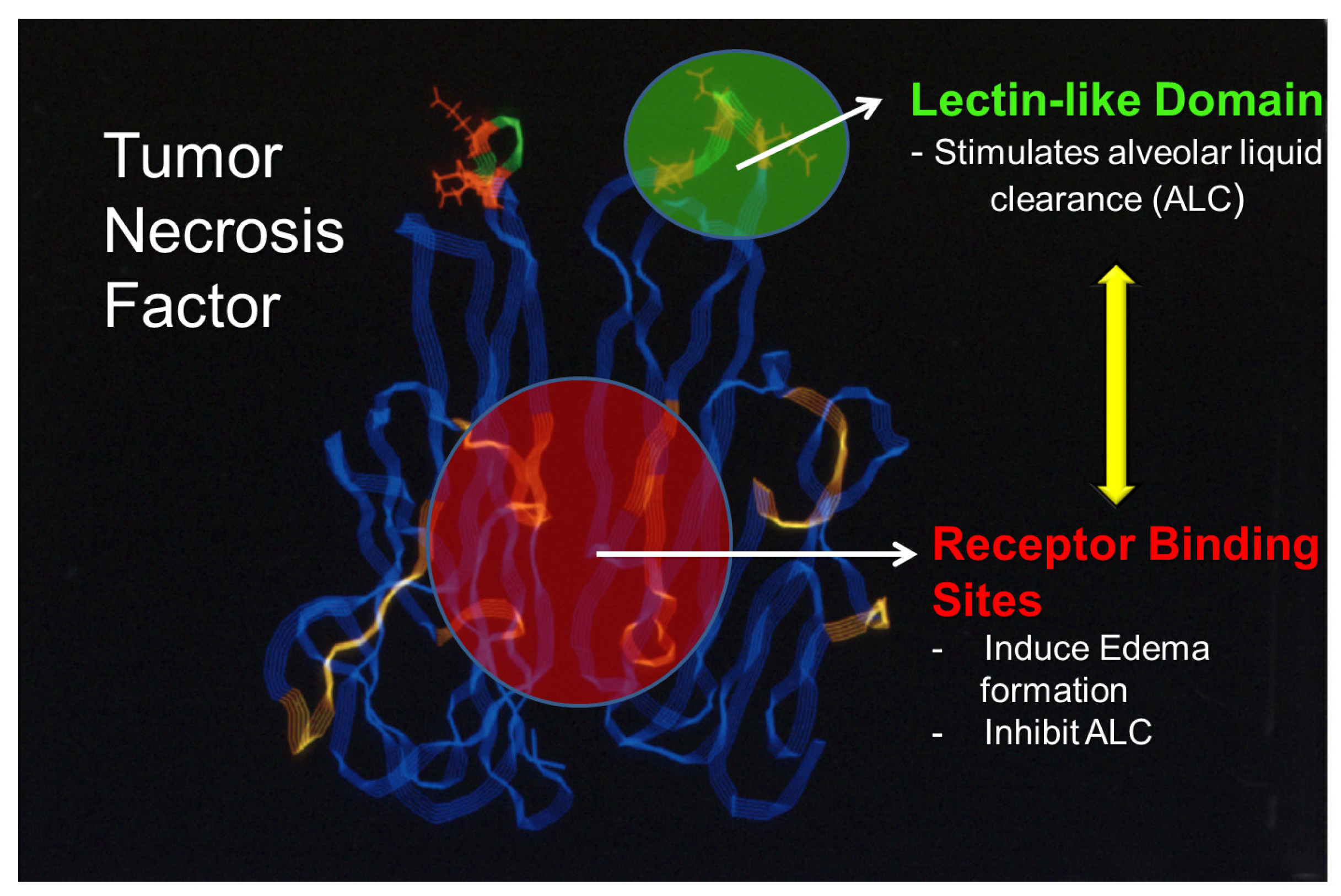
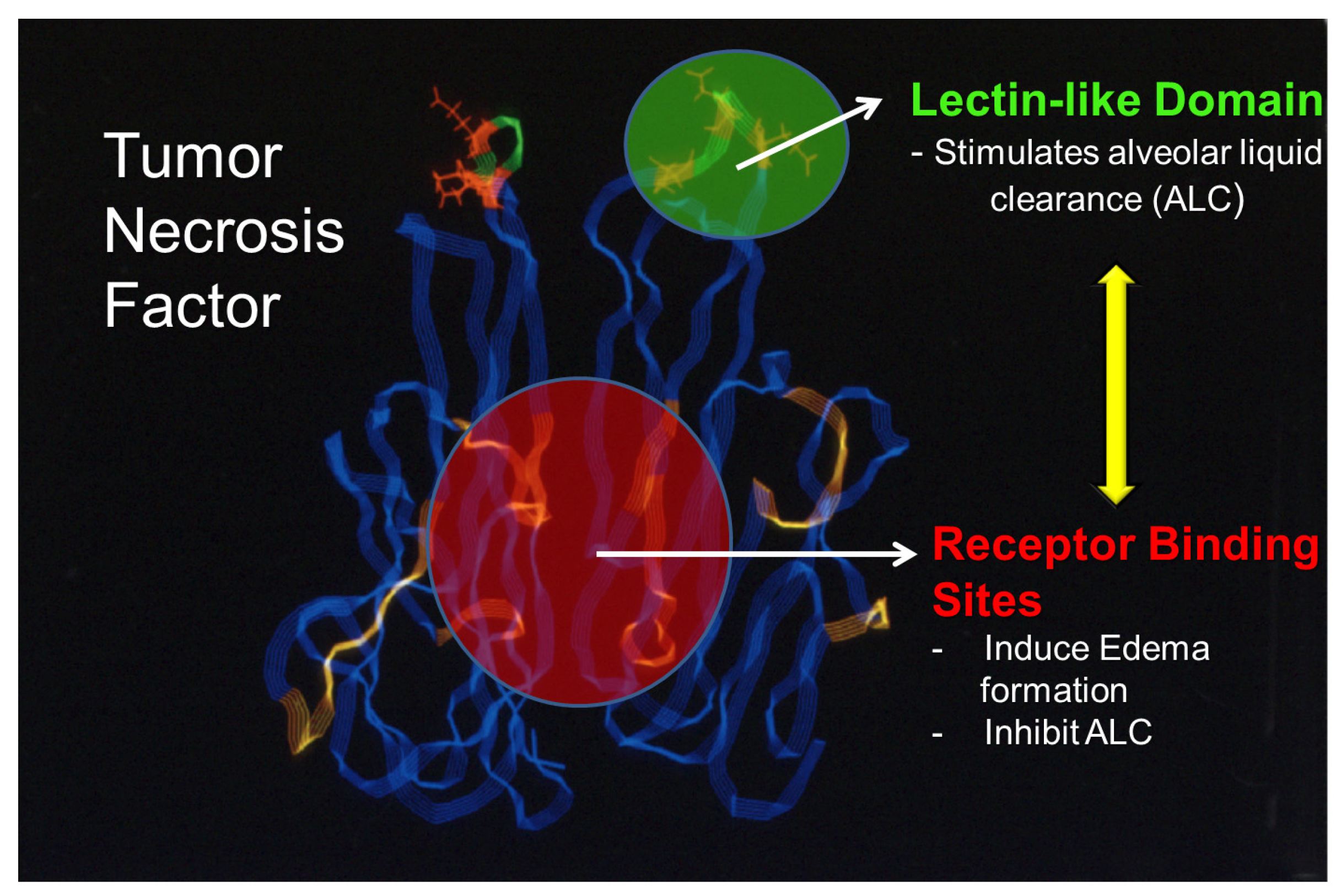
7. The Lectin-like Domain of Tumor Necrosis Factor: A Promising Therapeutic Candidate for the Treatment of Permeability Edema
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Rudan, I.; Boschi-Pinto, C.; Biloglav, Z.; Mulholland, K.; Campbell, H. Epidemiology and etiology of childhood pneumonia. Bull. World Health Organ. 2008, 86, 408–416. [Google Scholar] [CrossRef]
- Waterer, G.W.; Rello, J.; Wunderink, R.G. Management of community-acquired pneumonia in adults. Am. J. Respir. Crit. Care Med. 2011, 183, 157–164. [Google Scholar] [CrossRef]
- Rubins, J.B.; Charboneau, D.; Fasching, C.; Berry, A.M.; Paton, J.C.; Alexander, J.E.; Andrew, P.W.; Mitchell, T.J.; Janoff, E.N. Distinct roles for pneumolysin’s cytotoxic and complement activities in the pathogenesis of pneumococcal pneumonia. Am. J. Respir. Crit. Care Med. 1996, 153, 1339–1346. [Google Scholar] [CrossRef]
- Lucas, R.; Verin, A.D.; Black, S.M.; Catravas, J.D. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem. Pharmacol. 2009, 77, 1763–1772. [Google Scholar] [CrossRef]
- Repp, H.; Pamukçi, Z.; Koschinski, A.; Domann, E.; Darji, A.; Birringer, J.; Brockmeier, D.; Chakraborty, T.; Dreyer, F. Listeriolysin of Listeria monocytogenes forms Ca2+-permeable pores leading to intracellular Ca2+ oscillations. Cell Microbiol. 2002, 4, 483–491. [Google Scholar] [CrossRef]
- Ananthraman, A.; Israel, R.H.; Magnussen, C.R. Pleural-pulmonary aspects of Listeria monocytogenes infection. Respiration 1983, 44, 153–157. [Google Scholar] [CrossRef]
- Witzenrath, M.; Gutbier, B.; Hocke, A.C.; Schmeck, B.; Hippenstiel, S.; Berger, K.; Mitchell, T.J.; de los Toyos, J.R.; Rosseau, S.; Suttorp, N.; et al. Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit. Care Med. 2006, 34, 1947–1954. [Google Scholar] [CrossRef]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of endothelial junctional permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef]
- Dudek, S.M.; Garcia, J.G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar]
- Birukova, A.A.; Birukov, K.G.; Smurova, K.; Adyshev, D.; Kaibuchi, K.; Alieva, I.; Garcia, J.G.; Verin, A.D. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 2004, 18, 1879–1890. [Google Scholar]
- Birukova, A.A.; Birukov, K.G.; Adyshev, D.; Usatyuk, P.; Natarajan, V.; Garcia, J.G.; Verin, A.D. Involvement of microtubules and Rho pathway in TGF-beta1-induced lung vascular barrier dysfunction. J. Cell Physiol. 2005, 204, 934–947. [Google Scholar]
- Iliev, A.I.; Djannatian, J.R.; Nau, R.; Mitchell, T.J.; Wouters, F.S. Cholesterol-dependent actin remodeling via RhoA and Rac1 activation by the Streptococcus pneumoniae toxin pneumolysin. Proc. Natl. Acad. Sci. USA 2007, 104, 2897–2902. [Google Scholar]
- Siflinger-Birnboim, A.; Johnson, A. Protein kinase C modulates pulmonary endothelial permeability: A paradigm for acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L435–L451. [Google Scholar]
- Mehta, D.; Rahman, A.; Malik, A.B. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J. Biol. Chem. 2001, 276, 22614–22620. [Google Scholar] [CrossRef]
- Harrington, E.O.; Brunelle, J.L.; Shannon, C.J.; Kim, E.S.; Mennella, K.; Rounds, S. Role of protein kinase C isoforms in rat epididymal microvascular endothelial barrier function. Am. J. Respir. Cell Mol. Biol. 2003, 28, 626–636. [Google Scholar] [CrossRef]
- Dworakowski, R.; Alom-Ruiz, S.P.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol. Rep. 2008, 60, 21–28. [Google Scholar]
- Xu, H.; Goettsch, C.; Xia, N.; Horke, S.; Morawietz, H.; Förstermann, U.; Li, H. Differential roles of PKC-alpha and PKC-epsilon in controlling the gene expression of Nox4 in human endothelial cells. Free Radic. Biol. Med. 2008, 44, 1656–1667. [Google Scholar] [CrossRef]
- Predescu, D.; Predescu, S.; Shimizu, J.; Miyawaki-Shimizu, K.; Malik, A.B. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L371–L381. [Google Scholar]
- Min, J.K.; Cho, Y.L.; Choi, J.H.; Kim, Y.; Kim, J.H.; Yu, Y.S.; Rho, J.; Mochizuki, N.; Kim, Y.M.; Oh, G.T.; et al. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: Impaired permeability and angiogenesis in eNOS-deficient mice. Blood 2007, 109, 1495–1502. [Google Scholar] [CrossRef]
- Speyer, C.L.; Neff, T.A.; Warner, R.L.; Guo, R.F.; Sarma, J.V.; Riedemann, N.C.; Murphy, M.E.; Murphy, H.S.; Ward, P.A. Regulatory effects of iNOS on acute lung inflammatory responses in mice. Am. J. Pathol. 2003, 163, 2319–2328. [Google Scholar] [CrossRef]
- Paton, J.C.; Ferrante, A. Inhibition of human polymorphonuclear leukocyte respiratory burst, bactericidal activity, and migration by pneumolysin. Infect. Immun. 1983, 41, 1212–1216. [Google Scholar]
- Kanclerski, K.; Möllby, R. Production and purification of Streptococcus pneumoniae hemolysin (pneumolysin). J. Clin. Microbiol. 1987, 25, 222–225. [Google Scholar]
- Jefferies, J.M.; Johnston, C.H.; Kirkham, L.A.; Cowan, G.J.; Ross, K.S.; Smith, A.; Clarke, S.C.; Brueggemann, A.B.; George, R.C.; Pichon, B.; et al. Presence of non-hemolytic pneumolysin in serotypes of Streptococcus pneumoniae associated with disease outbreaks. J. Infect. Dis. 2007, 196, 936–944. [Google Scholar] [CrossRef]
- Libman, E. A Pneumococcus producing a peculiar form of hemolysis. Proc. N. Y. Pathol. Soc. 1905, 5, 168–175. [Google Scholar]
- Cole, R. Pneumococcus hemotoxin. J. Exp. Med. 1914, 20, 346–362. [Google Scholar] [CrossRef]
- Cohen, B.; Shwachman, H.; Perkins, M.E. Inactivation of pneumococcal hemolysin by certain sterols. Proc. Soc. Exp. Biol. Med. 1937, 35, 586–591. [Google Scholar] [CrossRef]
- Cohen, B.; Halbert, S.P.; Perkins, M.E. Pneumococcal hemolysin: The preparation of concentrates, and their action on red cells. J. Bacteriol. 1942, 43, 607–627. [Google Scholar]
- Avery, O.T.; Neill, J.M. Studies on oxidation and reduction by Pneumococcus: IV. Oxidation of hemotoxin in sterile extracts of Pneumococcus. J. Exp. Med. 1924, 39, 745–755. [Google Scholar]
- Cassidy, S.K.B.; O’Riordan, M.X.D. Review: More than a pore: The cellular response to cholesterol-dependent cytolysis. Toxins 2013, 5, 618–636. [Google Scholar] [CrossRef]
- Mitchell, T.J.; Walker, J.A.; Saunders, F.K.; Andrew, P.W.; Boulnois, G.J. Expression of the pneumolysin gene in Escherichia coli: Rapid purification and biological properties. Biochim. Biophys. Acta 1989, 1007, 67–72. [Google Scholar] [CrossRef]
- Walker, J.A.; Allen, R.L.; Falmagne, P.; Johnson, M.K.; Boulnois, G.J. Molecular cloning, characterization, and complete nucleotide sequence of the gene for pneumolysin, the sulfhydryl-activated toxin of Streptococcus pneumoniae. Infect. Immun. 1987, 55, 1184–1189. [Google Scholar]
- Paton, J.C.; Berry, A.M.; Lock, R.A.; Hansman, D.; Manning, P.A. Cloning and expression in Escherichia coli of the Streptococcus pneumoniae gene encoding pneumolysin. Infect. Immun. 1986, 54, 50–55. [Google Scholar]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20226–20231. [Google Scholar]
- Smyth, C.J.; Duncan, J.L. Thiol-activated (Oxygen Labile) Cytolysins. In Bacterial Toxins and Cell Membranes; Jeljaszewicz, J., Wadström, T., Eds.; Academic Press: New York, NY, USA, 1978; pp. 129–183. [Google Scholar]
- Sekino-Suzuki, N.; Nakamura, M.; Mitsui, K.I.; Ohno-Iwashita, Y. Contribution of individual tryptophan residues to the structure and activity of theta-toxin (perfringolysin O), a cholesterol-binding cytolysin. Eur. J. Biochem. FEBS 1996, 241, 941–947. [Google Scholar]
- Lim, J.E.; Park, S.A.; Bong, S.M.; Chi, Y.M.; Lee, K.S. Characterization of pneumolysin from Streptococcus pneumoniae, interacting with carbohydrate moiety and cholesterol as component of cell membrane. Biochem. Biophys. Res. Commun. 2013, 430, 659–663. [Google Scholar] [CrossRef]
- Darji, A.; Niebuhr, K.; Hense, M.; Wehland, J.; Chakraborty, T.; Weiss, S. Neutralizing monoclonal antibodies against listeriolysin: Mapping of epitopes involved in pore formation. Infect. Immun. 1996, 64, 2356–2358. [Google Scholar]
- Nato, F.; Reich, K.; L’hopital, S.; Rouyre, S.; Geoffroy, C.; Mazie, J.C.; Cossart, P. Production and characterization of neutralizing and non-neutralizing monoclonal antibodies against listeriolysin O. Infect. Immun. 1991, 59, 4641–4646. [Google Scholar]
- Fukuda, Y.; Yanagihara, K.; Higashiyama, Y.; Miyazaki, Y.; Hirakata, Y.; Mukae, H.; Tomono, K.; Mizuta, Y.; Tsukamoto, K.; Kohno, S. Effects of macrolides on pneumolysin of macrolide-resistant Streptococcus pneumoniae. Eur. Respir. J. 2006, 27, 1020–1025. [Google Scholar]
- Anderson, R.; Steel, H.C.; Cockeran, R.; von Gottberg, A.; de Gouveia, L.; Klugman, K.P.; Mitchell, T.J.; Feldman, C. Comparison of the effects of macrolides, amoxicillin, ceftriaxone, doxycycline, tobramycin and fluoroquinolones, on the production of pneumolysin by Streptococcus pneumoniae in vitro. J. Antimicrob. Chemother. 2007, 60, 1155–1158. [Google Scholar] [CrossRef]
- Berry, A.M.; Lock, R.A.; Hansman, D.; Paton, J.C. Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect. Immun. 1989, 57, 2324–2330. [Google Scholar]
- Berry, A.M.; Paton, J.C.; Hansman, D. Effect of insertional inactivation of the genes encoding pneumolysin and autolysin on the virulence of Streptococcus pneumonia type 3. Microb Pathog. 1992, 12, 87–93. [Google Scholar] [CrossRef]
- Howard, L.V.; Gooder, H. Specificity of the autolysin of Streptococcus (Diplococcus) pneumoniae. J. Bacteriol. 1974, 117, 796–804. [Google Scholar]
- Sanchez-Puelles, J.M.; Ronda, C.; Garcia, J.L.; Garcia, P.; Lopez, R.; Garcia, E. Searching for autolysin functions. Characterization of a pneumococcal mutant deleted in the lytA gene. Eur. J. Biochem. 1986, 158, 289–293. [Google Scholar] [CrossRef]
- Jedrzejas, M.J. Pneumococcal virulence factors: Structure and function. Microbiol. Mol. Biol. Rev. 2001, 65, 187–207. [Google Scholar] [CrossRef]
- Balachandran, P.; Hollingshead, S.K.; Paton, J.C.; Briles, D.E. The autolytic enzyme LytA of Streptococcus pneumoniae is not responsible for releasing pneumolysin. J. Bacteriol. 2001, 183, 3108–3116. [Google Scholar] [CrossRef]
- Ferrante, A.; Rowan-Kelly, B.; Paton, J.C. Inhibition of in vitro human lymphocyte response by the pneumococcal toxin pneumolysin. Infect. Immun. 1984, 46, 585–589. [Google Scholar]
- Nandoskar, M.; Ferrante, A.; Bates, E.J.; Hurst, N.; Paton, J.C. Inhibition of humanmonocyte respiratory burst, degranulation, phospholipid methylation and bactericidal activity by pneumolysin. Immunology 1986, 59, 515–520. [Google Scholar]
- Maus, U.; Srivastava, M.; Paton, J.C.; Mack, M.; Everhart, M.B.; Blackwell, T.S.; Christman, J.W.; Schlöndorff, D.; Seeger, W.; Lohmeyer, J. Pneumolysin-induced lung injury is independent of leukocyte trafficking into the alveolar space. J. Immunol. 2004, 173, 1307–1312. [Google Scholar]
- McNeela, E.A.; Burke, A.; Neill, D.R.; Baxter, C.; Fernandes, V.E.; Ferreira, D.; Smeaton, S.; El-Rachkidy, R.; McLoughlin, R.M.; Mori, A.; et al. Pneumolysin activates the NLRP3 inflammasome and promotes pro-inflammatory cytokines independently of TLR4. PLoS Pathog. 2010, 6, e1001191. [Google Scholar] [CrossRef]
- Koppe, U.; Högner, K.; Doehn, J.M.; Müller, H.C.; Witzenrath, M.; Gutbier, B.; Bauer, S.; Pribyl, T.; Hammerschmidt, S.; Lohmeyer, J.; et al. Steptococcus pneumonia stimulates a STING-and IFN regulatory factor 3-dependent type 1 IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells and mouse lungs. J. Immunol. 2012, 188, 811–817. [Google Scholar] [CrossRef]
- Feldman, C.; Munro, N.C.; Jeffery, P.K.; Mitchell, T.J.; Andrew, P.W.; Boulnois, G.J.; Guerreiro, D.; Rohde, J.A.; Todd, H.C.; Cole, P.J. Pneumolysin induces the salient histologic features of pneumococcal infection in the rat lung in vivo. Am. J. Respir. Crit. Care Med. 1991, 5, 416–423. [Google Scholar]
- Berry, A.M.; Yother, J.; Briles, D.E.; Hansman, D.; Paton, J.C. Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect. Immun. 1989, 57, 2037–2042. [Google Scholar]
- Kadioglu, A.; Taylor, S.; Iannelli, F.; Pozzi, G.; Mitchell, T.J.; Andrew, P.W. Upper and lower respiratory tract infection by Streptococcus pneumoniae is affected by pneumolysin deficiency and differences in capsule type. Infect. Immun. 2002, 70, 2886–2890. [Google Scholar] [CrossRef]
- Witzenrath, M.; Gutbier, B.; Owen, J.S.; Schmeck, B.; Mitchell, T.J.; Mayer, K.; Thomas, M.J.; Ishii, S.; Rosseau, S.; Suttorp, N.; et al. Role of platelet-activating factor in pneumolysin-induced acute lung injury. Crit. Care Med. 2007, 35, 1756–1762. [Google Scholar] [CrossRef]
- Romero, M.J.; Platt, D.; Tawfik, H.; Labazi, M.; El-Remessy, A.B.; Bartoli, M.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ. Res. 2008, 102, 95–102. [Google Scholar] [CrossRef]
- Hupp, S.; Förtsch, C.; Wippel, C.; Ma, J.; Mitchell, T.J.; Iliev, A.I. Direct transmembrane interaction between actin and the pore-competent, cholesterol-dependent cytolysin pneumolysin. J. Mol. Biol. 2013, 425, 636–646. [Google Scholar] [CrossRef]
- Lucas, R.; Yang, G.; Gorshkov, B.A.; Zemskov, E.A.; Sridhar, S.; Umapathy, N.S.; Jezierska-Drutel, A.; Alieva, I.B.; Leustik, M.; Hossain, H.; et al. Protein kinase C-α and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am. J. Respir. Cell Mol. Biol. 2012, 47, 445–453. [Google Scholar] [CrossRef]
- Rayner, C.F.; Jackson, A.D.; Rutman, A.; Dewar, A.; Mitchell, T.J.; Andrew, P.W.; Cole, P.J.; Wilson, R. Interaction of pneumolysin-sufficient and -deficient isogenic variants of Streptococcus pneumoniae with human respiratory mucosa. Infect. Immun. 1995, 63, 442–447. [Google Scholar]
- Steinfort, C.; Wilson, R.; Mitchell, T.J.; Feldman, C.; Rutman, A.; Todd, H.; Sykes, D.; Walker, J.; Saunders, K.; Andrew, P.W. Effect of Streptococcus pneumoniae on human respiratory epithelium in vitro. Infect. Immun. 1989, 57, 2006–2013. [Google Scholar]
- Feldman, C.; Anderson, R.; Cockeran, R.; Mitchell, T.J.; Cole, P.; Wilson, R. The effects of pneumolysin and hydrogen peroxide, alone and in combination, on human ciliated epithelium in vitro. Respir. Med. 2002, 96, 580–585. [Google Scholar] [CrossRef]
- Wray, C.; Mao, Y.; Pan, J.; Chandrasena, A.; Piasta, F.; Frank, J.A. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L219–L227. [Google Scholar] [CrossRef]
- Lucas, R.; Sridhar, S.; Rick, F.G.; Gorshkov, B.; Umapathy, N.S.; Yang, G.; Oseghale, A.; Verin, A.D.; Chakraborty, T.; Matthay, M.A.; et al. Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc. Natl. Acad. Sci. USA 2012, 109, 2084–2089. [Google Scholar] [CrossRef]
- Ware, L.B.; Matthay, M.A. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 163, 1376–1383. [Google Scholar] [CrossRef]
- Schally, A.V.; Kuroshima, A.; Ishida, Y.; Arimura, A.; Saito, T.; Bowers, C.Y.; Steelman, S.L. Purification of growthhormone-releasing factor from beef hypothalamus. Proc. Soc. Exp. Biol. Med. 1966, 122, 821–823. [Google Scholar] [CrossRef]
- Vance, M.L. Growth hormone releasing hormone. Clin. Chem. 1990, 36, 415–420. [Google Scholar]
- Rekasi, Z.; Czompoly, T.; Schally, A.V.; Halmos, G. Isolation and sequencing of cDNAs for splice variants of growth hormone-releasing hormone receptors from human cancers. Proc. Natl. Acad. Sci. USA 2000, 97, 10561–10566. [Google Scholar]
- Izdebski, J.; Pinski, J.; Horvath, J.E.; Halmos, G.; Groot, K.; Schally, A.V. Synthesis and biological evaluation of superactive agonists of growth hormone-releasing hormone. Proc. Natl. Acad. Sci. USA 1995, 92, 4872–4826. [Google Scholar] [CrossRef]
- Mayo, K.E.; Godfrey, P.A.; Suhr, S.T.; Kulik, D.J.; Rahal, J.O. Growth hormone-releasing hormone: Synthesis and signaling. Recent Prog. Horm. Res. 1995, 50, 35–73. [Google Scholar]
- Sayner, S.L. Emerging themes of cAMP regulation of the pulmonary endothelial barrier. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L667–L678. [Google Scholar] [CrossRef]
- Lawrence, D.W.; Comerford, K.M.; Colgan, S.P. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am. J. Physiol. Cell Physiol. 2002, 282, C1235–C1245. [Google Scholar] [CrossRef]
- Fukuhara, S.; Sakurai, A.; Sano, H.; Yamagishi, A.; Somekawa, S.; Takakura, N.; Saito, Y.; Kangawa, K.; Mochizuki, N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol. Cell. Biol. 2005, 25, 136–146. [Google Scholar] [CrossRef]
- Qiao, J.; Holian, O.; Lee, B.S.; Huang, F.; Zhang, J.; Lum, H. Phosphorylation of GTP dissociation inhibitor by PKA negatively regulates RhoA. Am. J. Physiol. Cell Physiol. 2008, 295, C1161–C1168. [Google Scholar] [CrossRef]
- Fang, X.; Song, Y.; Zemans, R.; Hirsch, J.; Matthay, M.A. Fluid transport across cultured rat alveolar epithelial cells: A novel in vitro system. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 87, L104–L110. [Google Scholar]
- Lazrak, A.; Matalon, S. cAMP-induced changes of apical membrane potentials of confluent H441 monolayers. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L443–L450. [Google Scholar]
- Mustafa, S.B.; Castro, R.; Falck, A.J.; Petershack, J.A.; Henson, B.M.; Mendoza, Y.M.; Choudary, A.; Seidner, S.R. Protein kinase and mitogen-activated protein kinase pathways mediate cAMP induction of alpha epithelial Na+ channels (alpha-ENaC). J. Cell Physiol. 2008, 215, 101–110. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, Y.W., Zhang; Xu, S.Q.; Liu, H.L.; Yang, W.Y.; Lou, J.N. Differential activations of PKC/PKA related to microvasculopathy in diabetic GK rats. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E173–E182. [Google Scholar]
- Schally, A.V.; Varga, J.L.; Engel, J.B. Antagonists of growth-hormone-releasing hormone: An emerging new therapy for cancer. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 33–43. [Google Scholar] [CrossRef]
- Lucas, R.; Magez, S.; de Leys, R.; Fransen, L.; Scheerlinck, J.P.; Rampelberg, M.; Sablon, E.; de Baetselier, P. Mapping the lectin-like activity of tumor necrosis factor. Science 1994, 263, 814–817. [Google Scholar]
- Hundsberger, H.; Verin, A.; Wiesner, C.; Pflueger, M.; Dulebo, A.; Schuett, W.; Maennel, D.; Wendel, A.; Lucas, R. TNF: A moonlighting protein at the interface between cancer and infection. Front. Biosc. 2008, 13, 5374–5386. [Google Scholar]
- Hession, C.; Decker, J.M.; Sherblom, A.P.; Kumar, S.; Yue, C.C.; Mattaliano, R.J.; Tizard, R.; Kawashima, E.; Schmeissner, U.; Heletky, S.; et al. Uromodulin (Tamm-Horsfall glycoprotein): A renal ligand for lymphokines. Science 1987, 237, 1479–1484. [Google Scholar]
- Magez, S.; Geuskens, M.; Beschin, A.; del Favero, H.; Verschueren, H.; Lucas, R.; Pays, E.; de Baetselier, P. Specific uptake of tumor necrosis factor-alpha is involved in growth control of Trypanosoma brucei. J. Cell. Biol. 1997, 137, 715–727. [Google Scholar] [CrossRef]
- Fukuda, N.; Jayr, C.; Lazrak, A.; Wang, Y.; Lucas, R.; Matalon, S.; Matthay, M.A. Mechanisms of TNF-stimulation of amiloride-sensitive sodium transport across the alveolar epithelium in vivo and epithelial cells in vitro. Am. J. Physiol. Lung Cell Physiol. 2001, 280, L1258–L1265. [Google Scholar]
- Hribar, M.; Bloc, A.; van der Goot, F.G.; Fransen, L.; de Baetselier, P.; Grau, G.E.; Bluethmann, H.; Matthay, M.A.; Dunant, Y.; Pugin, J.; et al. The lectin-like domain of tumor necrosis factor-α increases membrane conductance in microvascular endothelial cells and peritoneal macrophages. Eur. J. Immunol. 1999, 29, 3105–3111. [Google Scholar] [CrossRef]
- Elia, N.; Tapponnier, M.; Matthay, M.A.; Hamacher, J.; Pache, J.C.; Brundler, M.A.; Tötsch, M.; de Baetselier, P.; Fransen, L.; Fukuda, N.; et al. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am. J. Respir. Crit. Care Med. 2003, 168, 1043–1050. [Google Scholar] [CrossRef]
- Braun, C.; Hamacher, J.; Morel, D.; Wendel, A.; Lucas, R. Dichotomal role of TNF in experimental pulmonary edema reabsorption. J. Immunol. 2005, 75, 3402–3408. [Google Scholar]
- Dagenais, A.; Fréchette, R.; Yamagata, Y.; Yamagata, T.; Carmel, J.F.; Clermont, M.E.; Brochiero, E.; Massé, C.; Berthiaume, Y. Downregulation of ENac activity and expression by TNF-alpha in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L301–L311. [Google Scholar] [CrossRef]
- Yang, G.; Hamacher, J.; Gorshkov, B.; White, R.; Sridhar, S.; Verin, A.; Chakraborty, T.; Lucas, R. The dual role of TNF in pulmonary edema. J. Cardiovasc. Dis. Res. 2010, 1, 29–36. [Google Scholar]
- Rezaiguia, S.; Garat, C.; Delclaux, C.; Meignan, M.; Fleury, J.; Legrand, P.; Matthay, M.A.; Jayr, C. Acute bacterial pneumonia in rats increases alveolar epithelial fluid claearance by a tomor necrosis factor-alpha-dependent mechanism. J. Clin. Invest. 1997, 99, 325–335. [Google Scholar] [CrossRef]
- Wilson, M.R.; Goddard, M.E.; O’Dea, K.P.; Choudhury, S.; Takata, M. Differential roles of p55 and p75 tumor necrosis factor receptors on stretch-induced pulmonary edema in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L60–L68. [Google Scholar] [CrossRef]
- Vadász, I.; Schermuly, R.T.; Ghofrani, H.A.; Rummel, S.; Wehner, S.; Mühldorfer, I.; Schäfer, K.P.; Seeger, W.; Morty, R.E.; Grimminger, F. The lectin-like domain of tumor necrosis factor-alpha improves alveolar fluid balance in injured isolated rabbit lungs. Crit Care Med. 2008, 36, 1543–1550. [Google Scholar] [CrossRef]
- Hamacher, J.; Stammberger, U.; Roux, J.; Kumar, S.; Yang, G.; Xiong, C.; Schmid, R.A.; Fakin, R.M.; Chakraborty, T.; Hossain, H.M.D.; et al. The lectin-like domain of tumor necrosis factor improves lung function after rat lung transplantation—Potential role for a reduction in reactive oxygen species generation. Crit. Care Med. 2010, 38, 871–878. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lucas, R.; Czikora, I.; Sridhar, S.; Zemskov, E.; Gorshkov, B.; Siddaramappa, U.; Oseghale, A.; Lawson, J.; Verin, A.; Rick, F.G.; et al. Mini-Review: Novel Therapeutic Strategies to Blunt Actions of Pneumolysin in the Lungs. Toxins 2013, 5, 1244-1260. https://doi.org/10.3390/toxins5071244
Lucas R, Czikora I, Sridhar S, Zemskov E, Gorshkov B, Siddaramappa U, Oseghale A, Lawson J, Verin A, Rick FG, et al. Mini-Review: Novel Therapeutic Strategies to Blunt Actions of Pneumolysin in the Lungs. Toxins. 2013; 5(7):1244-1260. https://doi.org/10.3390/toxins5071244
Chicago/Turabian StyleLucas, Rudolf, Istvan Czikora, Supriya Sridhar, Evgeny Zemskov, Boris Gorshkov, Umapathy Siddaramappa, Aluya Oseghale, Jonathan Lawson, Alexander Verin, Ferenc G. Rick, and et al. 2013. "Mini-Review: Novel Therapeutic Strategies to Blunt Actions of Pneumolysin in the Lungs" Toxins 5, no. 7: 1244-1260. https://doi.org/10.3390/toxins5071244