Shiga Toxins and the Pathophysiology of Hemolytic Uremic Syndrome in Humans and Animals

Abstract
:1. Introduction
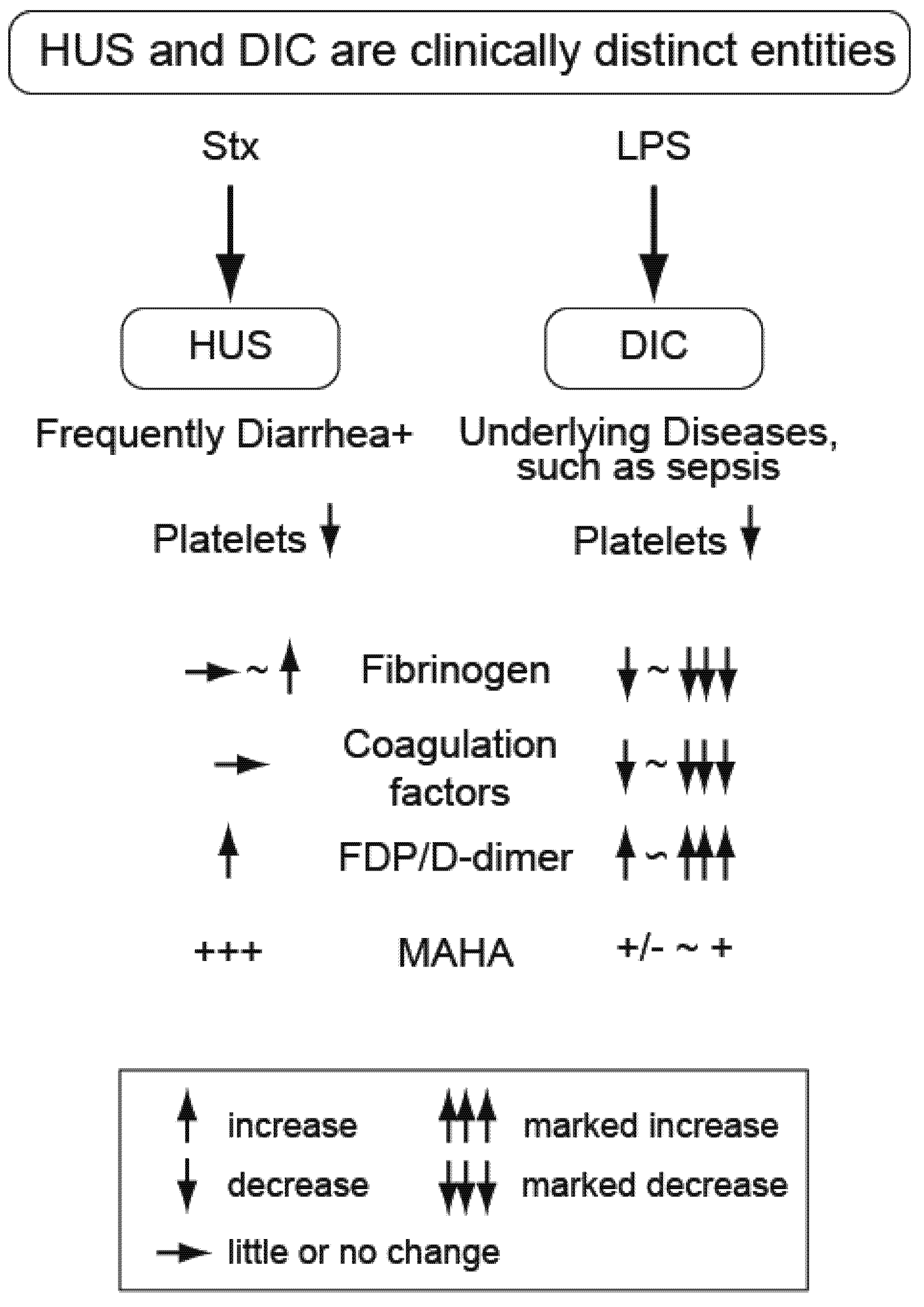
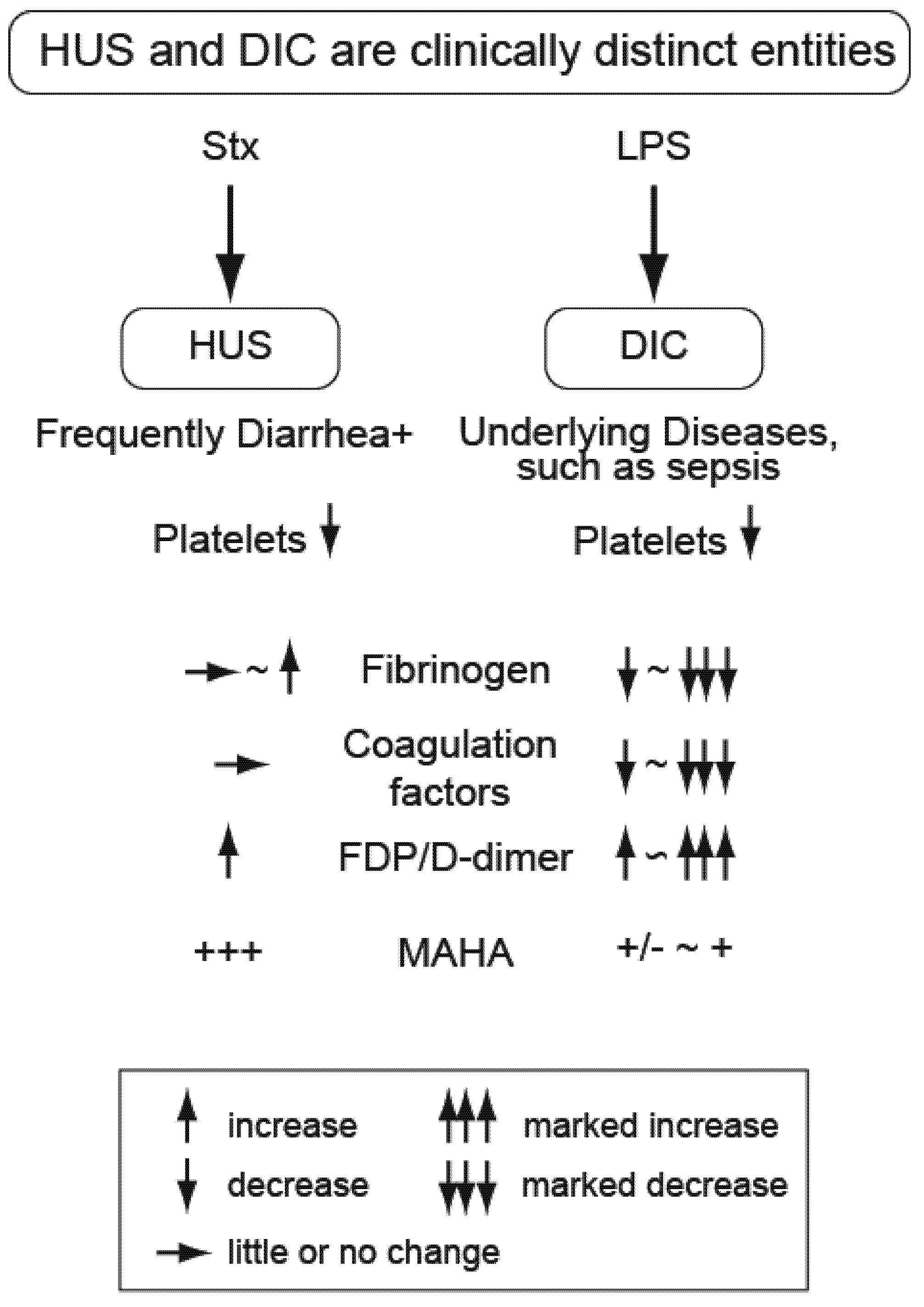
2. Distinguishing the Thrombotic Microangiopathy of EHEC-Related HUS
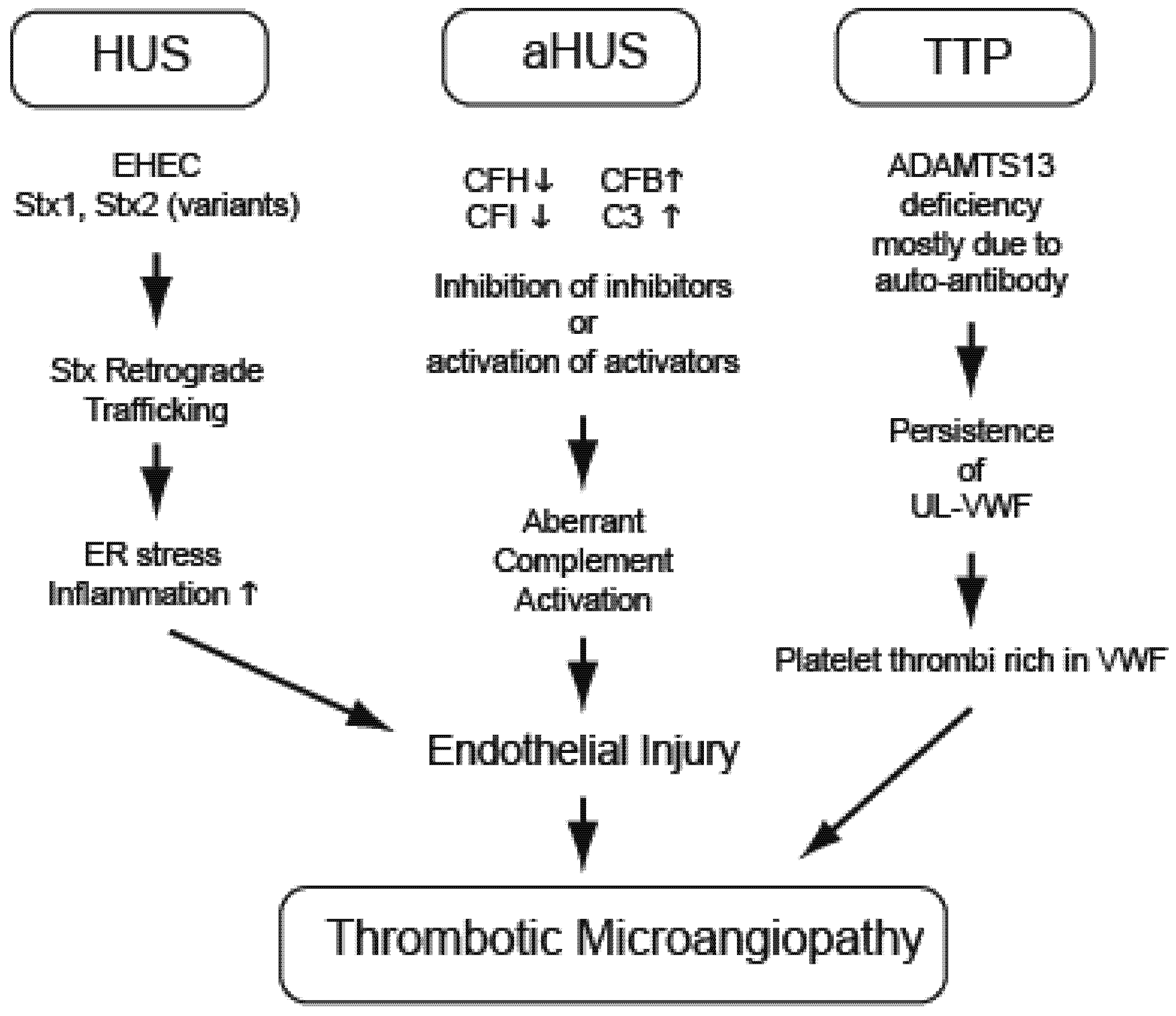
3. Hemolytic Uremic Syndrome (EHEC and Stx)
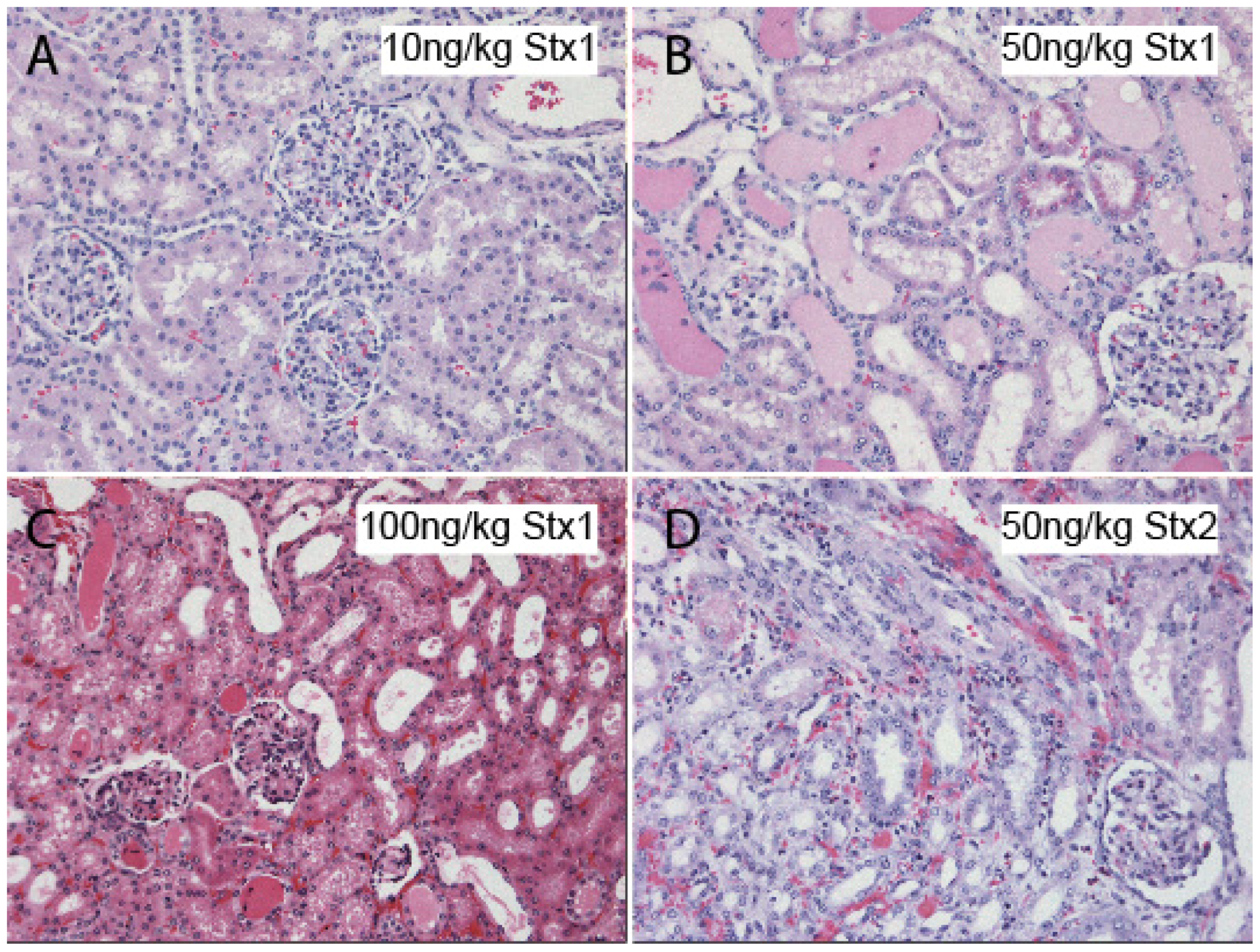
4. Atypical Hemolytic Uremic Syndrome (Non-EHEC)
5. Thrombotic Thrombocytopenic Purpura
6. Animal Models

{kind=link}
{kind=link}
{kind=link}
| Model | Pre-treatment | Animal | Features | Limitations | Reference |
|---|---|---|---|---|---|
| EHEC | none | Gnotobiotic piglet | Focal renal lesions, renal thrombotic microangiopathy | Normal serum creatinine; no thrombocytopenia | [103] |
| EHEC | streptomycin | CD-1 mice | Bacterial colonization; loose stools, anorexia, lethargy | No disease unless host-adapted strain used; no glomerular damage, coagulopathy or thrombocytopenia | [104,105] |
| EHEC | streptomycin and mitomycin C | ICR mice | Weakness, weight loss, microhemorrhages in brain and spinal cord, high BUN | Serum creatinine normal, unremarkable kidney pathology | [106] |
| EHEC | None or TNFα | Germ free IQI mice | Anorexia, renal tubular necrosis, thrombocytopenia, leukocytosis | No glomerular histopathology, inflammation only with TNFα pre-treatment | [107] |
| EHEC | 24 hour fast | C3H/HeJ mice | Gastrointestinal, neurologic and systemic symptoms, renal inflammation and necrosis, fragmented red blood cells | Kidney function and platelets not measure | [108] |
| EHEC | none | Newly weaned BALB/c mice | Renal damage, high urea concentrations, colon pathology | Thrombotic microangiopathy not evaluated | [109] |
| EHEC | Protein calorie malnutrition | C57Bl/6J mice | Systemic and neurologic symptoms, increased BUN, mild renal tubular degeneration | Normal serum creatinine, normal glomeruli, no significant platelet changes | [110] |
| EHEC | Host-adapted bacteria | Dutch Belted rabbits | Diarrhea, lethargy, anorexia, dehydration glomerular thrombi and renal congestion | No consistent thrombocytopenia | [111] |
| Stx1 | none | baboon | Thrombocytopenia, schistocytosis and hemolytic anemia, renal failure, GI injury, lesions in CNS, systemic inflammation | Non-bacterial | [42,48] |
| Stx2 | none | baboon | Thrombocytopenia, leukocytosis, acute renal failure, schistocytosis, hemolytic uremia, glomerular thrombotic microangiopathy, systemic inflammation | Non-bacterial | [42,112] |
| Stx2 | none | C57Bl/6J mice | Increased plasma BUN and creatinine, hemolysis, neutrophilia | Non-bacterial, no thrombocytopenia | [113] |
| Stx2+LPS | none | C57Bl/6J mice | Neutrophilia, thrombocytopenia, hemolysis, increased BUN and creatinine, renal histopathology | Non-bacterial, LPS effect depends on timing of administration, consumptive coagulopathy | [29] |
| EHEC culture supernatant | none | Sprague-Dawley rats | Increased BUN and creatinine, thrombocytopenia, hemolytic anemia and leukocytosis, renal histologic changes, watery diarrhea | Non-bacterial, crude bacterial supernatants rather than purified toxin(s). | [114] |
7. Gnotobiotic Piglet Models
8. Murine EHEC Models
9. Rabbit EHEC Model
10. Nonhuman Primate Toxin Models of HUS
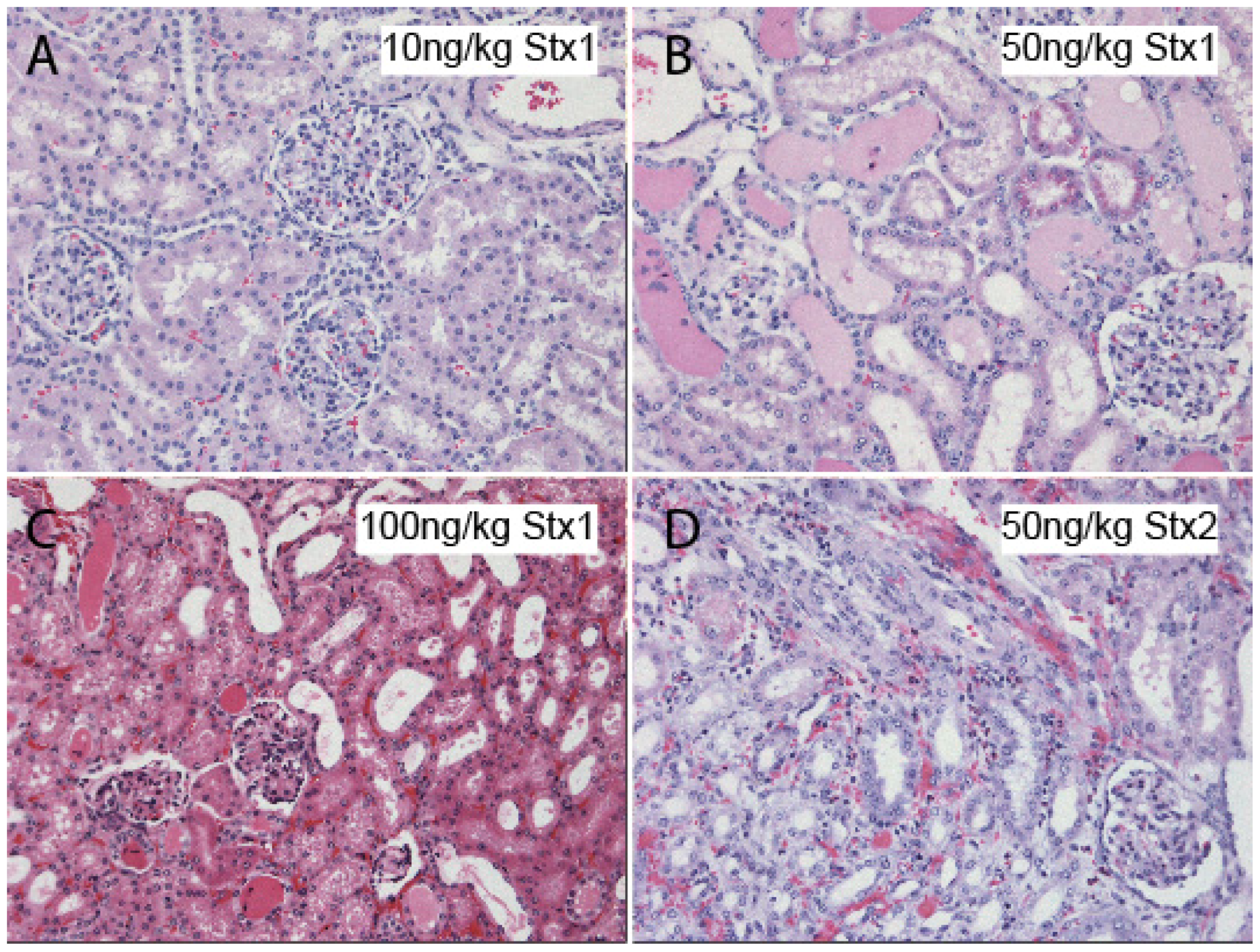
11. Rodent Models of Stx Challenge
12. Conclusions
Acknowledgements
Conflicts of Interest
References
- Riley, L.W.; Remis, R.S.; Helgerson, S.D.; McGee, H.B.; Wells, J.G.; Davis, B.R.; Hebert, R.J.; Olcott, E.S.; Johnson, L.M.; Hargrett, N.T.; et al. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 1983, 308, 681–685. [Google Scholar] [CrossRef]
- Bell, B.P.; Goldoft, M.; Griffin, P.M.; Davis, M.A.; Gordon, D.C.; Tarr, P.I.; Bartleson, C.A.; Lewis, J.H.; Barrett, T.J.; Wells, J.G.; et al. A multistate outbreak of Escherichia coli O157: H7-associated bloody diarrhea and hemolytic uremic syndrome from hamburgers. The Washington experience. J. Am. Med. Assoc. 1994, 272, 1349–1353. [Google Scholar]
- Rangel, J.M.; Sparling, P.H.; Crowe, C.; Griffin, P.M.; Swerdlow, D.L. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982-2002. Emerg. Infect. Dis. 2005, 11, 603–609. [Google Scholar] [CrossRef]
- Schmidt, H.; Geitz, C.; Tarr, P.I.; Frosch, M.; Karch, H. Non-O157:H7 pathogenic Shiga toxin-producing Escherichia coli: Phenotypic and genetic profiling of virulence traits and evidence for clonality. J. Infect. Dis. 1999, 179, 115–123. [Google Scholar] [CrossRef]
- Beutin, L.; Zimmermann, S.; Gleier, K. Human infections with Shiga toxin-producing Escherichia coli other than serogroup O157 in germany. Emerg. Infect. Dis. 1998, 4, 635–639. [Google Scholar] [CrossRef]
- Savage, P.J.; Campellone, K.G.; Leong, J.M. Interaction of enterohemorrhagic Escherichia coli (EHEC) with mammalian cells: Cell adhesion, type iii secretion, and actin pedestal formation. Curr. Protoc. Microbiol. 2007. [Google Scholar] [CrossRef]
- Farfan, M.J.; Torres, A.G. Molecular mechanisms that mediate colonization of Shiga toxin-producing Escherichia coli strains. Infect. Immun. 2012, 80, 903–913. [Google Scholar] [CrossRef]
- Melton-Celsa, A.; Mohawk, K.; Teel, L.; O’Brien, A. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr. Top Microbiol. Immunol. 2012, 357, 67–103. [Google Scholar]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar]
- Gould, L.H.; Demma, L.; Jones, T.F.; Hurd, S.; Vugia, D.J.; Smith, K.; Shiferaw, B.; Segler, S.; Palmer, A.; Zansky, S.; et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin. Infect. Dis. 2009, 49, 1480–1485. [Google Scholar] [CrossRef]
- Fukushima, H.; Hashizume, T.; Morita, Y.; Tanaka, J.; Azuma, K.; Mizumoto, Y.; Kaneno, M.; Matsuura, M.; Konma, K.; Kitani, T. Clinical experiences in Sakai City hospital during the massive outbreak of enterohemorrhagic Escherichia coli O157 infections in Sakai City, 1996. Pediatr. Int. 1999, 41, 213–217. [Google Scholar]
- Frank, C.; Faber, M.; Askar, M.; Bernard, H.; Fruth, A.; Gilsdorf, A.; Hohle, M.; Karch, H.; Krause, G.; Prager, R.; et al. Large and ongoing outbreak of haemolytic uraemic syndrome, Germany, May 2011. Euro. Surveill. 2011, 16, 1–3. [Google Scholar]
- Pan, D.; Das, A.; Liu, D.; Veazey, R.S.; Pahar, B. Isolation and characterization of intestinal epithelial cells from normal and SIV-infected rhesus macaques. PLoS One 2012, 7, e30247. [Google Scholar]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Köck, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Wscherichia coli O104:H4 outbreak in Germany-Preliminary report. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Steele, B.T. Escherichia coli cytotoxin, haemolytic-uraemic syndrome, and haemorrhagic colitis. Lancet 1983, 2, 1299–1300. [Google Scholar]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar] [CrossRef]
- Wong, C.S.; Mooney, J.C.; Brandt, J.R.; Staples, A.O.; Jelacic, S.; Boster, D.R.; Watkins, S.L.; Tarr, P.I. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: A multivariable analysis. Clin. Infect. Dis. 2012, 55, 33–41. [Google Scholar] [CrossRef]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by Shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef]
- Buchholz, U.; Bernard, H.; Werber, D.; Bohmer, M.M.; Remschmidt, C.; Wilking, H.; Deleré, Y.; an der Heiden, M.; Adlhoch, C.; Dreesman, J.; et al. German outbreak of Escherichia coli O104:H4 associated with sprouts. N. Engl. J. Med. 2011, 365, 1763–1770. [Google Scholar]
- Menne, J.; Nitschke, M.; Stingele, R.; Abu-Tair, M.; Beneke, J.; Bramstedt, J.; Bremer, J.P.; Brunkhorst, R.; Busch, V.; Dengler, R.; et al. Validation of treatment strategies for enterohaemorrhagic Escherichia coli O104:H4 induced haemolytic uraemic syndrome: Case-control study. Br. Med. J. 2012, 345, e4565. [Google Scholar] [CrossRef]
- Corogeanu, D.; Willmes, R.; Wolke, M.; Plum, G.; Utermohlen, O.; Kronke, M. Therapeutic concentrations of antibiotics inhibit Shiga toxin release from enterohemorrhagic E. coli O104:H4 from the 2011 German outbreak. BMC Microbiol. 2012, 12, 160. [Google Scholar] [CrossRef]
- Rivero, M.A.; Passucci, J.A.; Rodriguez, E.M.; Signorini, M.L.; Tarabla, H.D.; Parma, A.E. Factors associated with sporadic verotoxigenic Escherichia coli infection in children with diarrhea from the central eastern area of Argentina. Foodborne Pathog. Dis. 2011, 8, 901–906. [Google Scholar] [CrossRef]
- Garg, A.X.; Suri, R.S.; Barrowman, N.; Rehman, F.; Matsell, D.; Rosas-Arellano, M.P.; Salvadori, M.; Haynes, R.B.; Clark, W.F. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: A systematic review, meta-analysis, and meta-regression. JAMA 2003, 290, 1360–1370. [Google Scholar] [CrossRef]
- Oakes, R.S.; Kirkham, J.K.; Nelson, R.D.; Siegler, R.L. Duration of oliguria and anuria as predictors of chronic renal-related sequelae in post-diarrheal hemolytic uremic syndrome. Pediatr. Nephrol. 2008, 23, 1303–1308. [Google Scholar] [CrossRef]
- Obrig, T.G.; Louise, C.B.; Lingwood, C.A.; Boyd, B.; Barley-Maloney, L.; Daniel, T.O. Endothelial heterogeneity in Shiga toxin receptors and responses. J. Biol. Chem. 1993, 268, 15484–15488. [Google Scholar]
- Zoja, C.; Angioletti, S.; Donadelli, R.; Zanchi, C.; Tomasoni, S.; Binda, E.; Imberti, B.; te Loo, M.; Monnens, L.; Remuzzi, G.; et al. Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via NF-κb dependent up-regulation of IL-8 and MCP-1. Kidney Int. 2002, 62, 846–856. [Google Scholar] [CrossRef]
- Reitsma, P.H.; Versteeg, H.H.; Middeldorp, S. Mechanistic view of risk factors for venous thromboembolism. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 563–568. [Google Scholar] [CrossRef]
- Keepers, T.R.; Psotka, M.A.; Gross, L.K.; Obrig, T.G. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J. Am. Soc. Nephrol. 2006, 17, 3404–3414. [Google Scholar] [CrossRef]
- Camerer, E.; Cornelissen, I.; Kataoka, H.; Duong, D.N.; Zheng, Y.-W.; Coughlin, S.R. Roles of protease-activated receptors in a mouse model of endotoxemia. Blood 2006, 107, 3912–3921. [Google Scholar] [CrossRef]
- Constantinescu, A.R.; Bitzan, M.; Weiss, L.S.; Christen, E.; Kaplan, B.S.; Cnaan, A.; Trachtman, H. Non-enteropathic hemolytic uremic syndrome: Causes and short-term course. Am. J. Kidney Dis. 2004, 43, 976–982. [Google Scholar] [CrossRef]
- Banerjee, R.; Hersh, A.L.; Newland, J.; Beekmann, S.E.; Polgreen, P.M.; Bender, J.; Shaw, J.; Copelovitch, L.; Kaplan, B.S.; Shah, S.S.; et al. Streptococcus pneumoniae-associated hemolytic uremic syndrome among children in North America. Pediatr. Infect. Dis. J. 2011, 30, 736–739. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2005, 16, 1035–1050. [Google Scholar] [CrossRef]
- Obrig, T.G. Escherichia coli Shiga toxin mechanisms of action in renal disease. Toxins 2010, 2, 2769–2794. [Google Scholar] [CrossRef]
- Karch, H.; Tarr, P.I.; Bielaszewska, M. Enterohaemorrhagic Escherichia coli in human medicine. Int. J. Med. Microbiol. 2005, 295, 405–418. [Google Scholar] [CrossRef]
- Paton, J.C.; Paton, A.W. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 1998, 11, 450–479. [Google Scholar]
- O’Brien, A.O.; Lively, T.A.; Chen, M.E.; Rothman, S.W.; Formal, S.B. Escherichia coli O157:H7 strains associated with haemorrhagic colitis in the United States produce a shigella dysenteriae 1 (Shiga) like cytotoxin. Lancet 1983, 1, 702. [Google Scholar]
- Okuda, T.; Tokuda, N.; Numata, S.; Ito, M.; Ohta, M.; Kawamura, K.; Wiels, J.; Urano, T.; Tajima, O.; Furukawa, K.; et al. Targeted disruption of Gb3/CD77 synthase gene resulted in the complete deletion of globo-series glycosphingolipids and loss of sensitivity to verotoxins. J. Biol. Chem. 2006, 281, 10230–10235. [Google Scholar]
- Saxena, S.; O’Brien, A.; Ackerman, E. Shiga toxin, Shiga-like toxin II variant, and ricin are all single-site RNA N-glycosidases of 28S RNA when microinjected into Xenopus oocytes. J. Biol. Chem. 1989, 264, 596–601. [Google Scholar]
- Mukhopadhyay, S.; Linstedt, A.D. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 2012, 335, 332–335. [Google Scholar] [CrossRef]
- Proulx, F.; Turgeon, J.P.; Litalien, C.; Mariscalco, M.M.; Robitaille, P.; Seidman, E. Inflammatory mediators in Escherichia coli O157:H7 hemorrhagic colitis and hemolytic-uremic syndrome. Pediatr. Infect. Dis. J. 1998, 17, 899–904. [Google Scholar] [CrossRef]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Tesh, V.L.; Kurosawa, S. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect. Immun. 2010, 78, 2497–2504. [Google Scholar] [CrossRef]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.K.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef]
- Karpman, D.; Håkansson, A.; Perez, M.T.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of renal cortical cells in the hemolytic-uremic syndrome: In vivo and in vitro studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar]
- Inward, C.D.; Howie, A.J.; Fitzpatrick, M.M.; Rafaat, F.; Milford, D.V.; Taylor, C.M. Renal histopathology in fatal cases of diarrhoea-associated haemolytic uraemic syndrome. Pediatr. Nephrol 1997, 11, 556–559. [Google Scholar] [CrossRef]
- Chaisri, U.; Nagata, M.; Kurazono, H.; Horie, H.; Tongtawe, P.; Hayashi, H.; Watanabe, T.; Tapchaisri, P.; Chongsa-nguan, M.; Chaicumpa, W. Localization of Shiga toxins of enterohaemorrhagic Escherichia coli in kidneys of paediatric and geriatric patients with fatal haemolytic uraemic syndrome. Microb. Pathog. 2001, 31, 59–67. [Google Scholar] [CrossRef]
- Taylor, F.B., Jr.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the baboon responses to Shiga-like toxin: Descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar] [CrossRef]
- Stearns-Kurosawa, D.J.; Oh, S.-Y.; Cherla, R.P.; Lee, M.-S.; Tesh, V.L.; Papin, J.; Henderson, J.; Kurosawa, S. Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic E. coli Shiga toxins. 2012. submitted for publication. [Google Scholar]
- Morigi, M.; Micheletti, G.; Figliuzzi, M.; Imberti, B.; Karmali, M.A.; Remuzzi, A.; Remuzzi, G.; Zoja, C. Verotoxin-1 promotes leukocyte adhesion to cultured endothelial cells under physiologic flow conditions. Blood 1995, 86, 4553–4558. [Google Scholar]
- Van Setten, P.A.; Monnens, L.A.; Verstraten, R.G.; van den Heuvel, L.P.; van Hinsbergh, V.W. Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine release. Blood 1996, 88, 174–183. [Google Scholar]
- Richardson, S.E.; Karmali, M.A.; Becker, L.E.; Smith, C.R. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections. Hum. Pathol. 1988, 19, 1102–1108. [Google Scholar] [CrossRef]
- King, A.J. Acute inflammation in the pathogenesis of hemolytic-uremic syndrome. Kidney Int. 2002, 61, 1553–1564. [Google Scholar] [CrossRef]
- Thorpe, C.M.; Hurley, B.P.; Lincicome, L.L.; Jacewicz, M.S.; Keusch, G.T.; Acheson, D.W. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999, 67, 5985–5993. [Google Scholar]
- Tesh, V.L.; Ramegowda, B.; Samuel, J.E. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect. Immun. 1994, 62, 5085–5094. [Google Scholar]
- Thorpe, C.M.; Smith, W.E.; Hurley, B.P.; Acheson, D.W.K. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 2001, 69, 6140–6147. [Google Scholar] [CrossRef]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar] [CrossRef]
- Fernandez, G.C.; te Loo, M.W.; van der Velden, T.J.; van der Heuvel, L.P.; Palermo, M.S.; Monnens, L.L. Decrease of thrombomodulin contributes to the procoagulant state of endothelium in hemolytic uremic syndrome. Pediatr. Nephrol. 2003, 18, 1066–1068. [Google Scholar] [CrossRef]
- Weiler, H. Regulation of inflammation by the protein c system. Crit. Care Med. 2010, 38, S18–S25. [Google Scholar] [CrossRef]
- Esmon, C.T. The interactions between inflammation and coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef]
- Lin, S.M.; Wang, Y.M.; Lin, H.C.; Lee, K.Y.; Huang, C.D.; Liu, C.Y.; Wang, C.H.; Kuo, H.P. Serum thrombomodulin level relates to the clinical course of disseminated intravascular coagulation, multiorgan dysfunction syndrome, and mortality in patients with sepsis. Crit. Care Med. 2008, 36, 683–689. [Google Scholar] [CrossRef]
- Kurosawa, S.; Stearns-Kurosawa, D.J.; Kinasewitz, G.T. Soluble thrombomodulin: A sign of bad times. Crit. Care Med. 2008, 36, 985–987. [Google Scholar]
- Karpman, D.; Manea, M.; Vaziri-Sani, F.; Stahl, A.L.; Kristoffersson, A.C. Platelet activation in hemolytic uremic syndrome. Semin. Thromb. Hemost. 2006, 32, 128–145. [Google Scholar] [CrossRef]
- Morigi, M.; Galbusera, M.; Binda, E.; Imberti, B.; Gastoldi, S.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Verotoxin-1-induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood 2001, 98, 1828–1835. [Google Scholar] [CrossRef] [Green Version]
- Nolasco, L.H.; Turner, N.A.; Bernardo, A.; Tao, Z.; Cleary, T.G.; Dong, J.; Moake, J.L. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von willebrand factor multimers. Blood 2005, 106, 4199–4209. [Google Scholar] [CrossRef]
- Walters, M.D.; Matthei, I.U.; Kay, R.; Dillon, M.J.; Barratt, T.M. The polymorphonuclear leucocyte count in childhood haemolytic uraemic syndrome. Pediatr. Nephrol. 1989, 3, 130–134. [Google Scholar] [CrossRef]
- Te Loo, D.M.; Monnens, L.A.; der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; van den Heuvel, L.P.; van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar]
- Brigotti, M. The interactions of human neutrophils with Shiga toxins and related plant toxins: Danger or safety? Toxins 2012, 4, 157–190. [Google Scholar] [CrossRef]
- Tazzari, P.L.; Ricci, F.; Carnicelli, D.; Caprioli, A.; Tozzi, A.E.; Rizzoni, G.; Conte, R.; Brigotti, M. Flow cytometry detection of Shiga toxins in the blood from children with hemolytic uremic syndrome. Cytometry B Clin. Cytom. 2004, 61, 40–44. [Google Scholar]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of Shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar]
- Geelen, J.M.; van der Velden, T.J.A.M.; te Loo, D.M.W.M.; Boerman, O.C.; van den Heuvel, L.P.W.J.; Monnens, L.A.H. Lack of specific binding of Shiga-like toxin (verocytotoxin) and non-specific interaction of Shiga-like toxin 2 antibody with human polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2007, 22, 749–755. [Google Scholar] [CrossRef]
- Abe, H.; Okajima, K.; Okabe, H.; Takatsuki, K.; Binder, B.R. Granulocyte proteases and hydrogen peroxide synergistically inactivate thrombomodulin of endothelial cells in vitro. J. Lab. Clin. Med. 1994, 123, 874–881. [Google Scholar]
- Zoja, C.; Locatelli, M.; Pagani, C.; Corna, D.; Zanchi, C.; Isermann, B.; Remuzzi, G.; Conway, E.M.; Noris, M. Lack of the lectin-like domain of thrombomodulin worsens Shiga toxin-associated hemolytic uremic syndrome in mice. J. Immunol. 2012.
- Magnus, T.; Rother, J.; Simova, O.; Meier-Cillien, M.; Repenthin, J.; Moller, F.; Gbadamosi, J.; Panzer, U.; Wengenroth, M.; Hagel, C.; et al. The neurological syndrome in adults during the 2011 northern German E. coli serotype O104:H4 outbreak. Brain 2012, 135, 1850–1859. [Google Scholar] [CrossRef]
- Landoni, V.I.; Schierloh, P.; de Campos Nebel, M.; Fernandez, G.C.; Calatayud, C.; Lapponi, M.J.; Isturiz, M.A. Shiga toxin 1 induces on lipopolysaccharide-treated astrocytes the release of tumor necrosis factor-alpha that alter brain-like endothelium integrity. PLoS Pathog. 2012, 8, e1002632. [Google Scholar] [CrossRef]
- Greinacher, A.; Friesecke, S.; Abel, P.; Dressel, A.; Stracke, S.; Fiene, M.; Ernst, F.; Selleng, K.; Weissenborn, K.; Schmidt, B.M.; et al. Treatment of severe neurological deficits with igg depletion through immunoadsorption in patients with Escherichia coli O104:H4-associated haemolytic uraemic syndrome: A prospective trial. Lancet 2011, 378, 1166–1173. [Google Scholar]
- Huang, J.; Motto, D.G.; Bundle, D.R.; Sadler, J.E. Shiga toxin B subunits induce vWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood 2010, 116, 3653–3659. [Google Scholar] [CrossRef]
- Zhu, Y.; Thangamani, S.; Ho, B.; Ding, J.L. The ancient origin of the complement system. EMBO J. 2005, 24, 382–394. [Google Scholar] [CrossRef]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef]
- Stahl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef]
- Orth, D.; Khan, A.B.; Naim, A.; Grif, K.; Brockmeyer, J.; Karch, H.; Joannidis, M.; Clark, S.J.; Day, A.J.; Fidanzi, S.; et al. Shiga toxin activates complement and binds factor H: Evidence for an active role of complement in hemolytic uremic syndrome. J. Immunol. 2009, 182, 6394–6400. [Google Scholar] [CrossRef]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef]
- Thurman, J.M.; Marians, R.; Emlen, W.; Wood, S.; Smith, C.; Akana, H.; Holers, V.M.; Lesser, M.; Kline, M.; Hoffman, C.; et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. 2009, 4, 1920–1924. [Google Scholar] [CrossRef]
- Lapeyraque, A.L.; Malina, M.; Fremeaux-Bacchi, V.; Boppel, T.; Kirschfink, M.; Oualha, M.; Proulx, F.; Clermont, M.J.; Le Deist, F.; Niaudet, P.; et al. Eculizumab in severe Shiga-toxin-associated HUS. N. Engl. J. Med. 2011, 364, 2561–2563. [Google Scholar]
- Nurnberger, J.; Philipp, T.; Witzke, O.; Opazo Saez, A.; Vester, U.; Baba, H.A.; Kribben, A.; Zimmerhackl, L.B.; Janecke, A.R.; Nagel, M.; et al. Eculizumab for atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 360, 542–544. [Google Scholar] [CrossRef]
- Artunc, F. Treating Shiga toxin induced haemolytic uraemic syndrome. Br. J. Haematol. 2012, 345, e4598. [Google Scholar]
- Ruggenenti, P.; Remuzzi, G. Thrombotic microangiopathy: E. coli O104:H4 German outbreak: A missed opportunity. Nat. Rev. Nephrol. 2012, 8, 558–560. [Google Scholar] [CrossRef]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. 2010, 5, 1844–1859. [Google Scholar] [CrossRef]
- Kavanagh, D.; Goodship, T. Genetics and complement in atypical HUS. Pediatr. Nephrol. 2010, 25, 2431–2442. [Google Scholar] [CrossRef]
- Caprioli, J.; Noris, M.; Brioschi, S.; Pianetti, G.; Castelletti, F.; Bettinaglio, P.; Mele, C.; Bresin, E.; Cassis, L.; Gamba, S.; et al. Genetics of HUS: The impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006, 108, 1267–1279. [Google Scholar]
- Loirat, C.; Fremeaux-Bacchi, V. Atypical hemolytic uremic syndrome. Orphanet J. Rare Dis. 2011, 6, 60. [Google Scholar] [CrossRef]
- Gruppo, R.A.; Rother, R.P. Eculizumab for congenital atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 360, 544–546. [Google Scholar]
- Delvaeye, M.; Noris, M.; de Vriese, A.; Esmon, C.T.; Esmon, N.L.; Ferrell, G.; Del-Favero, J.; Plaisance, S.; Claes, B.; Lambrechts, D.; et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 361, 345–357. [Google Scholar]
- Warwicker, P.; Goodship, T.H.; Donne, R.L.; Pirson, Y.; Nicholls, A.; Ward, R.M.; Turnpenny, P.; Goodship, J.A. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998, 53, 836–844. [Google Scholar] [CrossRef]
- Richards, A.; Kathryn Liszewski, M.; Kavanagh, D.; Fang, C.J.; Moulton, E.; Fremeaux-Bacchi, V.; Remuzzi, G.; Noris, M.; Goodship, T.H.; Atkinson, J.P. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol. Immunol. 2007, 44, 111–122. [Google Scholar] [CrossRef]
- Goicoechea de Jorge, E.; Harris, C.L.; Esparza-Gordillo, J.; Carreras, L.; Arranz, E.A.; Garrido, C.A.; Lopez-Trascasa, M.; Sanchez-Corral, P.; Morgan, B.P.; Rodriguez de Cordoba, S. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 240–245. [Google Scholar]
- Fremeaux-Bacchi, V.; Miller, E.C.; Liszewski, M.K.; Strain, L.; Blouin, J.; Brown, A.L.; Moghal, N.; Kaplan, B.S.; Weiss, R.A.; Lhotta, K.; et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 2008, 112, 4948–4952. [Google Scholar]
- Fang, C.J.; Fremeaux-Bacchi, V.; Liszewski, M.K.; Pianetti, G.; Noris, M.; Goodship, T.H.; Atkinson, J.P. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (ahus), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood 2008, 111, 624–632. [Google Scholar] [CrossRef]
- Tsai, H.M. Pathophysiology of thrombotic thrombocytopenic purpura. Int. J. Hematol. 2010, 91, 1–19. [Google Scholar] [CrossRef]
- Saland, J.M.; Ruggenenti, P.; Remuzzi, G. Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2009, 20, 940–949. [Google Scholar] [CrossRef]
- Motto, D.G.; Chauhan, A.K.; Zhu, G.; Homeister, J.; Lamb, C.B.; Desch, K.C.; Zhang, W.; Tsai, H.M.; Wagner, D.D.; Ginsburg, D. Shiga toxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J. Clin. Invest. 2005, 115, 2752–2761. [Google Scholar]
- Melton-Celsa, A.R.; O’Brien, A.D. Animal models for STEC-mediated disease. Methods Mol. Med. 2003, 73, 291–305. [Google Scholar]
- Gunzer, F.; Hennig-Pauka, I.; Waldmann, K.-H.; Sandhoff, R.; Grone, H.-J.; Kreipe, H.-H.; Matussek, A.; Mengel, M. Gnotobiotic piglets develop thrombotic microangiopathy after oral infection with enterohemorrhagic Escherichia coli. Am. J. Clin. Pathol. 2002, 118, 364–375. [Google Scholar] [CrossRef]
- Myhal, M.L.; Laux, D.C.; Cohen, P.S. Relative colonizing abilities of human fecal and K 12 strains of Escherichia coli in the large intestines of streptomycin-treated mice. Eur. J. Clin. Microbiol. 1982, 1, 186–192. [Google Scholar] [CrossRef]
- Wadolkowski, E.A.; Burris, J.A.; O’Brien, A.D. Mouse model for colonization and disease caused by enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 1990, 58, 2438–2445. [Google Scholar]
- Fujii, J.; Kita, T.; Yoshida, S.; Takeda, T.; Kobayashi, H.; Tanaka, N.; Ohsato, K.; Mizuguchi, Y. Direct evidence of neuron impairment by oral infection with verotoxin-producing Escherichia coli O157:H- in mitomycin-treated mice. Infect. Immun. 1994, 62, 3447–3453. [Google Scholar]
- Isogai, E.; Isogai, H.; Kimura, K.; Hayashi, S.; Kubota, T.; Fujii, N.; Takeshi, K. Role of tumor necrosis factor alpha in gnotobiotic mice infected with an Escherichia coli O157:H7 strain. Infect. Immun. 1998, 66, 197–202. [Google Scholar]
- Karpman, D.; Connell, H.; Svensson, M.; Scheutz, F.; Alm, P.; Svanborg, C. The role of lipopolysaccharide and Shiga-like toxin in a mouse model of Escherichia coli O157:H7 infection. J. Infect. Dis. 1997, 175, 611–620. [Google Scholar] [CrossRef]
- Brando, R.J.; Miliwebsky, E.; Bentancor, L.; Deza, N.; Baschkier, A.; Ramos, M.V.; Fernandez, G.C.; Meiss, R.; Rivas, M.; Palermo, M.S. Renal damage and death in weaned mice after oral infection with Shiga toxin 2-producing Escherichia coli strains. Clin. Exp. Immunol. 2008, 153, 297–306. [Google Scholar]
- Kurioka, T.; Yunou, Y.; Kita, E. Enhancement of susceptibility to Shiga toxin-producing Escherichia coli O157:H7 by protein calorie malnutrition in mice. Infect. Immun. 1998, 66, 1726–1734. [Google Scholar]
- Garcia, A.; Bosques, C.J.; Wishnok, J.S.; Feng, Y.; Karalius, B.J.; Butterton, J.R.; Schauer, D.B.; Rogers, A.B.; Fox, J.G. Renal injury is a consistent finding in Dutch Belted rabbits experimentally infected with enterohemorrhagic Escherichia coli. J. Infect. Dis. 2006, 193, 1125–1134. [Google Scholar] [CrossRef]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Debord, D.; Nishikawa, K.; Oh, S.Y.; Leibowitz, C.S.; Kurosawa, S. Rescue from lethal Shiga toxin 2-induced renal failure with a cell-permeable peptide. Pediatr. Nephrol. 2011, 26, 2031–2039. [Google Scholar] [CrossRef]
- Sauter, K.A.; Melton-Celsa, A.R.; Larkin, K.; Troxell, M.L.; O’Brien, A.D.; Magun, B.E. Mouse model of hemolytic-uremic syndrome caused by endotoxin-free Shiga toxin 2 (Stx2) and protection from lethal outcome by anti-stx2 antibody. Infect. Immun. 2008, 76, 4469–4478. [Google Scholar] [CrossRef]
- Zotta, E.; Lago, N.; Ochoa, F.; Repetto, H.A.; Ibarra, C. Development of an experimental hemolytic uremic syndrome in rats. Pediatr. Nephrol. 2008, 23, 559–567. [Google Scholar] [CrossRef]
- Moon, H.W.; Whipp, S.C.; Argenzio, R.A.; Levine, M.M.; Giannella, R.A. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 1983, 41, 1340–1351. [Google Scholar]
- Tzipori, S.; Wachsmuth, I.K.; Chapman, C.; Birden, R.; Brittingham, J.; Jackson, C.; Hogg, J. The pathogenesis of hemorrhagic colitis caused by Escherichia coli O157:H7 in gnotobiotic piglets. J. Infect. Dis. 1986, 154, 712–716. [Google Scholar] [CrossRef]
- Mohawk, K.L.; O’Brien, A.D. Mouse models of Escherichia coli O157:H7 infection and shiga toxin injection. J. Biomed. Biotechnol. 2011, 2011, 258185. [Google Scholar]
- Al-Jumaili, I.; Burke, D.A.; Scotland, S.M.; al-Mardini, H.; Record, C.O. A method of enhancing verocytotoxin production by Escherichia coli. FEMS Microbiol. Lett. 1992, 72, 121–125. [Google Scholar]
- MacLeod, D.L.; Gyles, C.L. Effects of culture conditions on yield of Shiga-like toxin-II from Escherichia coli. Can. J. Microbiol. 1989, 35, 623–629. [Google Scholar] [CrossRef]
- Lomas-Neira, J.; Perl, M.; Venet, F.; Chung, C.S.; Ayala, A. The role and source of tumor necrosis factor-alpha in hemorrhage-induced priming for septic lung injury. Shock 2012, 37, 611–620. [Google Scholar] [CrossRef]
- Landoni, V.I.; de Campos-Nebel, M.; Schierloh, P.; Calatayud, C.; Fernandez, G.C.; Ramos, M.V.; Rearte, B.; Palermo, M.S.; Isturiz, M.A. Shiga toxin 1-induced inflammatory response in lipopolysaccharide-sensitized astrocytes is mediated by endogenous tumor necrosis factor alpha. Infect. Immun. 2010, 78, 1193–1201. [Google Scholar] [CrossRef]
- Landoni, V.I.; Schierloh, P.; de Campos Nebel, M.; Fernández, G.C.; Calatayud, C.; Lapponi, M.J.; Isturiz, M.A. Shiga toxin 1 induces on lipopolysaccharide-treated astrocytes the release of tumor necrosis factor-alpha that alter brain-like endothelium integrity. PLoS Pathog. 2012, 8, e1002632. [Google Scholar]
- Barrett, T.J.; Potter, M.E.; Wachsmuth, I.K. Bacterial endotoxin both enhances and inhibits the toxicity of Shiga-like toxin II in rabbits and mice. Infect. Immun. 1989, 57, 3434–3437. [Google Scholar]
- Taylor, F.B.; Toh, C.H.; Hoots, W.K.; Wada, H.; Levi, M. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. On behalf of the scientific subcommittee on disseminated intravascular coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Thromb. Haemost. 2001, 86, 1327–1330. [Google Scholar]
- Stearns-Kurosawa, D.J.; Osuchowski, M.F.; Valentine, C.; Kurosawa, S.; Remick, D.G. The pathogenesis of sepsis. Annu. Rev. Pathol. 2011, 6, 19–48. [Google Scholar] [CrossRef]
- Remick, D.G.; Ward, P.A. Evaluation of endotoxin models for the study of sepsis. Shock 2005, 24, 7–11. [Google Scholar] [CrossRef]
- Chart, H.; Smith, H.R.; Scotland, S.M.; Rowe, B.; Milford, D.V.; Taylor, C.M. Serological identification of Escherichia coli O157:H7 infection in haemolytic uraemic syndrome. Lancet 1991, 337, 138–140. [Google Scholar]
- Bitzan, M.; Moebius, E.; Ludwig, K.; Muller-Wiefel, D.E.; Heesemann, J.; Karch, H. High incidence of serum antibodies to Escherichia coli O157 lipopolysaccharide in children with hemolytic-uremic syndrome. J. Pediatr. 1991, 119, 380–385. [Google Scholar] [CrossRef]
- Chart, H.; Jenkins, C. The serodiagnosis of infections caused by verocytotoxin-producing Escherichia coli. J. Appl. Microbiol. 1999, 86, 731–740. [Google Scholar] [CrossRef]
- Ziegler, E.J.; Fisher, C.J., Jr.; Sprung, C.L.; Straube, R.C.; Sadoff, J.C.; Foulke, G.E.; Wortel, C.H.; Fink, M.P.; Dellinger, R.P.; Teng, N.N.; et al. Treatment of gram-negative bacteremia and septic shock with HA-1a human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1a sepsis study group. N. Engl. J. Med. 1991, 324, 429–436. [Google Scholar] [CrossRef]
- Angus, D.C.; Birmingham, M.C.; Balk, R.A.; Scannon, P.J.; Collins, D.; Kruse, J.A.; Graham, D.R.; Dedhia, H.V.; Homann, S.; MacIntyre, N. E5 murine monoclonal antiendotoxin antibody in gram-negative sepsis: A randomized controlled trial. E5 study investigators. J. Am. Med. Asso. 2000, 283, 1723–1730. [Google Scholar]
- Deitch, E.A. Gut-origin sepsis: Evolution of a concept. Surgeon 2012. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Wolfson, J.J.; May, K.L.; Thorpe, C.M.; Jandhyala, D.M.; Paton, J.C.; Paton, A.W. Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell Microbiol. 2008, 10, 1775–1786. [Google Scholar] [CrossRef]
Appendix
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mayer, C.L.; Leibowitz, C.S.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga Toxins and the Pathophysiology of Hemolytic Uremic Syndrome in Humans and Animals. Toxins 2012, 4, 1261-1287. https://doi.org/10.3390/toxins4111261
Mayer CL, Leibowitz CS, Kurosawa S, Stearns-Kurosawa DJ. Shiga Toxins and the Pathophysiology of Hemolytic Uremic Syndrome in Humans and Animals. Toxins. 2012; 4(11):1261-1287. https://doi.org/10.3390/toxins4111261
Chicago/Turabian StyleMayer, Chad L., Caitlin S. Leibowitz, Shinichiro Kurosawa, and Deborah J. Stearns-Kurosawa. 2012. "Shiga Toxins and the Pathophysiology of Hemolytic Uremic Syndrome in Humans and Animals" Toxins 4, no. 11: 1261-1287. https://doi.org/10.3390/toxins4111261