The Discodermia calyx Toxin Calyculin A Enhances Cyclin D1 Phosphorylation and Degradation, and Arrests Cell Cycle Progression in Human Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Culture
2.2. Inhibitor Treatment
2.3. Antibodies and Immunoblotting
2.4. Cell Cycle Analysis
2.5. Reverse Phase Protein Array (RPPA)
3. Results and Discussion
3.1. Results
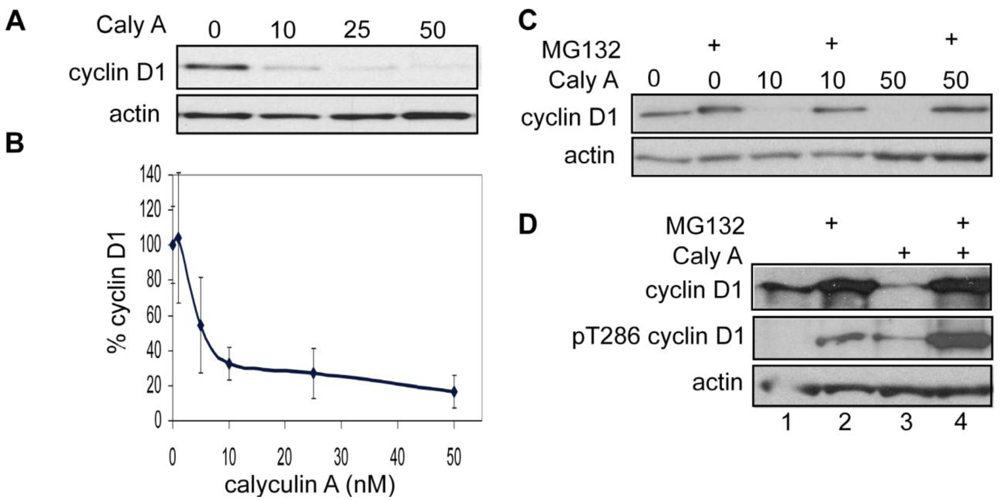
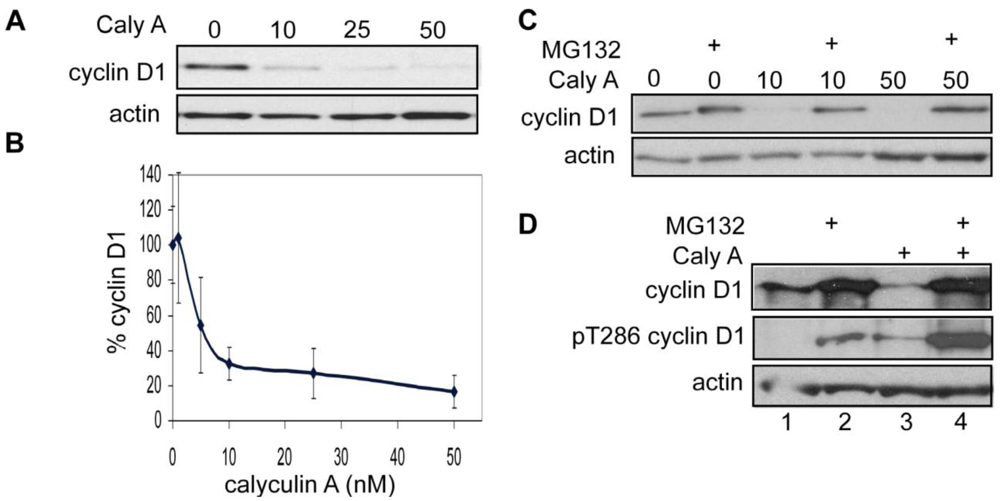
3.1.1. Calyculin A Induces Depletion of Cyclin D1 in Human Breast Cancer MDA-MB-468 and MDA-MB-231 Cells
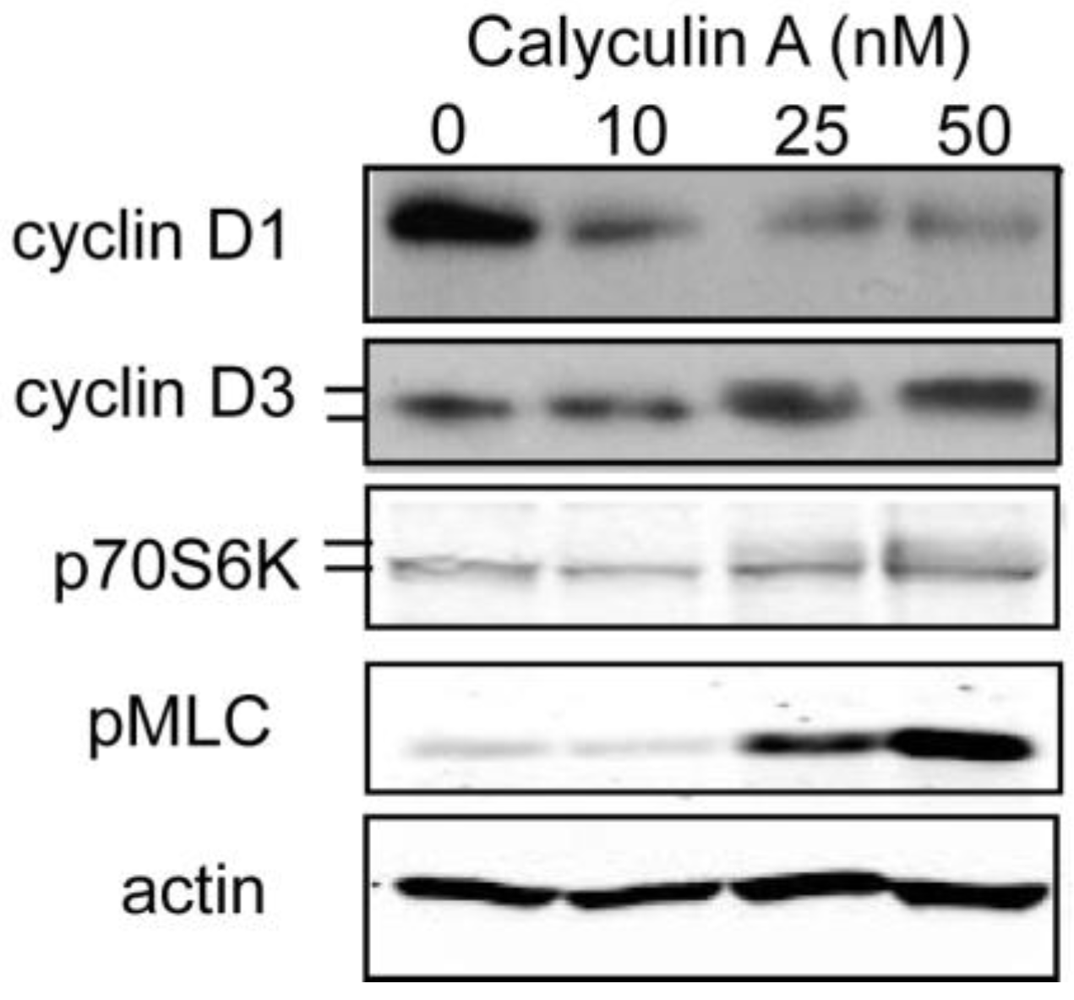
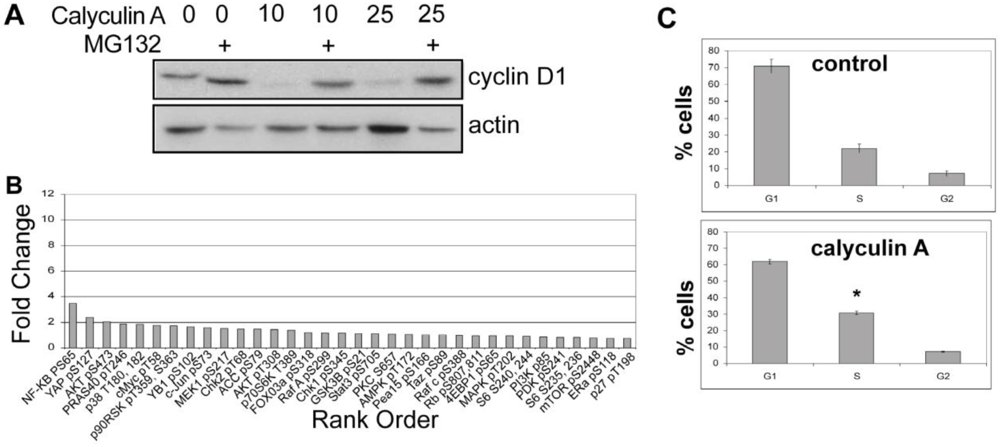
3.1.2. Effect of Calyculin A on Other Endogenous Phosphoproteins
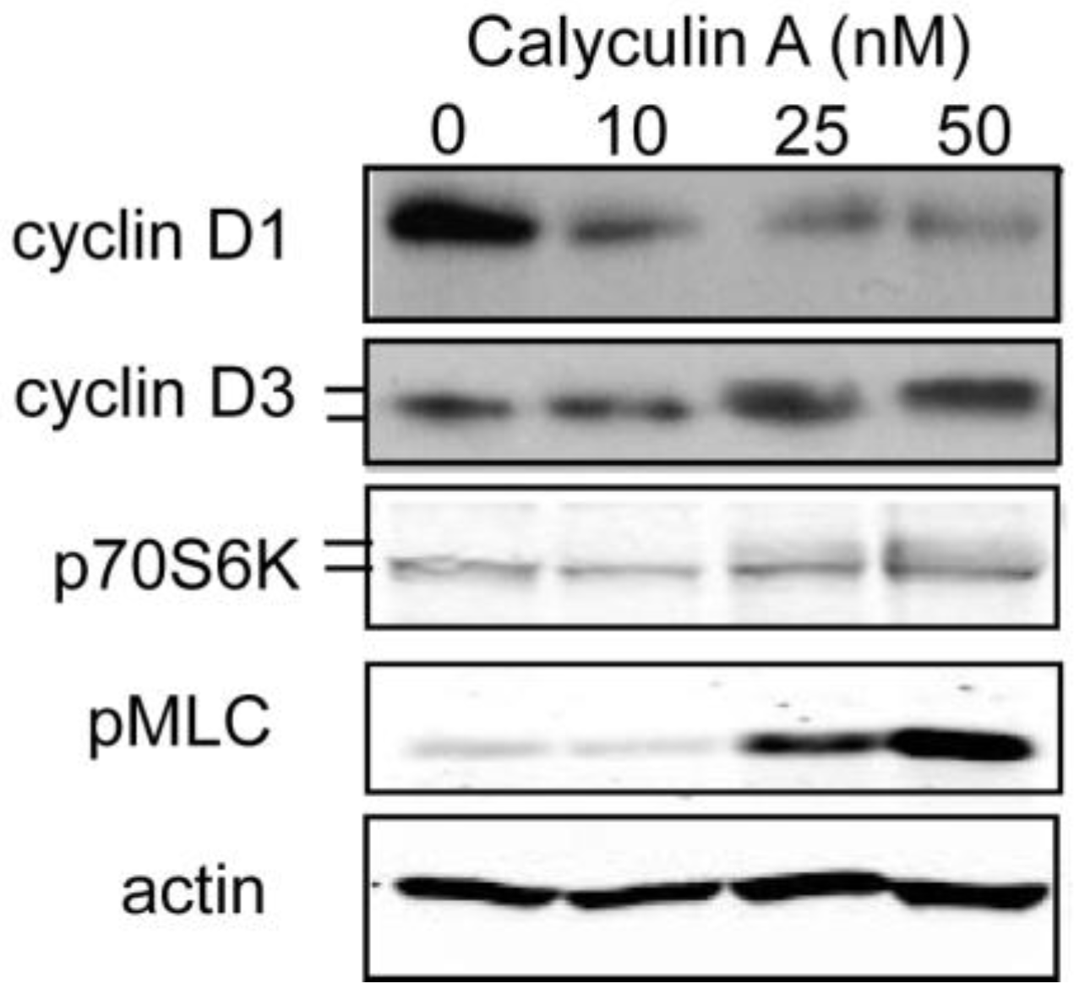
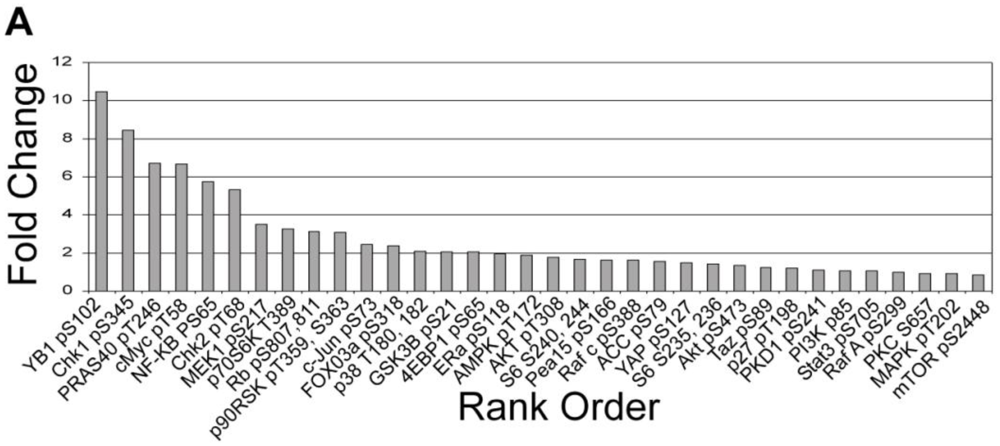
3.1.3. Analysis of Calyculin A Effects on Protein Phosphorylation In Breast Cancer Cells by Reverse Phase Protein Array
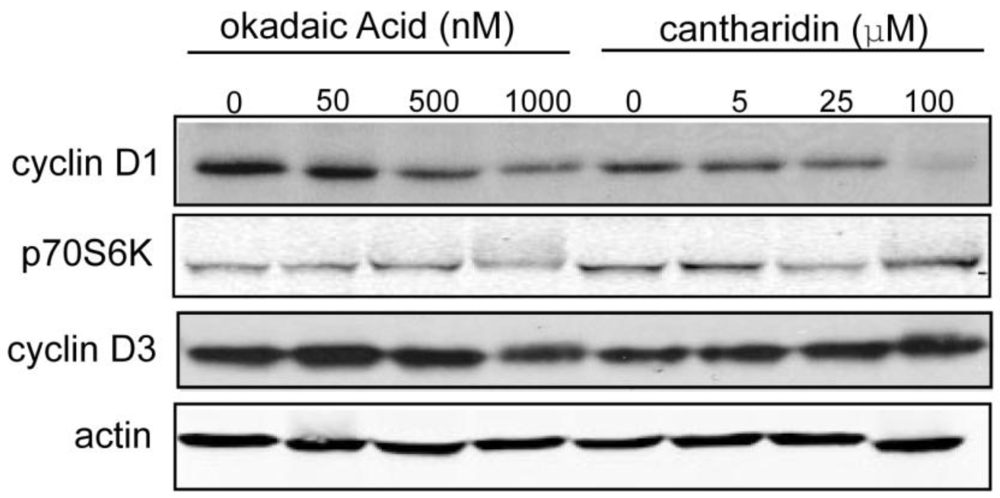
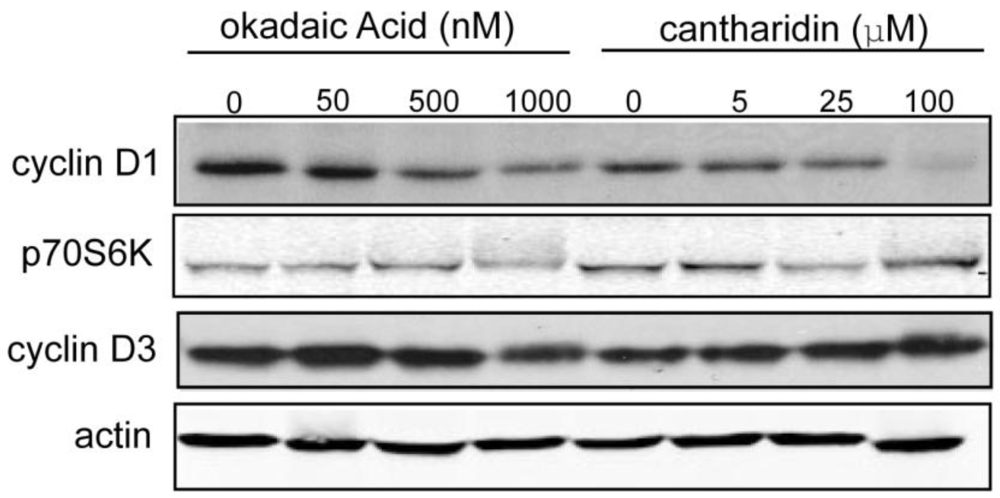
3.1.4. Effect of Okadaic Acid and Cantharidin on Cyclin D1 and Endogenous Phosphoproteins in MDA-MB-468 Breast Cancer Cells

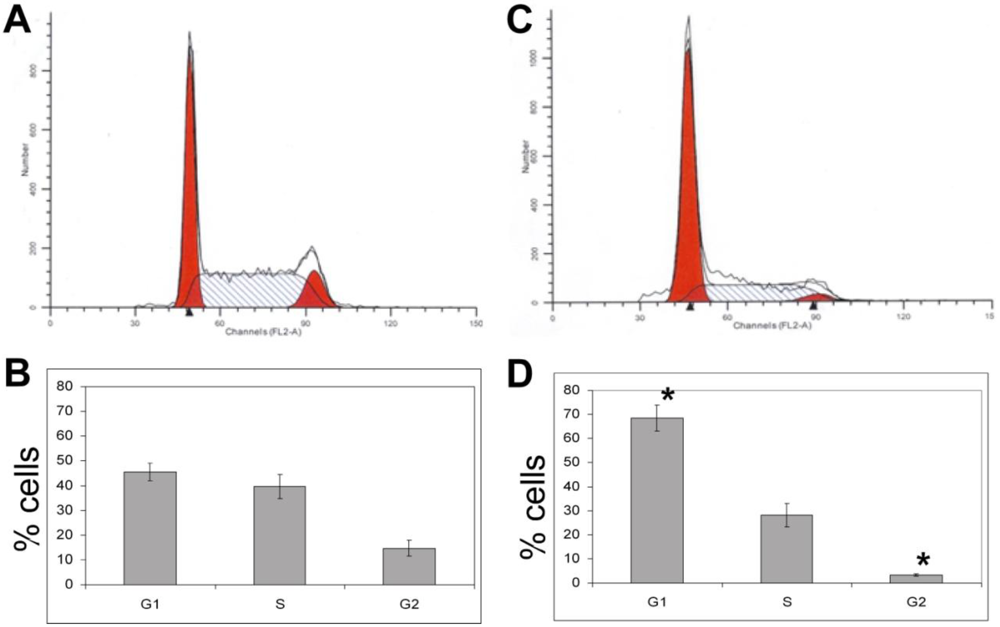
3.1.5. The Effects of Calyculin A on Cell Cycle in MDA-MB-468 Breast Cancer Cells
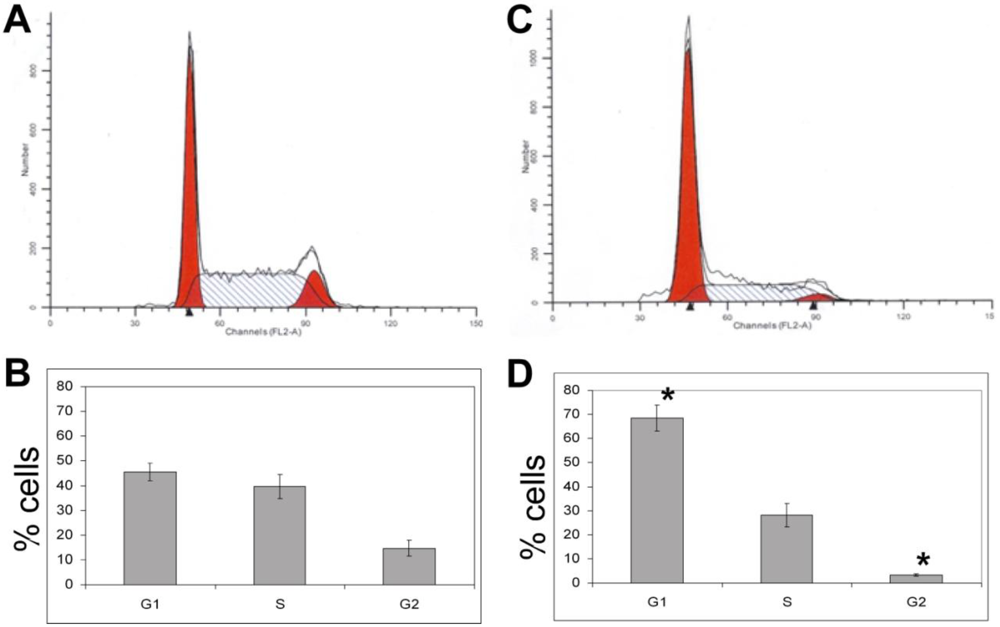
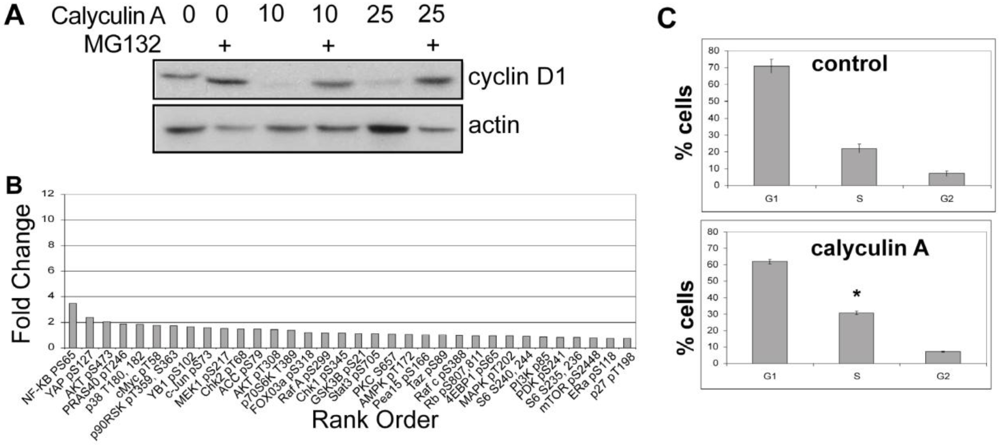
3.1.6. Calyculin A Induces Proteasome Degradation of Cyclin D1 and Cell Cycle Arrest in MCF-7 cells
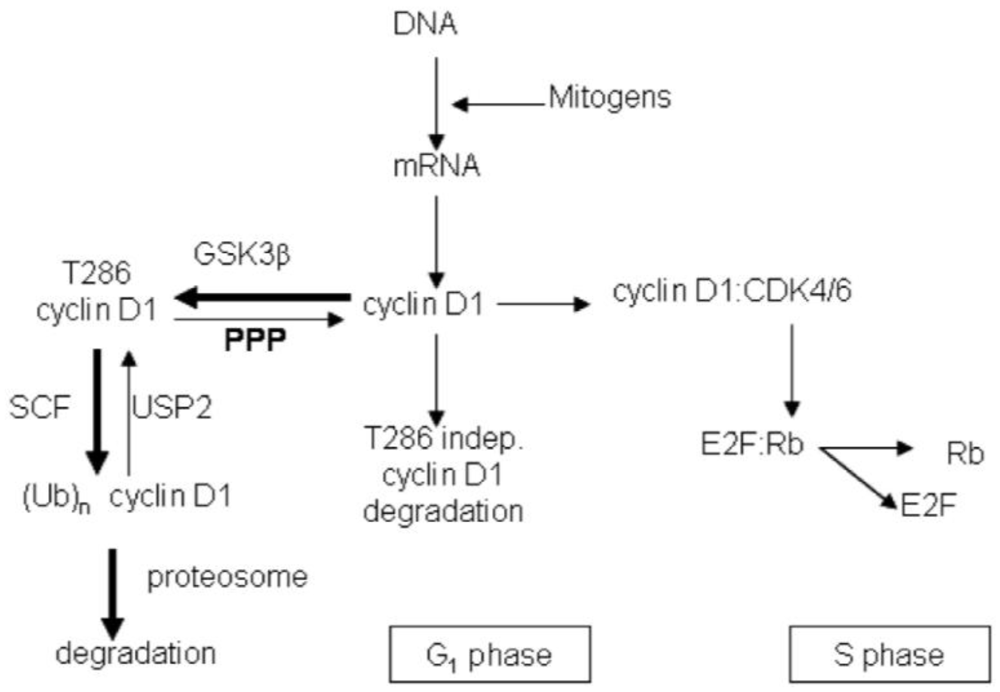
3.2. Discussion
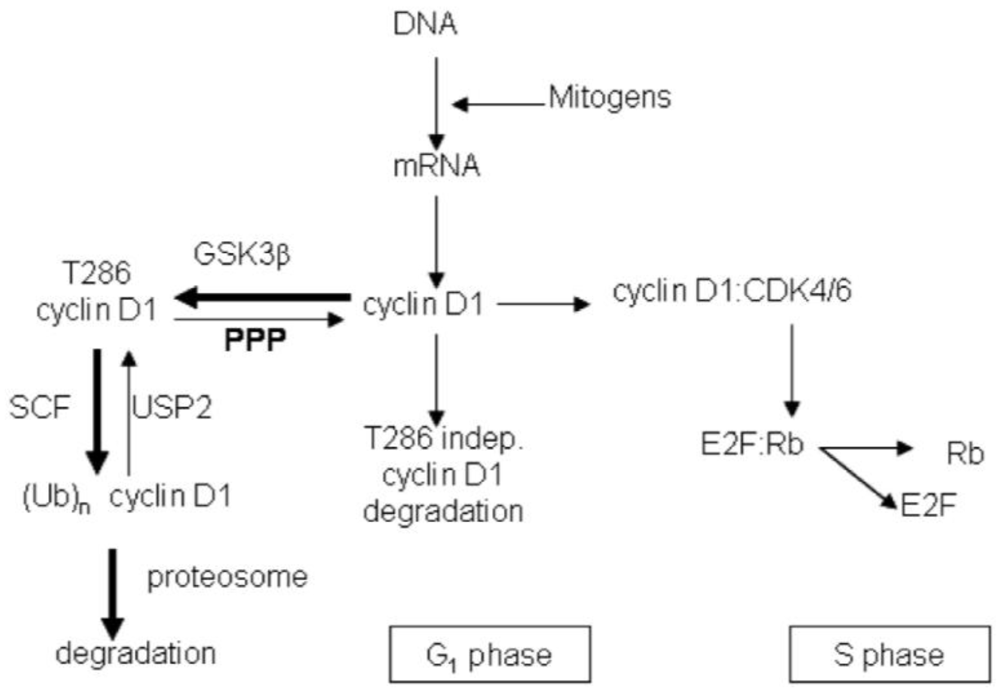
4. Conclusions
Acknowledgements
References
- Gillett, C.; Smith, P.; Gregory, W.; Richards, M.; Millis, R.; Peters, G.; Barnes, D. Cyclin D1 and prognosis in human breast cancer. Int. J. Cancer 1996, 69, 92–99. [Google Scholar] [CrossRef]
- Gille, H.; Downward, J. Multiple ras effector pathways contribute to G(1) cell cycle progression. J. Biol. Chem. 1999, 274, 22033–22040. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Sherr, C.J. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol. Cell Biol. 1997, 17, 7362–7374. [Google Scholar] [PubMed]
- Kato, J.Y.; Matsuoka, M.; Strom, D.K.; Sherr, C.J. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol. Cell Biol. 1994, 14, 2713–2721. [Google Scholar] [PubMed]
- Alt, J.R.; Cleveland, J.L.; Hannink, M.; Diehl, J.A. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000, 14, 3102–3114. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yang, K.; Harwalkar, J.; Nye, J.M.; Mason, D.R.; Garrett, M.D.; Hitomi, M.; Stacey, D.W. Phosphorylation of cyclin D1 at Thr 286 during S phase leads to its proteasomal degradation and allows efficient DNA synthesis. Oncogene 2005, 24, 2599–2612. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.; Thompson, M.A.; Hendley, J.; Trute, L.; Armes, J.; Germain, D. Cyclin D1 and D3 associate with the SCF complex and are coordinately elevated in breast cancer. Oncogene 1999, 18, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.; Russell, A.; Thompson, A.; Hendley, J. Ubiquitination of free cyclin D1 is independent of phosphorylation on threonine 286. J. Biol. Chem. 2000, 275, 12074–12079. [Google Scholar] [PubMed]
- Solomon, D.A.; Wang, Y.; Fox, S.R.; Lambeck, T.C.; Giesting, S.; Lan, Z.; Senderowicz, A.M.; Conti, C.J.; Knudsen, E.S. Cyclin D1 splice variants. Differential effects on localization, RB phosphorylation, and cellular transformation. J. Biol. Chem. 2003, 278, 30339–30347. [Google Scholar] [PubMed]
- Shan, J.; Zhao, W.; Gu, W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol. Cell 2009, 36, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Sarabia, M.J.; Sutton, A.; Zhong, T. Arndt, KT: SIT4 protein phosphatase is required for the normal accumulation of SWI4, CLN1, CLN2, and HCS26 RNAs during late. Genes Dev. 1992, 6, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, B.; Brautigan, D.L. Protein phosphatase PP6 N terminal domain restricts G1 to S phase progression in human cancer cells. Cell Cycle 2007, 6, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Bielinski, V.A.; Mumby, M.C. Functional analysis of the PP2A subfamily of protein phosphatases in regulating Drosophila S6 kinase. Exp. Cell Res. 2007, 313, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Casida, J.E. Cantharidin-binding protein: Identification as protein phosphatase 2A. Proc. Natl. Acad. Sci. USA 1992, 89, 11867–11870. [Google Scholar] [CrossRef]
- Lin, T.A.; Lawrence, J.C., Jr. Activation of ribosomal protein S6 kinases does not increase glycogen synthesis or glucose transport in rat adipocytes. J. Biol. Chem. 1994, 269, 21255–21261. [Google Scholar] [PubMed]
- Lahne, H.U.; Kloster, M.M.; Lefdal, S.; Blomhoff, H.K.; Naderi, S. Degradation of cyclin D3 independent of Thr-283 phosphorylation. Oncogene 2006, 25, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.T.; Desai, B.N.; Hardwick, J.S.; Schreiber, S.L. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc. Natl. Acad. Sci. USA 1999, 96, 4438–4442. [Google Scholar]
- Senba, S.; Eto, M.; Yazawa, M. Identification of trimeric myosin phosphatase (PP1M) as a target for a novel PKC-potentiated protein phosphatase-1 inhibitory protein (CPI17) in porcine aorta smooth muscle. J. Biochem. 1999, 125, 354–362. [Google Scholar] [PubMed]
- Gregory, M.A.; Hann, S.R. c-Myc proteolysis by the ubiquitin-proteasome pathway: Stabilization of c-Myc in Burkitt's lymphoma cells. Mol. Cell Biol. 2000, 20, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Virshup, D.M.; Shenolikar, S. From promiscuity to precision: Protein phosphatases get a makeover. Mol. Cell 2009, 33, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Maynes, J.T.; Bateman, K.S.; Cherney, M.M.; Das, A.K.; Luu, H.A.; Holmes, C.F.; James, M.N. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J. Biol. Chem. 2001, 276, 44078–44082. [Google Scholar] [PubMed]
- Kita, A.; Matsunaga, S.; Takai, A.; Kataiwa, H.; Wakimoto, T.; Fusetani, N.; Isobe, M.; Miki, K. Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure 2002, 10, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Cho, U.S.; Xu, W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2007, 445, 53–57. [Google Scholar] [PubMed]
- Ishihara, H.; Martin, B.L.; Brautigan, D.L.; Karaki, H.; Ozaki, H.; Kato, Y.; Fusetani, N.; Watabe, S.; Hashimoto, K.; Uemura, D.; et al. Calyculin A and okadaic acid: Inhibitors of protein phosphatase activity. Biochem. Biophys. Res. Commun. 1989, 159, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small-molecule inhibitors of ser/thr protein phosphatases: Specificity, use and common forms of abuse. Methods Mol. Biol. 2007, 365, 23–38. [Google Scholar] [PubMed]
- Cohen, P.; Holmes, C.F.; Tsukitani, Y. Okadaic acid: A new probe for the study of cellular regulation. Trends Biochem. Sci. 1990, 15, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Favre, B.; Turowski, P.; Hemmings, B.A. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J. Biol. Chem. 1997, 272, 13856–13863. [Google Scholar] [PubMed]
- Prickett, T.D.; Brautigan, D.L. The alpha4 regulatory subunit exerts opposing allosteric effects on protein phosphatases PP6 and PP2A. J. Biol. Chem. 2006, 281, 30503–30511. [Google Scholar] [PubMed]
- den Engelsman, J.; Keijsers, V.; de Jong, W.W.; Boelens, W.C. The small heat-shock protein alpha B-crystallin promotes FBX4-dependent ubiquitination. J. Biol. Chem. 2003, 278, 4699–4704. [Google Scholar] [PubMed]
- Barbash, O.; Zamfirova, P.; Lin, D.I.; Chen, X.; Yang, K.; Nakagawa, H.; Lu, F.; Rustgi, A.K.; Diehl, J.A. Mutations in Fbx4 inhibit dimerization of the SCF(Fbx4) ligase and contribute to cyclin D1 overexpression in human cancer. Cancer Cell 2008, 14, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Hoon, S.; Smith, A.M.; Wallace, I.M.; Suresh, S.; Miranda, M.; Fung, E.; Proctor, M.; Shokat, K.M.; Zhang, C.; Davis, R.W.; et al. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol. 2008, 4, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Kelker, M.S.; Page, R.; Peti, W. Crystal structures of protein phosphatase-1 bound to nodularin-R and tautomycin: A novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors. J. Mol. Biol. 2009, 385, 11–21. [Google Scholar] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Edelson, J.R.; Brautigan, D.L. The Discodermia calyx Toxin Calyculin A Enhances Cyclin D1 Phosphorylation and Degradation, and Arrests Cell Cycle Progression in Human Breast Cancer Cells. Toxins 2011, 3, 105-119. https://doi.org/10.3390/toxins3010105
Edelson JR, Brautigan DL. The Discodermia calyx Toxin Calyculin A Enhances Cyclin D1 Phosphorylation and Degradation, and Arrests Cell Cycle Progression in Human Breast Cancer Cells. Toxins. 2011; 3(1):105-119. https://doi.org/10.3390/toxins3010105
Chicago/Turabian StyleEdelson, Jessica R., and David L. Brautigan. 2011. "The Discodermia calyx Toxin Calyculin A Enhances Cyclin D1 Phosphorylation and Degradation, and Arrests Cell Cycle Progression in Human Breast Cancer Cells" Toxins 3, no. 1: 105-119. https://doi.org/10.3390/toxins3010105