Toxin Mediated Diarrhea in the 21st Century: The Pathophysiology of Intestinal Ion Transport in the Course of ETEC, V. cholerae and Rotavirus Infection

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Physiology of Intestinal Ion Transport
2.1. Enterocyte Electrolyte Absorption
2.2. Enterocyte Electrolyte Secretion
3. The Pathophysiology of Enterotoxin Mediated Diarrhea
3.1. Enterotoxigenic E. coli
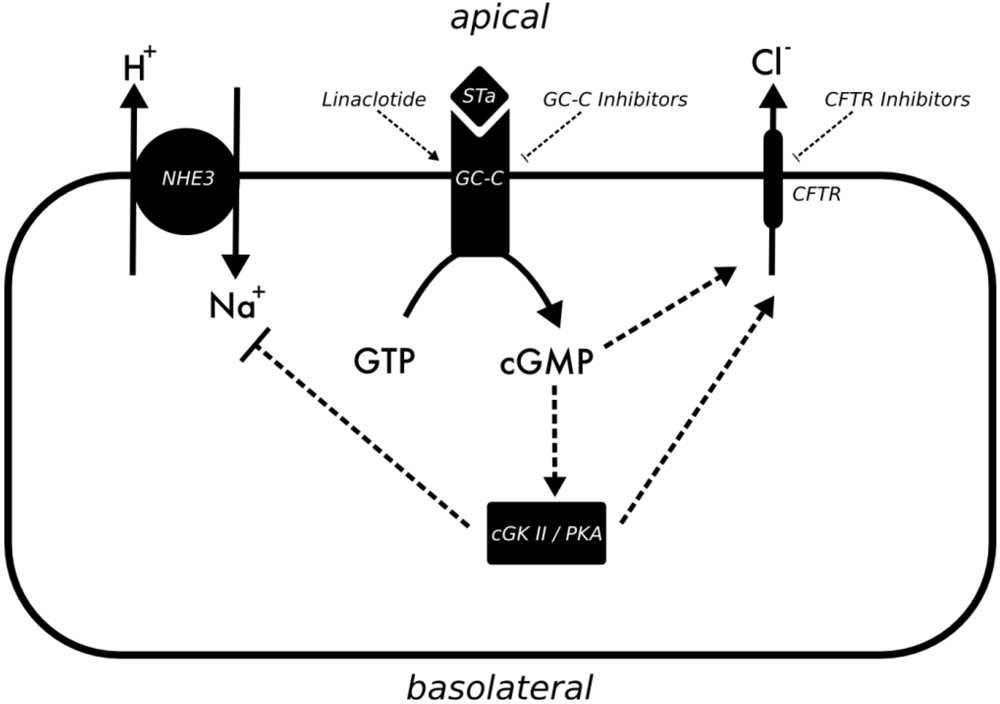
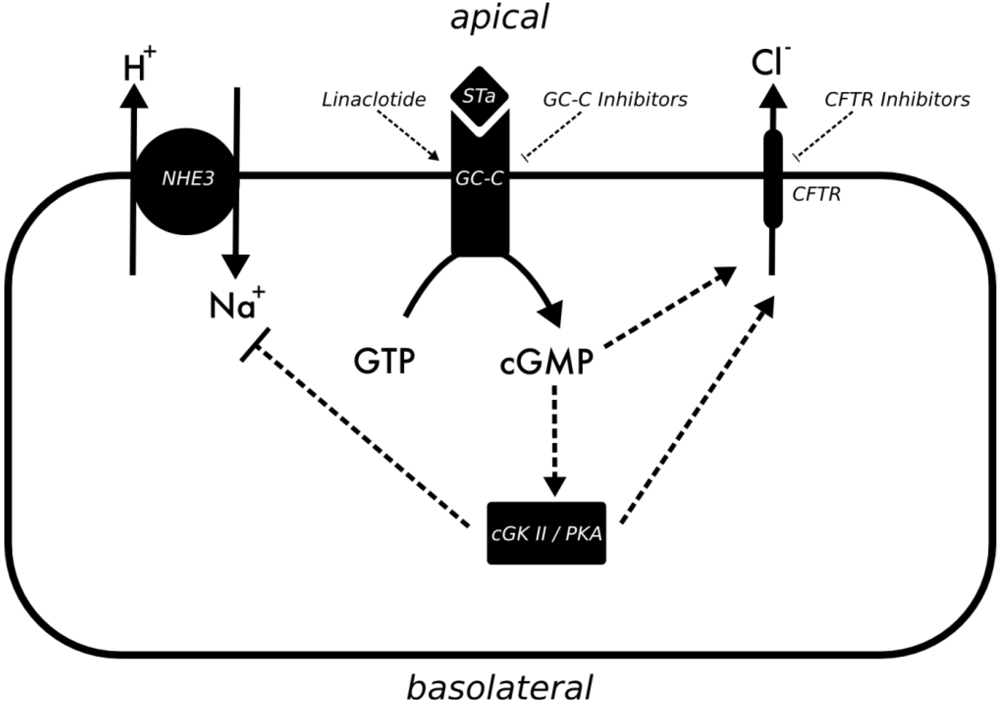
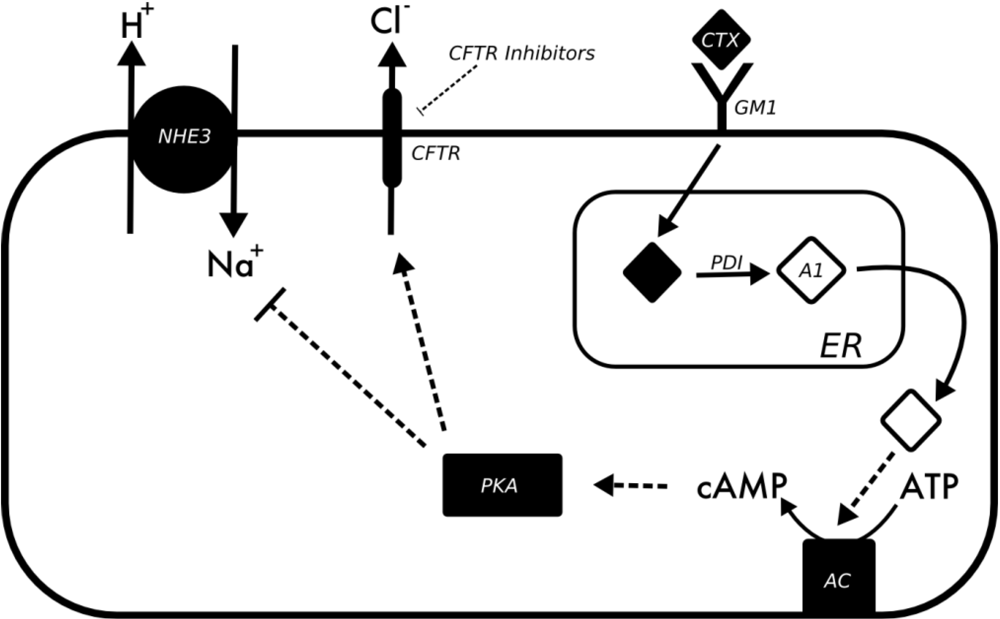
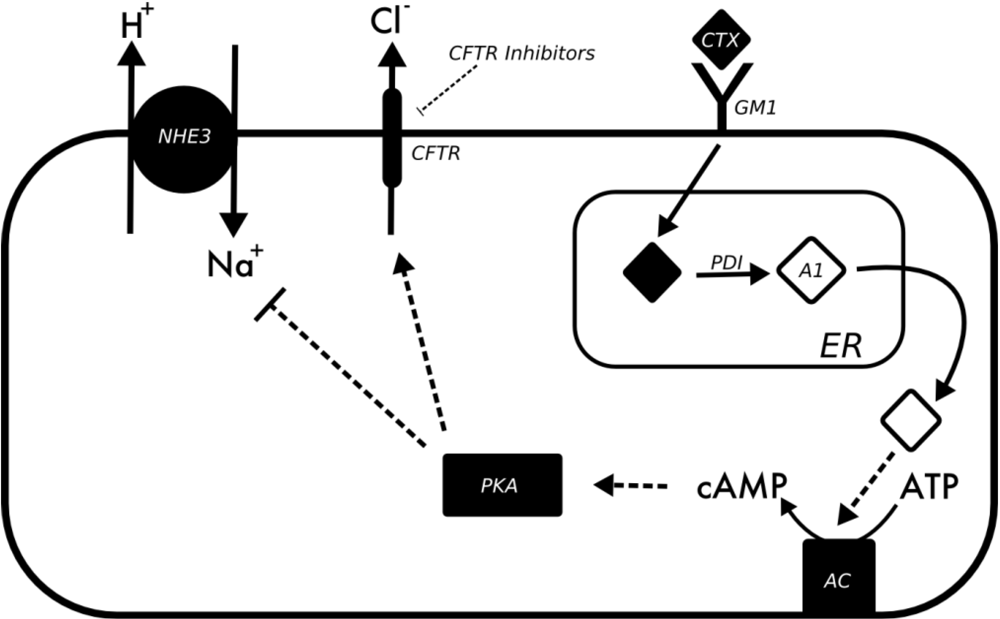
3.2. Vibrio Cholerae
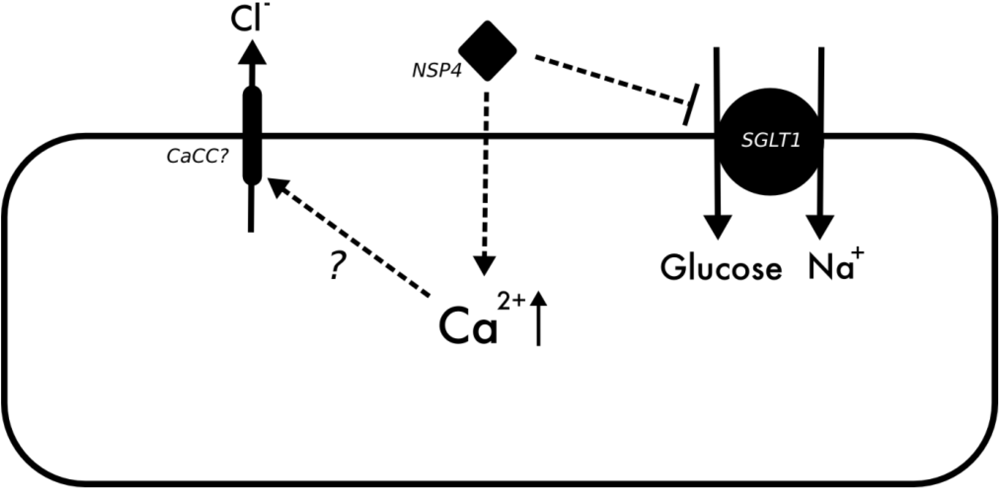
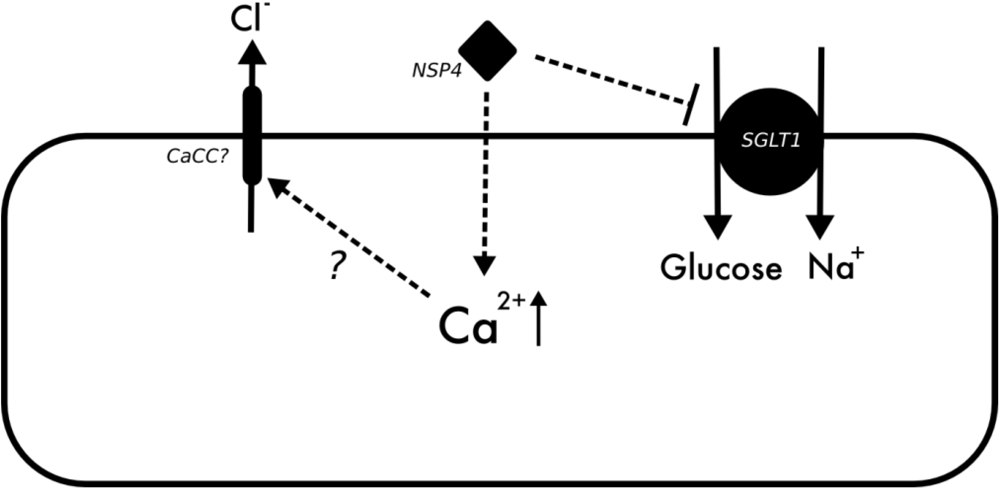
3.3. Rotavirus
4. Conclusions
References
- Kosek, M.; Bern, C.; Guerrant, R.L. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull. World Health Organ. 2003, 81, 197–204. [Google Scholar] [Green Version]
- Barrett, K.E.; Keely, S.J. Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu. Rev. Physiol. 2000, 62, 535–572. [Google Scholar] [Green Version]
- Singh, S.K.; Binder, H.J.; Boron, W.F.; Geibel, J.P. Fluid absorption in isolated perfused colonic crypts. J. Clin. Invest. 1995, 96, 2373–2379. [Google Scholar] [Green Version]
- Strong, T.V.; Boehm, K.; Collins, F.S. Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J. Clin. Invest. 1994, 93, 347–354. [Google Scholar] [Green Version]
- Kunzelmann, K.; Mall, M. Electrolyte transport in the mammalian colon: Mechanisms and implications for disease. Physiol. Rev. 2002, 82, 245–289. [Google Scholar] [Green Version]
- Kockerling, A.; Fromm, M. Origin of cAMP-dependent Cl− secretion from both crypts and surface epithelia of rat intestine. Am. J. Physiol. 1993, 264, C1294–C1301. [Google Scholar] [Green Version]
- Zachos, N.C.; Tse, M.; Donowitz, M. Molecular physiology of intestinal Na+/H+ exchange. Annu. Rev. Physiol. 2005, 67, 411–443. [Google Scholar] [Green Version]
- Wakabayashi, S.; Shigekawa, M.; Pouyssegur, J. Molecular physiology of vertebrate Na+/H+ exchangers. Physiol. Rev. 1997, 77, 51–74. [Google Scholar] [Green Version]
- Bookstein, C.; DePaoli, A.M.; Xie, Y.; Niu, P.; Musch, M.W.; Rao, M.C.; Chang, E.B. Na+/H+ exchangers, NHE-1 and NHE-3, of rat intestin. Expression and localization. J. Clin. Invest. 1994, 93, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.M.; Ma, A.I.; Yang, V.W.; Watson, A.J.; Levine, S.; Montrose, M.H.; Potter, J.; Sardet, C.; Pouyssegur, J.; Donowitz, M. Molecular cloning and expression of a cDNA encoding the rabbit ileal villus cell basolateral membrane Na+/H+ exchanger. EMBO J. 1991, 10, 1957–1967. [Google Scholar]
- Collins, J.F.; Honda, T.; Knobel, S.; Bulus, N.M.; Conary, J.; DuBois, R.; Ghishan, F.K. Molecular cloning, sequencing, tissue distribution, and functional expression of a Na+/H+ exchanger (NHE-2). Proc. Natl. Acad. Sci. USA 1993, 90, 3938–3942. [Google Scholar]
- Brant, S.R.; Yun, C.H.; Donowitz, M.; Tse, C.M. Cloning, tissue distribution, and functional analysis of the human Na+/N+ exchanger isoform, NHE3. Am. J. Physiol. 1995, 269, C198–C206. [Google Scholar]
- Bookstein, C.; Xie, Y.; Rabenau, K.; Musch, M.W.; McSwine, R.L.; Rao, M.C.; Chang, E.B. Tissue distribution of Na+/H+ exchanger isoforms NHE2 and NHE4 in rat intestine and kidney. Am. J. Physiol. 1997, 273, C1496–C1505. [Google Scholar]
- Gawenis, L.R.; Stien, X.; Shull, G.E.; Schultheis, P.J.; Woo, A.L.; Walker, N.M.; Clarke, L.L. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G776–G784. [Google Scholar]
- Xu, H.; Chen, H.; Dong, J.; Lynch, R.; Ghishan, F.K. Gastrointestinal distribution and kinetic characterization of the sodium-hydrogen exchanger isoform 8 (NHE8). Cell. Physiol. Biochem. 2008, 21, 109–116. [Google Scholar]
- Xu, H.; Chen, R.; Ghishan, F.K. Subcloning, localization, and expression of the rat intestinal sodium-hydrogen exchanger isoform 8. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G36–G41. [Google Scholar]
- Donowitz, M.; Mohan, S.; Zhu, C.X.; Chen, T.E.; Lin, R.; Cha, B.; Zachos, N.C.; Murtazina, R.; Sarker, R.; Li, X. NHE3 regulatory complexes. J. Exp. Biol. 2009, 212, 1638–1646. [Google Scholar]
- Yun, C.H.; Oh, S.; Zizak, M.; Steplock, D.; Tsao, S.; Tse, C.M.; Weinman, E.J.; Donowitz, M. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc. Natl. Acad. Sci. USA 1997, 94, 3010–3015. [Google Scholar]
- Cha, B.; Donowitz, M. The epithelial brush border Na+/H+ exchanger NHE3 associates with the actin cytoskeleton by binding to ezrin directly and via PDZ domain-containing Na+/H+ exchanger regulatory factor (NHERF) proteins. Clin. Exp. Pharmacol. Physiol. 2008, 35, 863–871. [Google Scholar]
- Rajendran, V.M.; Geibel, J.; Binder, H.J. Role of Cl channels in Cl-dependent Na/H exchange. Am. J. Physiol. 1999, 276, G73–G78. [Google Scholar]
- Rajendran, V.M.; Geibel, J.; Binder, H.J. Chloride-dependent Na-H exchange.A novel mechanism of sodium transport in colonic crypts. J. Biol. Chem. 1995, 270, 11051–11054. [Google Scholar] [PubMed]
- Ahn, W.; Kim, K.H.; Lee, J.A.; Kim, J.Y.; Choi, J.Y.; Moe, O.W.; Milgram, S.L.; Muallem, S.; Lee, M.G. Regulatory interaction between the cystic fibrosis transmembrane conductance regulator and HCO3- salvage mechanisms in model systems and the mouse pancreatic duct. J. Biol. Chem. 2001, 276, 17236–17243. [Google Scholar]
- Mount, D.B.; Romero, M.F. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch. 2004, 447, 710–721. [Google Scholar]
- Wang, Z.; Petrovic, S.; Mann, E.; Soleimani, M. Identification of an apical Cl(-)/HCO3(-) exchanger in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G573–G579. [Google Scholar]
- Simpson, J.E.; Schweinfest, C.W.; Shull, G.E.; Gawenis, L.R.; Walker, N.M.; Boyle, K.T.; Soleimani, M.; Clarke, L.L. PAT-1 (Slc26a6) is the predominant apical membrane Cl-/HCO3- exchanger in the upper villous epithelium of the murine duodenum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1079–1088. [Google Scholar]
- Xie, Q.; Welch, R.; Mercado, A.; Romero, M.F.; Mount, D.B. Molecular characterization of the murine Slc26a6 anion exchanger: Functional comparison with Slc26a1. Am. J. Physiol. Renal Physiol. 2002, 283, F826–F838. [Google Scholar]
- Ko, S.B.; Shcheynikov, N.; Choi, J.Y.; Luo, X.; Ishibashi, K.; Thomas, P.J.; Kim, J.Y.; Kim, K.H.; Lee, M.G.; Naruse, S.; Muallem, S. A molecular mechanism for aberrant CFTR-dependent HCO(3)(-) transport in cystic fibrosis. EMBO J. 2002, 21, 5662–5672. [Google Scholar]
- Hoglund, P.; Haila, S.; Socha, J.; Tomaszewski, L.; Saarialho-Kere, U.; Karjalainen-Lindsberg, M.L.; Airola, K.; Holmberg, C.; de la Chapelle, A.; Kere, J. Mutations of the Down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nat. Genet. 1996, 14, 316–319. [Google Scholar]
- Makela, S.; Kere, J.; Holmberg, C.; Hoglund, P. SLC26A3 mutations in congenital chloride diarrhea. Hum. Mutat. 2002, 20, 425–438. [Google Scholar]
- Walker, N.M.; Simpson, J.E.; Brazill, J.M.; Gill, R.K.; Dudeja, P.K.; Schweinfest, C.W.; Clarke, L.L. Role of down-regulated in adenoma anion exchanger in HCO3- secretion across murine duodenum. Gastroenterology 2009, 136, 893–901. [Google Scholar]
- Lamprecht, G.; Hsieh, C.J.; Lissner, S.; Nold, L.; Heil, A.; Gaco, V.; Schafer, J.; Turner, J.R.; Gregor, M. Intestinal anion exchanger down-regulated in adenoma (DRA) is inhibited by intracellular calcium. J. Biol. Chem. 2009, 284, 19744–19753. [Google Scholar]
- Jacob, P.; Rossmann, H.; Lamprecht, G.; Kretz, A.; Neff, C.; Lin-Wu, E.; Gregor, M.; Groneberg, D.A.; Kere, J.; Seidler, U. Down-regulated in adenoma mediates apical Cl-/HCO3- exchange in rabbit, rat, and human duodenum. Gastroenterology 2002, 122, 709–724. [Google Scholar]
- Walker, N.M.; Simpson, J.E.; Yen, P.F.; Gill, R.K.; Rigsby, E.V.; Brazill, J.M.; Dudeja, P.K.; Schweinfest, C.W.; Clarke, L.L. Down-regulated in adenoma Cl/HCO3 exchanger couples with Na/H exchanger 3 for NaCl absorption in murine small intestine. Gastroenterology 2008, 135 e1643, 1645–1653. [Google Scholar] [PubMed]
- Charney, A.N.; Egnor, R.W.; Henner, D.; Rashid, H.; Cassai, N.; Sidhu, G.S. Acid-base effects on intestinal Cl- absorption and vesicular trafficking. Am. J. Physiol. Cell Physiol. 2004, 286, C1062–C1070. [Google Scholar]
- Rajendran, V.M.; Black, J.; Ardito, T.A.; Sangan, P.; Alper, S.L.; Schweinfest, C.; Kashgarian, M.; Binder, H.J. Regulation of DRA and AE1 in rat colon by dietary Na depletion. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G931–G942. [Google Scholar]
- Alrefai, W.A.; Tyagi, S.; Nazir, T.M.; Barakat, J.; Anwar, S.S.; Hadjiagapiou, C.; Bavishi, D.; Sahi, J.; Malik, P.; Goldstein, J.; Layden, T.J.; Ramaswamy, K.; Dudeja, P.K. Human intestinal anion exchanger isoforms: Expression, distribution, and membrane localization. Biochim. Biophys. Acta. 2001, 1511, 17–27. [Google Scholar]
- Xu, J.; Barone, S.; Petrovic, S.; Wang, Z.; Seidler, U.; Riederer, B.; Ramaswamy, K.; Dudeja, P.K.; Shull, G.E.; Soleimani, M. Identification of an apical Cl-/HCO3- exchanger in gastric surface mucous and duodenal villus cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G1225–G1234. [Google Scholar]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.D.; Rossier, B.C. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994, 367, 463–467. [Google Scholar]
- Barbry, P.; Hofman, P. Molecular biology of Na+ absorption. Am. J. Physiol. 1997, 273, G571–G585. [Google Scholar]
- Fukushima, K.; Sato, S.; Naito, H.; Funayama, Y.; Haneda, S.; Shibata, C.; Sasaki, I. Comparative study of epithelial gene expression in the small intestine among total proctocolectomized, dietary sodium-depleted, and aldosterone-infused rats. J. Gastrointest. Surg. 2005, 9, 236–244. [Google Scholar]
- Will, P.C.; Cortright, R.N.; Groseclose, R.G.; Hopfer, U. Amiloride-sensitive salt and fluid absorption in small intestine of sodium-depleted rats. Am. J. Physiol. 1985, 248, G133–G141. [Google Scholar]
- Berdiev, B.K.; Qadri, Y.J.; Benos, D.J. Assessment of the CFTR and ENaC association. Mol. Biosyst. 2009, 5, 123–127. [Google Scholar]
- Konig, J.; Schreiber, R.; Voelcker, T.; Mall, M.; Kunzelmann, K. The cystic fibrosis transmembrane conductance regulator (CFTR) inhibits ENaC through an increase in the intracellular Cl- concentration. EMBO Rep. 2001, 2, 1047–1051. [Google Scholar]
- Nagel, G.; Barbry, P.; Chabot, H.; Brochiero, E.; Hartung, K.; Grygorczyk, R. CFTR fails to inhibit the epithelial sodium channel ENaC expressed in Xenopus laevis oocytes. J. Physiol. 2005, 564, 671–682. [Google Scholar]
- Mall, M.; Bleich, M.; Kuehr, J.; Brandis, M.; Greger, R.; Kunzelmann, K. CFTR-mediated inhibition of epithelial Na+ conductance in human colon is defective in cystic fibrosis. Am. J. Physiol. 1999, 277, G709–G716. [Google Scholar]
- Hediger, M.A.; Coady, M.J.; Ikeda, T.S.; Wright, E.M. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature 1987, 330, 379–381. [Google Scholar]
- Wright, E.M.; Hirayama, B.A.; Loo, D.F. Active sugar transport in health and disease. J. Intern. Med. 2007, 261, 32–43. [Google Scholar]
- Chen, X.Z.; Coady, M.J.; Jackson, F.; Berteloot, A.; Lapointe, J.Y. Thermodynamic determination of the Na+: Glucose coupling ratio for the human SGLT1 cotransporter. Biophys. J. 1995, 69, 2405–2414. [Google Scholar]
- Hirschhorn, N.; Kinzie, J.L.; Sachar, D.B.; Northrup, R.S.; Taylor, J.O.; Ahmad, S.Z.; Phillips, R.A. Decrease in net stool output in cholera during intestinal perfusion with glucose-containing solutions. N. Engl. J. Med. 1968, 279, 176–181. [Google Scholar]
- Turk, E.; Zabel, B.; Mundlos, S.; Dyer, J.; Wright, E.M. Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature 1991, 350, 354–356. [Google Scholar]
- Santer, R.; Calado, J. Familial renal glucosuria and SGLT2: From a mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. 2009, 5, 133–141. [Google Scholar]
- Broer, S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol. Rev. 2008, 88, 249–286. [Google Scholar]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar]
- Ameen, N.A.; Ardito, T.; Kashgarian, M.; Marino, C.R. A unique subset of rat and human intestinal villus cells express the cystic fibrosis transmembrane conductance regulator. Gastroenterology 1995, 108, 1016–1023. [Google Scholar]
- Sheppard, D.N.; Welsh, M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999, 79, S23–S45. [Google Scholar]
- Chao, A.C.; de Sauvage, F.J.; Dong, Y.J.; Wagner, J.A.; Goeddel, D.V.; Gardner, P. Activation of intestinal CFTR Cl- channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J. 1994, 13, 1065–1072. [Google Scholar]
- Cheng, S.H.; Rich, D.P.; Marshall, J.; Gregory, R.J.; Welsh, M.J.; Smith, A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 1991, 66, 1027–1036. [Google Scholar]
- Jia, Y.; Mathews, C.J.; Hanrahan, J.W. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J. Biol. Chem. 1997, 272, 4978–4984. [Google Scholar]
- Tabcharani, J.A.; Chang, X.B.; Riordan, J.R.; Hanrahan, J.W. Phosphorylation-regulated Cl- channel in CHO cells stably expressing the cystic fibrosis gene. Nature 1991, 352, 628–631. [Google Scholar]
- Vaandrager, A.B.; Tilly, B.C.; Smolenski, A.; Schneider-Rasp, S.; Bot, A.G.; Edixhoven, M.; Scholte, B.J.; Jarchau, T.; Walter, U.; Lohmann, S.M.; Poller, W.C.; de Jonge, H.R. cGMP stimulation of cystic fibrosis transmembrane conductance regulator Cl- channels co-expressed with cGMP-dependent protein kinase type II but not type Ibeta. J. Biol. Chem. 1997, 272, 4195–4200. [Google Scholar]
- Wagner, J.A.; Cozens, A.L.; Schulman, H.; Gruenert, D.C.; Stryer, L.; Gardner, P. Activation of chloride channels in normal and cystic fibrosis airway epithelial cells by multifunctional calcium/calmodulin-dependent protein kinase. Nature 1991, 349, 793–796. [Google Scholar]
- Fischer, H.; Machen, T.E. The tyrosine kinase p60c-src regulates the fast gate of the cystic fibrosis transmembrane conductance regulator chloride channel. Biophys. J. 1996, 71, 3073–3082. [Google Scholar]
- Howard, M.; Jiang, X.; Stolz, D.B.; Hill, W.G.; Johnson, J.A.; Watkins, S.C.; Frizzell, R.A.; Bruton, C.M.; Robbins, P.D.; Weisz, O.A. Forskolin-induced apical membrane insertion of virally expressed, epitope-tagged CFTR in polarized MDCK cells. Am. J. Physiol. Cell Physiol. 2000, 279, C375–C382. [Google Scholar]
- Lewarchik, C.M.; Peters, K.W.; Qi, J.; Frizzell, R.A. Regulation of CFTR trafficking by its R domain. J. Biol. Chem. 2008, 283, 28401–28412. [Google Scholar]
- Bradbury, N.A.; Jilling, T.; Berta, G.; Sorscher, E.J.; Bridges, R.J.; Kirk, K.L. Regulation of plasma membrane recycling by CFTR. Science 1992, 256, 530–532. [Google Scholar]
- Bertrand, C.A.; Frizzell, R.A. The role of regulated CFTR trafficking in epithelial secretion. Am. J. Physiol. Cell Physiol. 2003, 285, C1–C18. [Google Scholar]
- Eggermont, E. Gastrointestinal manifestations in cystic fibrosis. Eur. J. Gastroenterol. Hepatol. 1996, 8, 731–738. [Google Scholar]
- Grubb, B.R.; Gabriel, S.E. Intestinal physiology and pathology in gene-targeted mouse models of cystic fibrosis. Am. J. Physiol. 1997, 273, G258–266. [Google Scholar]
- Clarke, L.L.; Grubb, B.R.; Gabriel, S.E.; Smithies, O.; Koller, B.H.; Boucher, R.C. Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science 1992, 257, 1125–1128. [Google Scholar]
- Ratcliff, R.; Evans, M.J.; Cuthbert, A.W.; MacVinish, L.J.; Foster, D.; Anderson, J.R.; Colledge, W.H. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat. Genet. 1993, 4, 35–41. [Google Scholar]
- Bronsveld, I.; Mekus, F.; Bijman, J.; Ballmann, M.; Greipel, J.; Hundrieser, J.; Halley, D.J.; Laabs, U.; Busche, R.; De Jonge, H.R.; Tummler, B.; Veeze, H.J. Residual chloride secretion in intestinal tissue of deltaF508 homozygous twins and siblings with cystic fibrosis.The European CF Twin and Sibling Study Consortium. Gastroenterology 2000, 119, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Barrett, K.E.; Smitham, J.; Traynor-Kaplan, A.; Uribe, J.M. Inhibition of Ca(2+)-dependent Cl- secretion in T84 cells: Membrane target(s) of inhibition is agonist specific. Am. J. Physiol. 1998, 274, C958–C965. [Google Scholar]
- Kachintorn, U.; Vajanaphanich, M.; Traynor-Kaplan, A.E.; Dharmsathaphorn, K.; Barrett, K.E. Activation by calcium alone of chloride secretion in T84 epithelial cells. Br. J. Pharmacol. 1993, 109, 510–517. [Google Scholar]
- McEwan, G.T.; Hirst, B.H.; Simmons, N.L. Carbachol stimulates Cl- secretion via activation of two distinct apical Cl- pathways in cultured human T84 intestinal epithelial monolayers. Biochim. Biophys. Acta 1994, 1220, 241–247. [Google Scholar]
- Wagner, J.A.; McDonald, T.V.; Nghiem, P.T.; Lowe, A.W.; Schulman, H.; Gruenert, D.C.; Stryer, L.; Gardner, P. Antisense oligodeoxynucleotides to the cystic fibrosis transmembrane conductance regulator inhibit cAMP-activated but not calcium-activated chloride currents. Proc. Natl. Acad. Sci. USA 1992, 89, 6785–6789. [Google Scholar]
- Berschneider, H.M.; Knowles, M.R.; Azizkhan, R.G.; Boucher, R.C.; Tobey, N.A.; Orlando, R.C.; Powell, D.W. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988, 2, 2625–2629. [Google Scholar]
- Hardcastle, J.; Hardcastle, P.T.; Taylor, C.J.; Goldhill, J. Failure of cholinergic stimulation to induce a secretory response from the rectal mucosa in cystic fibrosis. Gut 1991, 32, 1035–1039. [Google Scholar]
- Taylor, C.J.; Baxter, P.S.; Hardcastle, J.; Hardcastle, P.T. Failure to induce secretion in jejunal biopsies from children with cystic fibrosis. Gut 1988, 29, 957–962. [Google Scholar]
- Rozmahel, R.; Wilschanski, M.; Matin, A.; Plyte, S.; Oliver, M.; Auerbach, W.; Moore, A.; Forstner, J.; Durie, P.; Nadeau, J.; Bear, C.; Tsui, L.C. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat. Genet. 1996, 12, 280–287. [Google Scholar]
- Gibson, A.; Lewis, A.P.; Affleck, K.; Aitken, A.J.; Meldrum, E.; Thompson, N. hCLCA1 and mCLCA3 are secreted non-integral membrane proteins and therefore are not ion channels. J. Biol. Chem. 2005, 280, 27205–27212. [Google Scholar]
- Suzuki, M.; Mizuno, A. A novel human Cl(-) channel family related to Drosophila flightless locus. J. Biol. Chem. 2004, 279, 22461–22468. [Google Scholar]
- Pifferi, S.; Pascarella, G.; Boccaccio, A.; Mazzatenta, A.; Gustincich, S.; Menini, A.; Zucchelli, S. Bestrophin-2 is a candidate calcium-activated chloride channel involved in olfactory transduction. Proc. Natl. Acad. Sci. USA 2006, 103, 12929–12934. [Google Scholar]
- Suzuki, M. The Drosophila tweety family: Molecular candidates for large-conductance Ca2+-activated Cl- channels. Exp. Physiol. 2006, 91, 141–147. [Google Scholar]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; Raouf, R.; Shin, Y.K.; Oh, U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar]
- Ousingsawat, J.; Martins, J.R.; Schreiber, R.; Rock, J.R.; Harfe, B.D.; Kunzelmann, K. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J. Biol. Chem. 2009, 284, 28698–28703. [Google Scholar]
- Gyomorey, K.; Garami, E.; Galley, K.; Rommens, J.M.; Bear, C.E. Non-CFTR chloride channels likely contribute to secretion in the murine small intestine. Pflugers Arch. 2001, 443 (Suppl. 1), S103–S106. [Google Scholar] [PubMed]
- Gyomorey, K.; Yeger, H.; Ackerley, C.; Garami, E.; Bear, C.E. Expression of the chloride channel ClC-2 in the murine small intestine epithelium. Am. J. Physiol. Cell Physiol. 2000, 279, C1787–C1794. [Google Scholar]
- Zdebik, A.A.; Cuffe, J.E.; Bertog, M.; Korbmacher, C.; Jentsch, T.J. Additional disruption of the ClC-2 Cl(-) channel does not exacerbate the cystic fibrosis phenotype of cystic fibrosis transmembrane conductance regulator mouse models. J. Biol. Chem. 2004, 279, 22276–22283. [Google Scholar]
- Lipecka, J.; Bali, M.; Thomas, A.; Fanen, P.; Edelman, A.; Fritsch, J. Distribution of ClC-2 chloride channel in rat and human epithelial tissues. Am. J. Physiol. Cell Physiol. 2002, 282, C805–C816. [Google Scholar]
- Bijvelds, M.J.; Bot, A.G.; Escher, J.C.; De Jonge, H.R. Activation of intestinal Cl- secretion by lubiprostone requires the cystic fibrosis transmembrane conductance regulator. Gastroenterology 2009, 137, 976–985. [Google Scholar]
- Wenneras, C.; Erling, V. Prevalence of enterotoxigenic Escherichia coli-associated diarrhoea and carrier state in the developing world. J. Health Popul. Nutr. 2004, 22, 370–382. [Google Scholar]
- Gascon, J.; Vargas, M.; Quinto, L.; Corachan, M.; Jimenez de Anta, M.T.; Vila, J. Enteroaggregative Escherichia coli strains as a cause of traveler's diarrhea: A case-control study. J. Infect. Dis. 1998, 177, 1409–1412. [Google Scholar]
- Qadri, F.; Svennerholm, A.M.; Faruque, A.S.; Sack, R.B. Enterotoxigenic Escherichia coli in developing countries: Epidemiology, microbiology, clinical features, treatment, and prevention. Clin. Microbiol. Rev. 2005, 18, 465–483. [Google Scholar]
- Jiang, Z.D.; Lowe, B.; Verenkar, M.P.; Ashley, D.; Steffen, R.; Tornieporth, N.; von Sonnenburg, F.; Waiyaki, P.; DuPont, H.L. Prevalence of enteric pathogens among international travelers with diarrhea acquired in Kenya (Mombasa), India (Goa), or Jamaica (Montego Bay). J. Infect. Dis. 2002, 185, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar]
- Lockman, H.A.; Galen, J.E.; Kaper, J.B. Vibrio cholerae enterotoxin genes: Nucleotide sequence analysis of DNA encoding ADP-ribosyltransferase. J. Bacteriol. 1984, 159, 1086–1089. [Google Scholar]
- Zhang, R.G.; Scott, D.L.; Westbrook, M.L.; Nance, S.; Spangler, B.D.; Shipley, G.G.; Westbrook, E.M. The three-dimensional crystal structure of cholera toxin. J. Mol. Biol. 1995, 251, 563–573. [Google Scholar]
- Sixma, T.K.; Pronk, S.E.; Kalk, K.H.; Wartna, E.S.; van Zanten, B.A.; Witholt, B.; Hol, W.G. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E coli. Nature 1991, 351, 371–377. [Google Scholar]
- Sixma, T.K.; Kalk, K.H.; van Zanten, B.A.; Dauter, Z.; Kingma, J.; Witholt, B.; Hol, W.G. Refined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J. Mol. Biol. 1993, 230, 890–918. [Google Scholar]
- Sears, C.L.; Kaper, J.B. Enteric bacterial toxins: Mechanisms of action and linkage to intestinal secretion. Microbiol. Rev. 1996, 60, 167–215. [Google Scholar]
- Aimoto, S.; Takao, T.; Shimonishi, Y.; Hara, S.; Takeda, T.; Takeda, Y.; Miwatani, T. Amino-acid sequence of a heat-stable enterotoxin produced by human enterotoxigenic Escherichia coli. Eur. J. Biochem. 1982, 129, 257–263. [Google Scholar]
- Ozaki, H.; Sato, T.; Kubota, H.; Hata, Y.; Katsube, Y.; Shimonishi, Y. Molecular structure of the toxin domain of heat-stable enterotoxin produced by a pathogenic strain of Escherichia coli. A putative binding site for a binding protein on rat intestinal epithelial cell membranes. J. Biol. Chem. 1991, 266, 5934–5941. [Google Scholar] [PubMed]
- Sukumar, M.; Rizo, J.; Wall, M.; Dreyfus, L.A.; Kupersztoch, Y.M.; Gierasch, L.M. The structure of Escherichia coli heat-stable enterotoxin b by nuclear magnetic resonance and circular dichroism. Protein Sci. 1995, 4, 1718–1729. [Google Scholar]
- Thompson, M.R.; Giannella, R.A. Revised amino acid sequence for a heat-stable enterotoxin produced by an Escherichia coli strain (18D) that is pathogenic for humans. Infect. Immun. 1985, 47, 834–836. [Google Scholar]
- Dreyfus, L.A.; Frantz, J.C.; Robertson, D.C. Chemical properties of heat-stable enterotoxins produced by enterotoxigenic Escherichia coli of different host origins. Infect. Immun. 1983, 42, 539–548. [Google Scholar]
- Gariepy, J.; Judd, A.K.; Schoolnik, G.K. Importance of disulfide bridges in the structure and activity of Escherichia coli enterotoxin ST1b. Proc. Natl. Acad. Sci. USA 1987, 84, 8907–8911. [Google Scholar]
- Okamoto, K.; Yukitake, J.; Kawamoto, Y.; Miyama, A. Substitutions of cysteine residues of Escherichia coli heat-stable enterotoxin by oligonucleotide-directed mutagenesis. Infect. Immun. 1987, 55, 2121–2125. [Google Scholar]
- Yoshimura, S.; Ikemura, H.; Watanabe, H.; Aimoto, S.; Shimonishi, Y.; Hara, S.; Takeda, T.; Miwatani, T.; Takeda, Y. Essential structure for full enterotoxigenic activity of heat-stable enterotoxin produced by enterotoxigenic Escherichia coli. FEBS Lett. 1985, 181, 138–142. [Google Scholar]
- Takao, T.; Shimonishi, Y.; Kobayashi, M.; Nishimura, O.; Arita, M.; Takeda, T.; Honda, T.; Miwatani, T. Amino acid sequence of heat-stable enterotoxin produced by Vibrio cholerae non-01. FEBS Lett. 1985, 193, 250–254. [Google Scholar]
- Takao, T.; Tominaga, N.; Yoshimura, S.; Shimonishi, Y.; Hara, S.; Inoue, T.; Miyama, A. Isolation, primary structure and synthesis of heat-stable enterotoxin produced by Yersinia enterocolitica. Eur. J. Biochem. 1985, 152, 199–206. [Google Scholar]
- Yoshino, K.; Miyachi, M.; Takao, T.; Bag, P.K.; Huang, X.; Nair, G.B.; Takeda, T.; Shimonishi, Y. Purification and sequence determination of heat-stable enterotoxin elaborated by a cholera toxin-producing strain of Vibrio cholerae O1. FEBS Lett. 1993, 326, 83–86. [Google Scholar]
- Cohen, M.B.; Guarino, A.; Shukla, R.; Giannella, R.A. Age-related differences in receptors for Escherichia coli heat-stable enterotoxin in the small and large intestine of children. Gastroenterology 1988, 94, 367–373. [Google Scholar]
- Schulz, S.; Green, C.K.; Yuen, P.S.; Garbers, D.L. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell 1990, 63, 941–948. [Google Scholar]
- de Sauvage, F.J.; Camerato, T.R.; Goeddel, D.V. Primary structure and functional expression of the human receptor for Escherichia coli heat-stable enterotoxin. J. Biol. Chem. 1991, 266, 17912–17918. [Google Scholar]
- Field, M.; Graf, L.H., Jr.; Laird, W.J.; Smith, P.L. Heat-stable enterotoxin of Escherichia coli: In vitro effects on guanylate cyclase activity, cyclic GMP concentration, and ion transport in small intestine. Proc. Natl. Acad. Sci. USA 1978, 75, 2800–2804. [Google Scholar]
- Pfeifer, A.; Aszodi, A.; Seidler, U.; Ruth, P.; Hofmann, F.; Fassler, R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science 1996, 274, 2082–2086. [Google Scholar]
- Cha, B.; Kim, J.H.; Hut, H.; Hogema, B.M.; Nadarja, J.; Zizak, M.; Cavet, M.; Lee-Kwon, W.; Lohmann, S.M.; Smolenski, A.; Tse, C.M.; Yun, C.; de Jonge, H.R.; Donowitz, M. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J. Biol. Chem. 2005, 280, 16642–16650. [Google Scholar]
- French, P.J.; Bijman, J.; Edixhoven, M.; Vaandrager, A.B.; Scholte, B.J.; Lohmann, S.M.; Nairn, A.C.; de Jonge, H.R. Isotype-specific activation of cystic fibrosis transmembrane conductance regulator-chloride channels by cGMP-dependent protein kinase II. J. Biol. Chem. 1995, 270, 26626–26631. [Google Scholar]
- Sullivan, S.K.; Agellon, L.B.; Schick, R. Identification and partial characterization of a domain in CFTR that may bind cyclic nucleotides directly. Curr. Biol. 1995, 5, 1159–1167. [Google Scholar]
- Cuthbert, A.W.; Hickman, M.E.; MacVinish, L.J.; Evans, M.J.; Colledge, W.H.; Ratcliff, R.; Seale, P.W.; Humphrey, P.P. Chloride secretion in response to guanylin in colonic epithelial from normal and transgenic cystic fibrosis mice. Br. J. Pharmacol. 1994, 112, 31–36. [Google Scholar]
- Goldstein, J.L.; Sahi, J.; Bhuva, M.; Layden, T.J.; Rao, M.C. Escherichia coli heat-stable enterotoxin-mediated colonic Cl- secretion is absent in cystic fibrosis. Gastroenterology 1994, 107, 950–956. [Google Scholar]
- Wada, A.; Hirayama, T.; Kitaura, H.; Fujisawa, J.; Hasegawa, M.; Hidaka, Y.; Shimonishi, Y. Identification of ligand recognition sites in heat-stable enterotoxin receptor, membrane-associated guanylyl cyclase C by site-directed mutational analysis. Infect. Immun. 1996, 64, 5144–5150. [Google Scholar]
- Hasegawa, M.; Hidaka, Y.; Matsumoto, Y.; Sanni, T.; Shimonishi, Y. Determination of the binding site on the extracellular domain of guanylyl cyclase C to heat-stable enterotoxin. J. Biol. Chem. 1999, 274, 31713–31718. [Google Scholar]
- Carpick, B.W.; Gariepy, J. Structural characterization of functionally important regions of the Escherichia coli heat-stable enterotoxin STIb. Biochemistry 1991, 30, 4803–4809. [Google Scholar]
- Hasegawa, M.; Shimonishi, Y. Recognition and signal transduction mechanism of Escherichia coli heat-stable enterotoxin and its receptor, guanylate cyclase C. J. Pept. Res. 2005, 65, 261–271. [Google Scholar]
- Waldman, S.A.; O'Hanley, P. Influence of a glycine or proline substitution on the functional properties of a 14-amino-acid analog of Escherichia coli heat-stable enterotoxin. Infect. Immun. 1989, 57, 2420–2424. [Google Scholar]
- Yamasaki, S.; Sato, T.; Hidaka, Y.; Ozaki, H.; Ito, H.; Hirayama, T.; Takeda, Y.; Sugimura, T.; Tai, A.; Shimonishi, Y. Structure-activity relationship of Escherichia coli heat-stable enterotoxin: Role of Ala residue at position 14 in toxin-receptor interaction. Bull Chem. Soc. Jpn. 1990, 63, 2063–2070. [Google Scholar]
- Mann, E.A.; Jump, M.L.; Wu, J.; Yee, E.; Giannella, R.A. Mice lacking the guanylyl cyclase C receptor are resistant to STa-induced intestinal secretion. Biochem. Biophys. Res. Commun. 1997, 239, 463–466. [Google Scholar]
- Schulz, S.; Lopez, M.J.; Kuhn, M.; Garbers, D.L. Disruption of the guanylyl cyclase-C gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J. Clin. Invest. 1997, 100, 1590–1595. [Google Scholar]
- Currie, M.G.; Fok, K.F.; Kato, J.; Moore, R.J.; Hamra, F.K.; Duffin, K.L.; Smith, C.E. Guanylin: An endogenous activator of intestinal guanylate cyclase. Proc. Natl. Acad. Sci. USA 1992, 89, 947–951. [Google Scholar]
- de Sauvage, F.J.; Keshav, S.; Kuang, W.J.; Gillett, N.; Henzel, W.; Goeddel, D.V. Precursor structure, expression, and tissue distribution of human guanylin. Proc. Natl. Acad. Sci. USA 1992, 89, 9089–9093. [Google Scholar]
- Nakazato, M. Guanylin family: New intestinal peptides regulating electrolyte and water homeostasis. J. Gastroenterol. 2001, 36, 219–225. [Google Scholar]
- Lorenz, J.N.; Nieman, M.; Sabo, J.; Sanford, L.P.; Hawkins, J.A.; Elitsur, N.; Gawenis, L.R.; Clarke, L.L.; Cohen, M.B. Uroguanylin knockout mice have increased blood pressure and impaired natriuretic response to enteral NaCl load. J. Clin. Invest. 2003, 112, 1244–1254. [Google Scholar]
- Joo, N.S.; London, R.M.; Kim, H.D.; Forte, L.R.; Clarke, L.L. Regulation of intestinal Cl- and HCO3-secretion by uroguanylin. Am. J. Physiol. 1998, 274, G633–644. [Google Scholar]
- Skelton, N.J.; Garcia, K.C.; Goeddel, D.V.; Quan, C.; Burnier, J.P. Determination of the solution structure of the peptide hormone guanylin: Observation of a novel form of topological stereoisomerism. Biochemistry 1994, 33, 13581–13592. [Google Scholar]
- van den Akker, F. Structural insights into the ligand binding domains of membrane bound guanylyl cyclases and natriuretic peptide receptors. J. Mol. Biol. 2001, 311, 923–937. [Google Scholar]
- Andresen, V.; Camilleri, M.; Busciglio, I.A.; Grudell, A.; Burton, D.; McKinzie, S.; Foxx-Orenstein, A.; Kurtz, C.B.; Sharma, V.; Johnston, J.M.; Currie, M.G.; Zinsmeister, A.R. Effect of 5 days linaclotide on transit and bowel function in females with constipation-predominant irritable bowel syndrome. Gastroenterology 2007, 133, 761–768. [Google Scholar]
- Lembo, A.J.; Kurtz, C.B.; Macdougall, J.E.; Lavins, B.J.; Currie, M.G.; Fitch, D.A.; Jeglinski, B.I.; Johnston, J.M. Efficacy of linaclotide for patients with chronic constipation. Gastroenterology 2010, 138, 886–895. [Google Scholar]
- Hakki, S.; Robertson, D.C.; Waldman, S.A. A 56 kDa binding protein for Escherichia coli heat-stable enterotoxin isolated from the cytoskeleton of rat intestinal membrane does not possess guanylate cyclase activity. Biochim. Biophys. Acta. 1993, 1152, 1–8. [Google Scholar]
- Hugues, M.; Crane, M.; Hakki, S.; O'Hanley, P.; Waldman, S.A. Identification and characterization of a new family of high-affinity receptors for Escherichia coli heat-stable enterotoxin in rat intestinal membranes. Biochemistry 1991, 30, 10738–10745. [Google Scholar]
- Ivens, K.; Gazzano, H.; O'Hanley, P.; Waldman, S.A. Heterogeneity of intestinal receptors for Escherichia coli heat-stable enterotoxin. Infect. Immun. 1990, 58, 1817–1820. [Google Scholar]
- Mann, E.A.; Cohen, M.B.; Giannella, R.A. Comparison of receptors for Escherichia coli heat-stable enterotoxin: Novel receptor present in IEC-6 cells. Am. J. Physiol. 1993, 264, G172–G178. [Google Scholar]
- Sellers, Z.M.; Mann, E.; Smith, A.; Ko, K.H.; Giannella, R.; Cohen, M.B.; Barrett, K.E.; Dong, H. Heat-stable enterotoxin of Escherichia coli (STa) can stimulate duodenal HCO3(-) secretion via a novel GC-C- and CFTR-independent pathway. FASEB J. 2008, 22, 1306–1316. [Google Scholar]
- Sellers, Z.M.; Childs, D.; Chow, J.Y.; Smith, A.J.; Hogan, D.L.; Isenberg, J.I.; Dong, H.; Barrett, K.E.; Pratha, V.S. Heat-stable enterotoxin of Escherichia coli stimulates a non-CFTR-mediated duodenal bicarbonate secretory pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G654–663. [Google Scholar]
- Seidler, U.; Blumenstein, I.; Kretz, A.; Viellard-Baron, D.; Rossmann, H.; Colledge, W.H.; Evans, M.; Ratcliff, R.; Gregor, M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca(2+)-dependent HCO3- secretion. J. Physiol. 1997, 505 (Pt. 2), 411–423. [Google Scholar] [PubMed]
- Kots, A.Y.; Choi, B.K.; Estrella-Jimenez, M.E.; Warren, C.A.; Gilbertson, S.R.; Guerrant, R.L.; Murad, F. Pyridopyrimidine derivatives as inhibitors of cyclic nucleotide synthesis: Application for treatment of diarrhea. Proc. Natl. Acad. Sci. USA 2008, 105, 8440–8445. [Google Scholar]
- Tanifum, E.A.; Kots, A.Y.; Choi, B.K.; Murad, F.; Gilbertson, S.R. Novel pyridopyrimidine derivatives as inhibitors of stable toxin a (STa) induced cGMP synthesis. Bioorg. Med. Chem. Lett. 2009, 19, 3067–3071. [Google Scholar]
- Parkinson, S.J.; Alekseev, A.E.; Gomez, L.A.; Wagner, F.; Terzic, A.; Waldman, S.A. Interruption of Escherichia coli heat-stable enterotoxin-induced guanylyl cyclase signaling and associated chloride current in human intestinal cells by 2-chloroadenosine. J. Biol. Chem. 1997, 272, 754–758. [Google Scholar]
- Zhang, W.; Mannan, I.; Schulz, S.; Parkinson, S.J.; Alekseev, A.E.; Gomez, L.A.; Terzic, A.; Waldman, S.A. Interruption of transmembrane signaling as a novel antisecretory strategy to treat enterotoxigenic diarrhea. FASEB J. 1999, 13, 913–922. [Google Scholar]
- Jaleel, M.; Shenoy, A.R.; Visweswariah, S.S. Tyrphostins are inhibitors of guanylyl and adenylyl cyclases. Biochemistry 2004, 43, 8247–8255. [Google Scholar]
- Thiagarajah, J.R.; Broadbent, T.; Hsieh, E.; Verkman, A.S. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology 2004, 126, 511–519. [Google Scholar]
- Bovee-Oudenhoven, I.M.; Lettink-Wissink, M.L.; Van Doesburg, W.; Witteman, B.J.; Van Der Meer, R. Diarrhea caused by enterotoxigenic Escherichia coli infection of humans is inhibited by dietary calcium. Gastroenterology 2003, 125, 469–476. [Google Scholar]
- Cholera: Global surveillance summary, 2008. Wkly Epidemiol. Rec. 2009, 84, 309–324. [PubMed]
- Sack, D.A.; Sack, R.B.; Nair, G.B.; Siddique, A.K. Cholera. Lancet 2004, 363, 223–233. [Google Scholar]
- Wolf, A.A.; Jobling, M.G.; Saslowsky, D.E.; Kern, E.; Drake, K.R.; Kenworthy, A.K.; Holmes, R.K.; Lencer, W.I. Attenuated endocytosis and toxicity of a mutant cholera toxin with decreased ability to cluster ganglioside GM1 molecules. Infect. Immun. 2008, 76, 1476–1484. [Google Scholar]
- Chinnapen, D.J.; Chinnapen, H.; Saslowsky, D.; Lencer, W.I. Rafting with cholera toxin: Endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007, 266, 129–137. [Google Scholar]
- Lencer, W.I.; Saslowsky, D. Raft trafficking of AB5 subunit bacterial toxins. Biochim. Biophys. Acta. 2005, 1746, 314–321. [Google Scholar]
- Badizadegan, K.; Wheeler, H.E.; Fujinaga, Y.; Lencer, W.I. Trafficking of cholera toxin-ganglioside GM1 complex into Golgi and induction of toxicity depend on actin cytoskeleton. Am. J. Physiol. Cell Physiol. 2004, 287, C1453–C1462. [Google Scholar]
- Field, M.; Fromm, D.; al-Awqati, Q.; Greenough, W.B., 3rd. Effect of cholera enterotoxin on ion transport across isolated ileal mucosa. J. Clin. Invest. 1972, 51, 796–804. [Google Scholar]
- Sharp, G.W.; Hynie, S. Stimulation of intestinal adenyl cyclase by cholera toxin. Nature 1971, 229, 266–269. [Google Scholar]
- Cassel, D.; Selinger, Z. Mechanism of adenylate cyclase activation by cholera toxin: Inhibition of GTP hydrolysis at the regulatory site. Proc. Natl. Acad. Sci. USA 1977, 74, 3307–3311. [Google Scholar]
- Freissmuth, M.; Gilman, A.G. Mutations of GS alpha designed to alter the reactivity of the protein with bacterial toxins. Substitutions at ARG187 result in loss of GTPase activity. J. Biol. Chem. 1989, 264, 21907–21914. [Google Scholar] [PubMed]
- Van Dop, C.; Tsubokawa, M.; Bourne, H.R.; Ramachandran, J. Amino acid sequence of retinal transducin at the site ADP-ribosylated by cholera toxin. J. Biol. Chem. 1984, 259, 696–698. [Google Scholar]
- Kato, J.; Zhu, J.; Liu, C.; Moss, J. Enhanced sensitivity to cholera toxin in ADP-ribosylarginine hydrolase-deficient mice. Mol. Cell Biol. 2007, 27, 5534–5543. [Google Scholar]
- Sundaram, U.; Knickelbein, R.G.; Dobbins, J.W. Mechanism of intestinal secretion: Effect of cyclic AMP on rabbit ileal crypt and villus cells. Proc. Natl. Acad. Sci. USA 1991, 88, 6249–6253. [Google Scholar]
- Subramanya, S.B.; Rajendran, V.M.; Srinivasan, P.; Nanda Kumar, N.S.; Ramakrishna, B.S.; Binder, H.J. Differential regulation of cholera toxin-inhibited Na-H exchange isoforms by butyrate in rat ileum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G857–G863. [Google Scholar]
- Bearcroft, C.P.; Perrett, D.; Farthing, M.J. 5-hydroxytryptamine release into human jejunum by cholera toxin. Gut 1996, 39, 528–531. [Google Scholar]
- Beubler, E.; Kollar, G.; Saria, A.; Bukhave, K.; Rask-Madsen, J. Involvement of 5-hydroxytryptamine, prostaglandin E2, and cyclic adenosine monophosphate in cholera toxin-induced fluid secretion in the small intestine of the rat in vivo. Gastroenterology 1989, 96, 368–376. [Google Scholar] [PubMed]
- Sundaram, U.; Knickelbein, R.G.; Dobbins, J.W. Mechanism of intestinal secretion. Effect of serotonin on rabbit ileal crypt and villus cells. J. Clin. Invest. 1991, 87, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Turvill, J.L.; Connor, P.; Farthing, M.J. The inhibition of cholera toxin-induced 5-HT release by the 5-HT(3) receptor antagonist, granisetron, in the rat. Br. J. Pharmacol. 2000, 130, 1031–1036. [Google Scholar]
- McGee, D.W.; Elson, C.O.; McGhee, J.R. Enhancing effect of cholera toxin on interleukin-6 secretion by IEC-6 intestinal epithelial cells: Mode of action and augmenting effect of inflammatory cytokines. Infect. Immun. 1993, 61, 4637–4644. [Google Scholar]
- McGee, D.W.; Beagley, K.W.; Aicher, W.K.; McGhee, J.R. The regulation of IL-6 secretion from IEC-6 intestinal epithelial cells by cytokines and mucosally important antigens. Adv. Exp. Med. Biol. 1995, 371A, 229–232. [Google Scholar]
- Flach, C.F.; Lange, S.; Jennische, E.; Lonnroth, I.; Holmgren, J. Cholera toxin induces a transient depletion of CD8+ intraepithelial lymphocytes in the rat small intestine as detected by microarray and immunohistochemistry. Infect. Immun. 2005, 73, 5595–5602. [Google Scholar]
- Elson, C.O.; Holland, S.P.; Dertzbaugh, M.T.; Cuff, C.F.; Anderson, A.O. Morphologic and functional alterations of mucosal T cells by cholera toxin and its B subunit. J. Immunol. 1995, 154, 1032–1040. [Google Scholar]
- Flach, C.F.; Qadri, F.; Bhuiyan, T.R.; Alam, N.H.; Jennische, E.; Lonnroth, I.; Holmgren, J. Broad up-regulation of innate defense factors during acute cholera. Infect. Immun. 2007, 75, 2343–2350. [Google Scholar]
- Peterson, J.W.; Ochoa, L.G. Role of prostaglandins and cAMP in the secretory effects of cholera toxin. Science 1989, 245, 857–859. [Google Scholar]
- Triadafilopoulos, G.; Pothoulakis, C.; Weiss, R.; Giampaolo, C.; Lamont, J.T. Comparative study of Clostridium difficile toxin A and cholera toxin in rabbit ileum. Gastroenterology 1989, 97, 1186–1192. [Google Scholar]
- Peterson, J.W.; Whipp, S.C. Comparison of the mechanisms of action of cholera toxin and the heat-stable enterotoxins of Escherichia coli. Infect. Immun. 1995, 63, 1452–1461. [Google Scholar]
- Hudson, N.; Hindi, S.E.; Wilson, D.E.; Poppe, L. Prostaglandin E in cholera toxin-induced intestinal secretion. Lack of an intermediary role. Am. J. Dig. Dis. 1975, 20, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Gessell-Lee, D.L.; Popov, V.L.; Boldogh, I.; Olano, J.P.; Peterson, J.W. Role of cyclooxygenase enzymes in a murine model of experimental cholera. Infect. Immun. 2003, 71, 6234–6242. [Google Scholar]
- Beubler, E.; Schuligoi, R.; Chopra, A.K.; Ribardo, D.A.; Peskar, B.A. Cholera toxin induces prostaglandin synthesis via post-transcriptional activation of cyclooxygenase-2 in the rat jejunum. J. Pharmacol. Exp. Ther. 2001, 297, 940–945. [Google Scholar]
- Peterson, J.W.; Lu, Y.; Duncan, S.; Cantu, J.; Chopra, A.K. Interactions of intestinal mediators in the mode of action of cholera toxin. J. Med. Microbiol. 1994, 41, 3–9. [Google Scholar]
- Hamabata, T.; Liu, C.; Takeda, Y. Positive and negative regulation of water channel aquaporins in human small intestine by cholera toxin. Microb. Pathog. 2002, 32, 273–277. [Google Scholar]
- Calamita, G.; Mazzone, A.; Bizzoca, A.; Cavalier, A.; Cassano, G.; Thomas, D.; Svelto, M. Expression and immunolocalization of the aquaporin-8 water channel in rat gastrointestinal tract. Eur. J. Cell Biol. 2001, 80, 711–719. [Google Scholar]
- Garcia, F.; Kierbel, A.; Larocca, M.C.; Gradilone, S.A.; Splinter, P.; LaRusso, N.F.; Marinelli, R.A. The water channel aquaporin-8 is mainly intracellular in rat hepatocytes, and its plasma membrane insertion is stimulated by cyclic AMP. J. Biol. Chem. 2001, 276, 12147–12152. [Google Scholar]
- Flach, C.F.; Lange, S.; Jennische, E.; Lonnroth, I. Cholera toxin induces expression of ion channels and carriers in rat small intestinal mucosa. FEBS Lett. 2004, 561, 122–126. [Google Scholar]
- Flach, C.F.; Qadri, F.; Bhuiyan, T.R.; Alam, N.H.; Jennische, E.; Holmgren, J.; Lonnroth, I. Differential expression of intestinal membrane transporters in cholera patients. FEBS Lett. 2007, 581, 3183–3188. [Google Scholar]
- Gabriel, S.E.; Brigman, K.N.; Koller, B.H.; Boucher, R.C.; Stutts, M.J. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 1994, 266, 107–109. [Google Scholar]
- Ma, T.; Thiagarajah, J.R.; Yang, H.; Sonawane, N.D.; Folli, C.; Galietta, L.J.; Verkman, A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 2002, 110, 1651–1658. [Google Scholar]
- Gabriel, S.E.; Davenport, S.E.; Steagall, R.J.; Vimal, V.; Carlson, T.; Rozhon, E.J. A novel plant-derived inhibitor of cAMP-mediated fluid and chloride secretion. Am. J. Physiol. 1999, 276, G58–G63. [Google Scholar]
- Fischer, H.; Machen, T.E.; Widdicombe, J.H.; Carlson, T.J.; King, S.R.; Chow, J.W.; Illek, B. A novel extract SB-300 from the stem bark latex of Croton lechleri inhibits CFTR-mediated chloride secretion in human colonic epithelial cells. J. Ethnopharmacol. 2004, 93, 351–357. [Google Scholar]
- Muanprasat, C.; Sonawane, N.D.; Salinas, D.; Taddei, A.; Galietta, L.J.; Verkman, A.S. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: Mechanism, structure-activity analysis, and in vivo efficacy. J. Gen. Physiol. 2004, 124, 125–137. [Google Scholar]
- Mahalanabis, D.; Choudhuri, A.B.; Bagchi, N.G.; Bhattacharya, A.K.; Simpson, T.W. Oral fluid therapy of cholera among Bangladesh refugees. Johns Hopkins Med. J. 1973, 132, 197–205. [Google Scholar]
- Saha, D.; Karim, M.M.; Khan, W.A.; Ahmed, S.; Salam, M.A.; Bennish, M.L. Single-dose azithromycin for the treatment of cholera in adults. N. Engl. J. Med. 2006, 354, 2452–2462. [Google Scholar]
- Kaushik, J.S.; Gupta, P.; Faridi, M.M.; Das, S. Single Dose Azithromycin Versus Ciprofloxacin for Cholera in Children: A Randomized Controlled Trial. Indian Pediatr. 2010, 47, 309–315. [Google Scholar]
- Bresee, J.; Fang, Z.Y.; Wang, B.; Nelson, E.A.; Tam, J.; Soenarto, Y.; Wilopo, S.A.; Kilgore, P.; Kim, J.S.; Kang, J.O.; Lan, W.S.; Gaik, C.L.; Moe, K.; Chen, K.T.; Jiraphongsa, C.; Ponguswanna, Y.; Nguyen, V.M.; Phan, V.T.; Le, T.L.; Hummelman, E.; Gentsch, J.R.; Glass, R. First report from the Asian Rotavirus Surveillance Network. Emerg. Infect. Dis. 2004, 10, 988–995. [Google Scholar]
- Nguyen, V.M.; Nguyen, V.T.; Huynh, P.L.; Dang, D.T.; Nguyen, T.H.; Phan, V.T.; Nguyen, T.L.; Le, T.L.; Ivanoff, B.; Gentsch, J.R.; Glass, R.I. The epidemiology and disease burden of rotavirus in Vietnam: Sentinel surveillance at 6 hospitals. J. Infect. Dis. 2001, 183, 1707–1712. [Google Scholar]
- Parashar, U.D.; Gibson, C.J.; Bresse, J.S.; Glass, R.I. Rotavirus and severe childhood diarrhea. Emerg. Infect. Dis. 2006, 12, 304–306. [Google Scholar]
- Fischer, T.K.; Viboud, C.; Parashar, U.; Malek, M.; Steiner, C.; Glass, R.; Simonsen, L. Hospitalizations and deaths from diarrhea and rotavirus among children <5 years of age in the United States, 1993-2003. J. Infect. Dis. 2007, 195, 1117–1125. [Google Scholar]
- Dong, Y.; Zeng, C.Q.; Ball, J.M.; Estes, M.K.; Morris, A.P. The rotavirus enterotoxin NSP4 mobilizes intracellular calcium in human intestinal cells by stimulating phospholipase C-mediated inositol 1,4,5-trisphosphate production. Proc. Natl. Acad. Sci. USA 1997, 94, 3960–3965. [Google Scholar]
- Ball, J.M.; Tian, P.; Zeng, C.Q.; Morris, A.P.; Estes, M.K. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science 1996, 272, 101–104. [Google Scholar]
- Lundgren, O.; Peregrin, A.T.; Persson, K.; Kordasti, S.; Uhnoo, I.; Svensson, L. Role of the enteric nervous system in the fluid and electrolyte secretion of rotavirus diarrhea. Science 2000, 287, 491–495. [Google Scholar]
- Rollo, E.E.; Kumar, K.P.; Reich, N.C.; Cohen, J.; Angel, J.; Greenberg, H.B.; Sheth, R.; Anderson, J.; Oh, B.; Hempson, S.J.; Mackow, E.R.; Shaw, R.D. The epithelial cell response to rotavirus infection. J. Immunol. 1999, 163, 4442–4452. [Google Scholar]
- Osborne, M.P.; Haddon, S.J.; Spencer, A.J.; Collins, J.; Starkey, W.G.; Wallis, T.S.; Clarke, G.J.; Worton, K.J.; Candy, D.C.; Stephen, J. An electron microscopic investigation of time-related changes in the intestine of neonatal mice infected with murine rotavirus. J. Pediatr. Gastroenterol. Nutr. 1988, 7, 236–248. [Google Scholar]
- Au, K.S.; Chan, W.K.; Burns, J.W.; Estes, M.K. Receptor activity of rotavirus nonstructural glycoprotein NS28. J. Virol. 1989, 63, 4553–4562. [Google Scholar]
- Bergmann, C.C.; Maass, D.; Poruchynsky, M.S.; Atkinson, P.H.; Bellamy, A.R. Topology of the non-structural rotavirus receptor glycoprotein NS28 in the rough endoplasmic reticulum. EMBO J. 1989, 8, 1695–1703. [Google Scholar]
- Lopez, T.; Camacho, M.; Zayas, M.; Najera, R.; Sanchez, R.; Arias, C.F.; Lopez, S. Silencing the morphogenesis of rotavirus. J. Virol. 2005, 79, 184–192. [Google Scholar]
- Cuadras, M.A.; Bordier, B.B.; Zambrano, J.L.; Ludert, J.E.; Greenberg, H.B. Dissecting rotavirus particle-raft interaction with small interfering RNAs: Insights into rotavirus transit through the secretory pathway. J. Virol. 2006, 80, 3935–3946. [Google Scholar]
- Zhang, M.; Zeng, C.Q.; Morris, A.P.; Estes, M.K. A functional NSP4 enterotoxin peptide secreted from rotavirus-infected cells. J. Virol. 2000, 74, 11663–11670. [Google Scholar]
- Storey, S.M.; Gibbons, T.F.; Williams, C.V.; Parr, R.D.; Schroeder, F.; Ball, J.M. Full-length, glycosylated NSP4 is localized to plasma membrane caveolae by a novel raft isolation technique. J. Virol. 2007, 81, 5472–5483. [Google Scholar]
- Bugarcic, A.; Taylor, J.A. Rotavirus nonstructural glycoprotein NSP4 is secreted from the apical surfaces of polarized epithelial cells. J. Virol. 2006, 80, 12343–12349. [Google Scholar]
- Morris, A.P.; Scott, J.K.; Ball, J.M.; Zeng, C.Q.; O'Neal, W.K.; Estes, M.K. NSP4 elicits age-dependent diarrhea and Ca(2+)mediated I(-) influx into intestinal crypts of CF mice. Am. J. Physiol. 1999, 277, G431–G444. [Google Scholar]
- Diaz, Y.; Chemello, M.E.; Pena, F.; Aristimuno, O.C.; Zambrano, J.L.; Rojas, H.; Bartoli, F.; Salazar, L.; Chwetzoff, S.; Sapin, C.; Trugnan, G.; Michelangeli, F.; Ruiz, M.C. Expression of nonstructural rotavirus protein NSP4 mimics Ca2+ homeostasis changes induced by rotavirus infection in cultured cells. J. Virol. 2008, 82, 11331–11343. [Google Scholar]
- Zambrano, J.L.; Diaz, Y.; Pena, F.; Vizzi, E.; Ruiz, M.C.; Michelangeli, F.; Liprandi, F.; Ludert, J.E. Silencing of rotavirus NSP4 or VP7 expression reduces alterations in Ca2+ homeostasis induced by infection of cultured cells. J. Virol. 2008, 82, 5815–5824. [Google Scholar]
- Hulst, M.; Kerstens, H.; de Wit, A.; Smits, M.; van der Meulen, J.; Niewold, T. Early transcriptional response in the jejunum of germ-free piglets after oral infection with virulent rotavirus. Arch. Virol. 2008, 153, 1311–1322. [Google Scholar]
- De Marco, G.; Bracale, I.; Buccigrossi, V.; Bruzzese, E.; Canani, R.B.; Polito, G.; Ruggeri, F.M.; Guarino, A. Rotavirus induces a biphasic enterotoxic and cytotoxic response in human-derived intestinal enterocytes, which is inhibited by human immunoglobulins. J. Infect. Dis. 2009, 200, 813–819. [Google Scholar]
- Halaihel, N.; Lievin, V.; Ball, J.M.; Estes, M.K.; Alvarado, F.; Vasseur, M. Direct inhibitory effect of rotavirus NSP4(114-135) peptide on the Na(+)-D-glucose symporter of rabbit intestinal brush border membrane. J. Virol. 2000, 74, 9464–9470. [Google Scholar]
- Tafazoli, F.; Zeng, C.Q.; Estes, M.K.; Magnusson, K.E.; Svensson, L. NSP4 enterotoxin of rotavirus induces paracellular leakage in polarized epithelial cells. J. Virol. 2001, 75, 1540–1546. [Google Scholar]
- Hyser, J.M.; Estes, M.K. Rotavirus vaccines and pathogenesis: 2008. Curr. Opin. Gastroenterol. 2009, 25, 36–43. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kopic, S.; Geibel, J.P. Toxin Mediated Diarrhea in the 21st Century: The Pathophysiology of Intestinal Ion Transport in the Course of ETEC, V. cholerae and Rotavirus Infection. Toxins 2010, 2, 2132-2157. https://doi.org/10.3390/toxins2082132
Kopic S, Geibel JP. Toxin Mediated Diarrhea in the 21st Century: The Pathophysiology of Intestinal Ion Transport in the Course of ETEC, V. cholerae and Rotavirus Infection. Toxins. 2010; 2(8):2132-2157. https://doi.org/10.3390/toxins2082132
Chicago/Turabian StyleKopic, Sascha, and John P. Geibel. 2010. "Toxin Mediated Diarrhea in the 21st Century: The Pathophysiology of Intestinal Ion Transport in the Course of ETEC, V. cholerae and Rotavirus Infection" Toxins 2, no. 8: 2132-2157. https://doi.org/10.3390/toxins2082132
APA StyleKopic, S., & Geibel, J. P. (2010). Toxin Mediated Diarrhea in the 21st Century: The Pathophysiology of Intestinal Ion Transport in the Course of ETEC, V. cholerae and Rotavirus Infection. Toxins, 2(8), 2132-2157. https://doi.org/10.3390/toxins2082132