Assessment of the Role of Renal Organic Anion Transporters in Drug-Induced Nephrotoxicity

Abstract
:1. Specific Renal Vulnerability
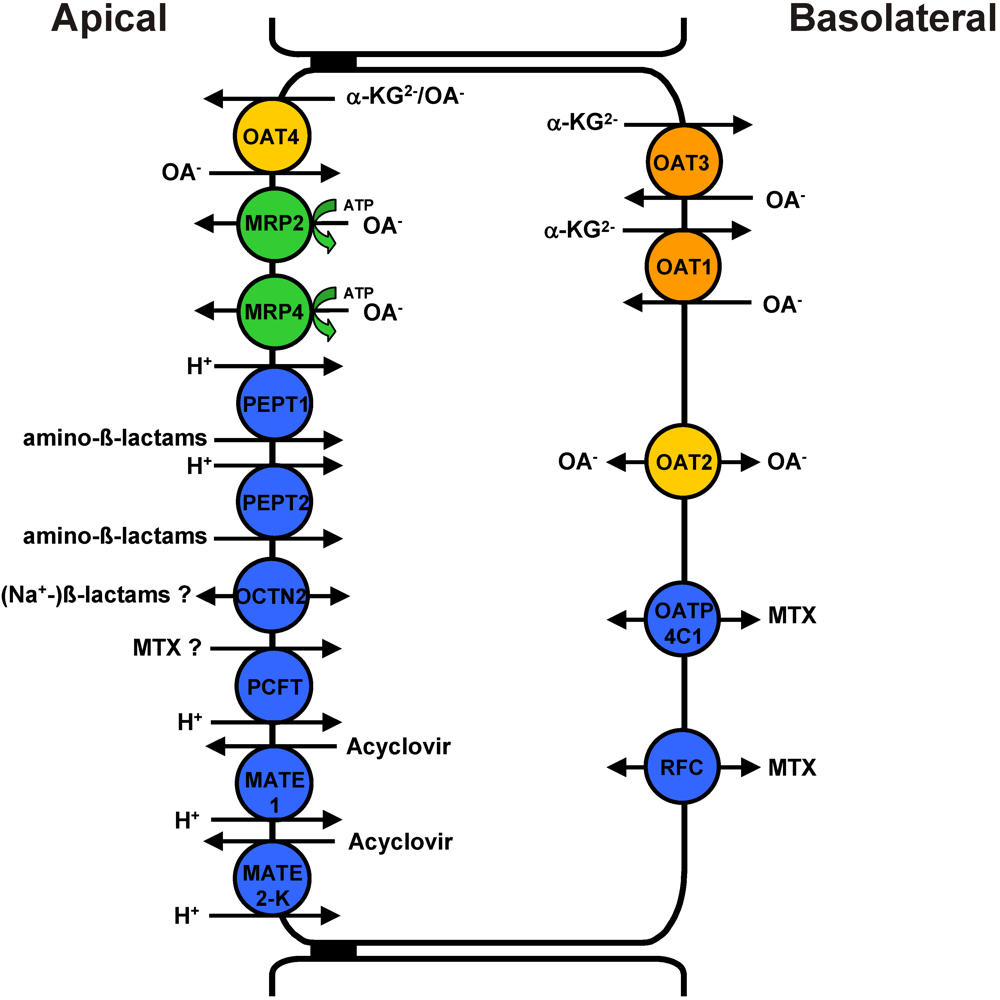
2. Involvement of OATs in Renal Proximal Tubular Solute Uptake and Transepithelial Secretion
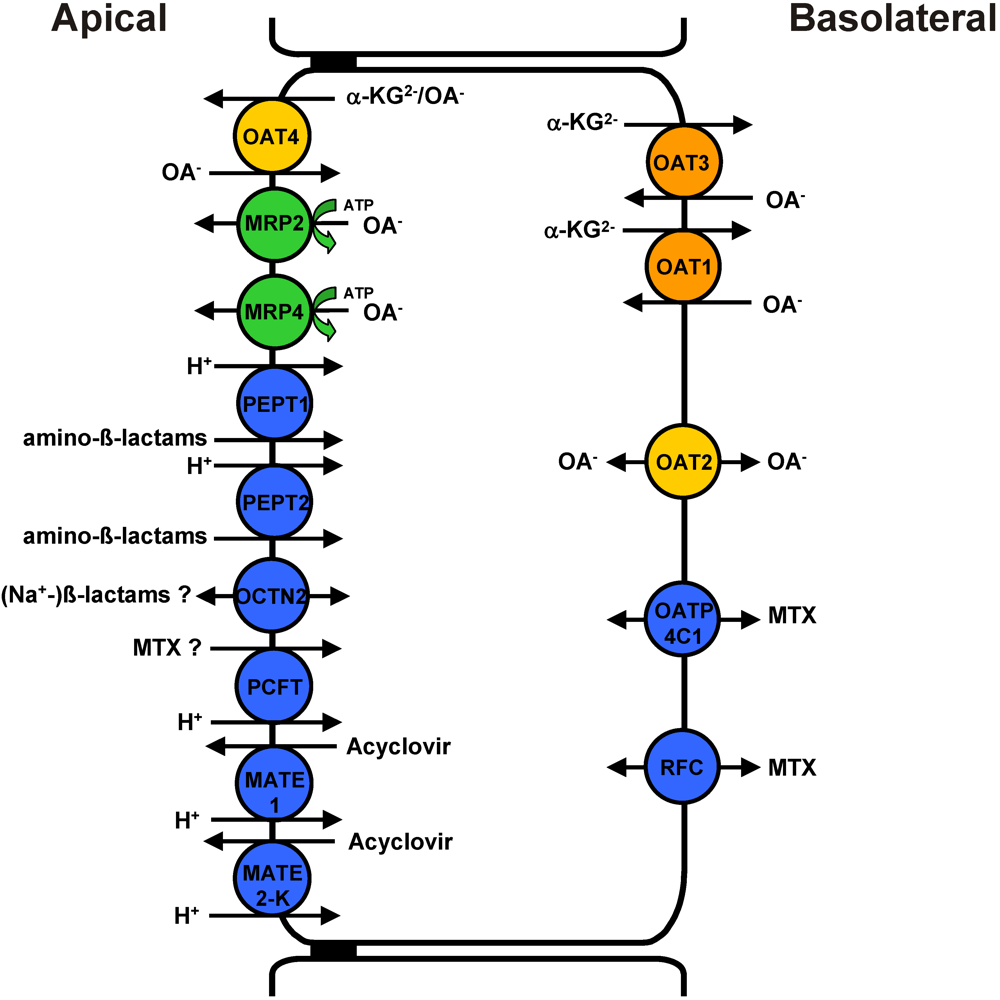
3. Direct Proximal Tubular Toxicity of OAT Drug Substrates
3.1. ß-Lactam antibiotics

{kind=link}
| Substrate | OAT1 | OAT2 | OAT3 | OAT4 | Other Transporters | References | ||
|---|---|---|---|---|---|---|---|---|
| MRP2 | MRP4 | Additional Carriers | ||||||
| β-Lactam Antibiotics | ||||||||
| Cefaclor | n.t.; ◊1096 | T; ◊120 | I | PEPT2: *70.2; ●65 | [34,36,41,42] | |||
| PEPT1: T, ●4520; ●~11000 | ||||||||
| Cefazolin | n.t.; ◊101 | ●5090 | T; ◊117 | n.t. | *81 | [29,33,34,42] | ||
| Cephaloridine | T; aT; | aT; | T; aT; | aT; | n.i. | PEPT2: n.i.; | [18,32,33,34,36,37,39,42] | |
| ●740; | ●2090 | ●2460; | ●3630 | PEPT1: n.i.; | ||||
| ◊2470; | ◊626 | OCTN2: T; ◊790; n.t. | ||||||
| ◊1250 | ||||||||
| Cephalothin | ●220 | ●1040 | ●40 | ●200 | I | PEPT2: ◊7500; PEPT1: ●14000 | [32,33,35,36,42] | |
| Imipenem | bn.t. | aT (770) | [43] | |||||
| Meropenem | T | *847 | [44] | |||||
| Antivirals | ||||||||
| Adefovir | *23.8; *30 | *1220 | n.t. | * >1 mM; cT | [12,18,45,46,47] | |||
| Cidofovir | *46; *58 | n.t. | n.t. | [12,46,47,48] | ||||
| Tenofovir | *33.8 | *770 | n.t. | * >1 mM; T | [45,46,49] | |||
| NSAIDs | ||||||||
| Acetylsalicylate | ◊769 | ◊ >2000 | ◊717 | ◊ >2000 | [50] | |||
| Ibuprofen | ◊55.6 | ◊692 | ◊6.00; | ◊103 | ◊930 | ◊26.3; I | [50,51,52,53] | |
| ●,d1170 | ||||||||
| Mefenamic acid | ◊0.83 | ◊21.7 | ◊0.78 | ◊61.7 | [50] | |||
| Phenacetin | ◊275 | ◊1878 | ◊19.4 | ◊ >2000 | [50] | |||
| Phenylbutazone | ●,d34.7 | ◊605 | ◊130 | [51,52] | ||||
| Aristolochic acid | ●0.6 | ●0.5 | ●20.6 | [54] | ||||
3.3. Non-steroidal anti-inflammatory drugs
3.4. Aristolochic acid
4. Nephrolithiasis
4.1. Methotrexate
| Substrate | OAT1 | OAT2 | OAT3 | OAT4 | Other Transporters | References | ||
|---|---|---|---|---|---|---|---|---|
| MRP2 | MRP4 | Additional Carriers | ||||||
| Cytostatics | ||||||||
| methotrexate (MTX) | *724; *554; n.t. | an.t.; T | *17.2; *10.9; *21.1 | *17.8 | *2500–3000; *250; *480 | *220; *220; *1300 | hOATP4C1: T | [10,13,19,51,52,103,129,131,132,133,134] |
| ß-Lactam Antibiotics | ||||||||
| Ceftriaxone | ●230 | ●6760 | ●4390 | ●2380 | T | PEPT1: n.i. | [29,32,33,36] | |
| Carboxyfluoroquinolones | ||||||||
| Ciprofloxacin | n.i. | I | MATE1: n.t.; ◊231; MATE2-K: n.t.; ◊98.7 | [135,136] | ||||
| Antivirals | ||||||||
| Acyclovir | *342 | n.i. | n.t.; I | n.i. | MATE1: *2640; MATE2-K: *4320 | [136,137] | ||
4.2. Crystal-nephropathy caused by ciprofloxacin and ceftriaxone
4.3. Acyclovir-induced nephrolithiasis
5. Concluding Remarks
References
- Perazella, M.A. Renal vulnerability to drug toxicity. Clin. J. Am. Soc. Nephrol. 2009, 4, 1275–1283. [Google Scholar]
- Nolin, T.D.; Himmelfarb, J. Mechanisms of drug-induced nephrotoxicity. Handb. Exp. Pharmacol. 2010, 196, 111–130. [Google Scholar]
- Perazella, M.A. Drug-induced nephropathy: an update. Expert Opin. Drug Saf. 2005, 4, 689–706. [Google Scholar]
- Yarlagadda, S.G.; Perazella, M.A. Drug-induced crystal nephropathy: an update. Expert Opin. Drug Saf. 2008, 7, 147–158. [Google Scholar]
- Sweet, D.H. Organic anion transporter (Slc22a) family members as mediators of toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 198–215. [Google Scholar]
- Rizwan, A.N.; Burckhardt, G. Organic anion transporters of the SLC22 family: biopharmaceutical, physiological, and pathological roles. Pharm. Res. 2007, 24, 450–470. [Google Scholar]
- VanWert, A.L.; Gionfriddo, M.R.; Sweet, D.H. Organic anion transporters: discovery, pharmacology, regulation and roles in pathophysiology. Biopharm. Drug Dispos. 2010, 31, 1–71. [Google Scholar]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar]
- Hosoyamada, M.; Sekine, T.; Kanai, Y.; Endou, H. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. Am. J. Physiol. 1999, 276, F122–F128. [Google Scholar]
- Cha, S.H.; Sekine, T.; Fukushima, J.I.; Kanai, Y.; Kobayashi, Y.; Goya, T.; Endou, H. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol. Pharmacol. 2001, 59, 1277–1286. [Google Scholar]
- Enomoto, A.; Takeda, M.; Shimoda, M.; Narikawa, S.; Kobayashi, Y.; Kobayashi, Y.; Yamamoto, T.; Sekine, T.; Cha, S.H.; Niwa, T.; Endou, H. Interaction of Human Organic Anion Transporters 2 and 4 with Organic Anion Transport Inhibitors. J. Pharmacol. Exp. Ther. 2002, 301, 797–802. [Google Scholar]
- Cihlar, T.; Lin, D.C.; Pritchard, J.B.; Fuller, M.D.; Mendel, D.B.; Sweet, D.H. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol. Pharmacol. 1999, 56, 570–580. [Google Scholar]
- Lu, R.; Chan, B.S.; Schuster, V.L. Cloning of the human kidney PAH transporter: narrow substrate specificity and regulation by protein kinase C. Am. J. Physiol. 1999, 276, F295–F303. [Google Scholar]
- Bakhiya, N.; Bahn, A.; Burckhardt, G.; Wolff, N.A. Human organic anion transporter 3 (hOAT3) can operate as an exchanger and mediate secretory urate flux. Cell. Physiol. Biochem. 2003, 13, 249–256. [Google Scholar]
- Shuprisha, A.; Lynch, R.M.; Wright, S.H.; Dantzler, W.H. Real-time assessment of α-ketoglutarate effect on organic anion secretion in perfused rabbit proximal tubules. Am. J. Physiol. 1999, 277, F513–F523. [Google Scholar]
- Pritchard, J.B.; Miller, D.S. Proximal tubular transport of organic anions and cations. In The Kidney: Physiology and Pathophysiology; Seldin, D.W., Giebisch, G., Eds.; Raven: New York, NY, USA, 1992; p. 2921. [Google Scholar]
- Pritchard, J.B. Intracellular α-ketoglutarate controls the efficacy of renal organic anion transport. J. Pharmacol. Exp. Ther. 1995, 274, 1278–1284. [Google Scholar]
- Cihlar, T.; Ho, E.S. Fluorescence-based assay for the interaction of small molecules with the human renal organic anion transporter 1. Anal. Biochem. 2000, 283, 49–55. [Google Scholar]
- Sun, W.; Wu, R.R.; van Poelje, P.D.; Erion, M.D. Isolation of a family of organic anion transporters from human liver and kidney. Biochem. Biophys. Res. Commun. 2001, 283, 417–422. [Google Scholar]
- Kobayashi, Y.; Sakai, R.; Ohshiro, N.; Ohbayashi, M.; Kohyama, N.; Yamamoto, T. Possible involvement of organic anion transporter 2 on the interaction of theophylline with erythromycin in the human liver. Drug Metab. Dispos. 2005, 33, 619–622. [Google Scholar]
- Ekaratanawong, S.; Anzai, N.; Jutabha, P.; Miyazaki, H.; Noshiro, R.; Takeda, M.; Kanai, Y.; Sophasan, S.; Endou, H. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J. Pharmacol. Sci. 2004, 94, 297–304. [Google Scholar]
- Hagos, Y.; Stein, D.; Ugele, B.; Burckhardt, G.; Bahn, A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J. Am. Soc. Nephrol. 2007, 18, 430–439. [Google Scholar]
- Burckhardt, B.C.; Burckhardt, G. Transport of organic anions across the basolateral membrane of proximal tubule cells. Rev. Physiol. Biochem. Pharmacol. 2003, 146, 95–158. [Google Scholar]
- van de Water, F.M.; Masereeuw, R.; Russel, F.G. Function and regulation of multidrug resistance proteins (MRPs) in the renal elimination of organic anions. Drug Metab. Rev. 2005, 37, 443–471. [Google Scholar]
- Sugiyama, D.; Kusuhara, H.; Shitara, Y.; Abe, T.; Meier, P.J.; Sekine, T.; Endou, H.; Suzuki, H.; Sugiyama, Y. Characterization of the efflux transport of 17beta-estradiol-D-17beta-glucuronide from the brain across the blood-brain barrier. J. Pharmacol. Exp. Ther. 2001, 298, 316–322. [Google Scholar]
- Tune, B.M. Nephrotoxicity of beta-lactam antibiotics: mechanisms and strategies for prevention. Pediatr. Nephrol. 1997, 11, 768–772. [Google Scholar]
- Cojocel, C.; Tolle, K.L.; El-Hajj, H.; Baumann, K. Protection against cephalosporin-induced lipid peroxidation and nephrotoxicity by (+)-cyanidanol-3 and vitamin E. Braz. J. Med. Biol. Res. 2007, 40, 867–875. [Google Scholar]
- Shitara, Y.; Sato, H.; Sugiyama, Y. Evaluation of drug-drug interaction in the hepatobiliary and renal transport of drugs. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 689–723. [Google Scholar]
- Kato, Y.; Takahara, S.; Kato, S.; Kubo, Y.; Sai, Y.; Tamai, I.; Yabuuchi, H.; Tsuji, A. Involvement of multidrug resistance-associated protein 2 (Abcc2) in molecular weight-dependent biliary excretion of beta-lactam antibiotics. Drug Metab. Dispos. 2008, 36, 1088–1096. [Google Scholar]
- Brown, G.R. Cephalosporin-probenecid drug interactions. Clin. Pharmacokinet. 1993, 24, 289–300. [Google Scholar]
- Tune, B.M. The nephrotoxicity of β-lactam antibiotics. In Toxicology of the Kidney; Hook, J.B., Goldstein, R.S., Eds.; Raven: New York, NY, USA, 1993; p. 257. [Google Scholar]
- Takeda, M.; Babu, E.; Narikawa, S.; Endou, H. Interaction of human organic anion transporters with various cephalosporin antibiotics. Eur. J. Pharmacol. 2002, 438, 137–142. [Google Scholar]
- Khamdang, S.; Takeda, M.; Babu, E.; Noshiro, R.; Onozato, M.L.; Tojo, A.; Enomoto, A.; Huang, X.L.; Narikawa, S.; Anzai, N.; Piyachaturawat, P.; Endou, H. Interaction of human and rat organic anion transporter 2 with various cephalosporin antibiotics. Eur. J. Pharmacol. 2003, 465, 1–7. [Google Scholar]
- Ueo, H.; Motohashi, H.; Katsura, T.; Inui, K. Human organic anion transporter hOAT3 is a potent transporter of cephalosporin antibiotics, in comparison with hOAT1. Biochem. Pharmacol. 2005, 70, 1104–1113. [Google Scholar]
- Ganapathy, M.E.; Brandsch, M.; Prasad, P.D.; Ganapathy, V.; Leibach, F.H. Differential recognition of beta -lactam antibiotics by intestinal and renal peptide transporters, PEPT 1 and PEPT 2. J. Biol. Chem. 1995, 270, 25672–25677. [Google Scholar]
- Bretschneider, B.; Brandsch, M.; Neubert, R. Intestinal transport of beta-lactam antibiotics: analysis of the affinity at the H+/peptide symporter (PEPT1), the uptake into Caco-2 cell monolayers and the transepithelial flux. Pharm. Res. 1999, 16, 55–61. [Google Scholar]
- Ganapathy, M.E.; Huang, W.; Rajan, D.P.; Carter, A.L.; Sugawara, M.; Iseki, K.; Leibach, F.H.; Ganapathy, V. beta-lactam antibiotics as substrates for OCTN2, an organic cation/carnitine transporter. J. Biol. Chem. 2000, 275, 1699–1707. [Google Scholar]
- Tamai, I.; China, K.; Sai, Y.; Kobayashi, D.; Nezu, J.; Kawahara, E.; Tsuji, A. Na(+)-coupled transport of L-carnitine via high-affinity carnitine transporter OCTN2 and its subcellular localization in kidney. Biochim. Biophys. Acta 2001, 1512, 273–284. [Google Scholar]
- Grigat, S.; Fork, C.; Bach, M.; Golz, S.; Geerts, A.; Schömig, E.; Gründemann, D. The carnitine transporter SLC22A5 is not a general drug transporter, but it efficiently translocates mildronate. Drug Metab. Dispos. 2009, 37, 330–337. [Google Scholar]
- Kano, T.; Kato, Y.; Ito, K.; Ogihara, T.; Kubo, Y.; Tsuji, A. Carnitine/organic cation transporter OCTN2 (Slc22a5) is responsible for renal secretion of cephaloridine in mice. Drug Metab. Dispos. 2009, 37, 1009–1016. [Google Scholar]
- Li, M.; Anderson, G.D.; Phillips, B.R.; Kong, W.; Shen, D.D.; Wang, J. Interactions of amoxicillin and cefaclor with human renal organic anion and peptide transporters. Drug Metab. Dispos. 2006, 34, 547–555. [Google Scholar]
- Ci, L.; Kusuhara, H.; Adachi, M.; Schuetz, J.D.; Takeuchi, K.; Sugiyama, Y. Involvement of MRP4 (ABCC4) in the luminal efflux of ceftizoxime and cefazolin in the kidney. Mol. Pharmacol. 2007, 71, 1591–1597. [Google Scholar]
- Lim, S.C.; Im, Y.B.; Bae, C.S.; Han, S.I.; Kim, S.E.; Han, H.K. Protective effect of morin on the imipenem-induced nephrotoxicity in rabbits. Arch. Pharm. Res. 2008, 31, 1060–1065. [Google Scholar]
- Shibayama, T.; Sugiyama, D.; Kamiyama, E.; Tokui, T.; Hirota, T.; Ikeda, T. Characterization of CS-023 (RO4908463), a novel parenteral carbapenem antibiotic, and meropenem as substrates of human renal transporters. Drug Metab. Pharmacokinet. 2007, 22, 41–47. [Google Scholar]
- Cihlar, T.; Laflamme, G.; Fisher, R.; Carey, A.C.; Vela, J.E.; Mackman, R.; Ray, A.S. Novel nucleotide human immunodeficiency virus reverse transcriptase inhibitor GS-9148 with a low nephrotoxic potential: characterization of renal transport and accumulation. Antimicrob. Agents Chemother. 2009, 53, 150–156. [Google Scholar]
- Imaoka, T.; Kusuhara, H.; Adachi, M.; Schuetz, J.D.; Takeuchi, K.; Sugiyama, Y. Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol. Pharmacol. 2007, 71, 619–627. [Google Scholar]
- Reid, G.; Wielinga, P.; Zelcer, N.; de Haas, M.; van Deemter, L.; Wijnholds, J.; Balzarini, J.; Borst, P. Characterization of the transport of nucleoside analog drugs by the human multidrug resistance proteins MRP4 and MRP5. Mol. Pharmacol. 2003, 63, 1094–1103. [Google Scholar]
- Ho, E.S.; Lin, D.C.; Mendel, D.B.; Cihlar, T. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J. Am. Soc. Nephrol. 2000, 11, 383–393. [Google Scholar]
- Ray, A.S.; Cihlar, T.; Robinson, K.L.; Tong, L.; Vela, J.E.; Fuller, M.D.; Wieman, L.M.; Eisenberg, E.J.; Rhodes, G.R. Mechanism of active renal tubular efflux of tenofovir. Antimicrob. Agents Chemother. 2006, 50, 3297–3304. [Google Scholar]
- Khamdang, S.; Takeda, M.; Noshiro, R.; Narikawa, S.; Enomoto, A.; Anzai, N.; Piyachaturawat, P.; Endou, H. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2002, 303, 534–539. [Google Scholar]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 302, 666–671. [Google Scholar]
- El-Sheikh, A.A.; van den Heuvel, J.J.; Koenderink, J.B.; Russel, F.G. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J. Pharmacol. Exp. Ther. 2007, 320, 229–235. [Google Scholar]
- Reid, G.; Wielinga, P.; Zelcer, N.; van der Heijden, I.; Kuil, A.; de Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249. [Google Scholar]
- Bakhiya, N.; Arlt, V.M.; Bahn, A.; Burckhardt, G.; Phillips, D.H.; Glatt, H. Molecular evidence for an involvement of organic anion transporters (OATs) in aristolochic acid nephropathy. Toxicology 2009, 264, 74–79. [Google Scholar]
- Uchida, Y.; Kamiie, J.; Ohtsuki, S.; Terasaki, T. Multichannel liquid chromatography-tandem mass spectrometry cocktail method for comprehensive substrate characterization of multidrug resistance-associated protein 4 transporter. Pharm. Res. 2007, 24, 2281–2296. [Google Scholar]
- Tune, B.M.; Fernholt, M.; Schwartz, A. Mechanism of cephaloridine transport in the kidney. J. Pharmacol. Exp. Ther. 1974, 191, 311–317. [Google Scholar]
- Yabuuchi, H.; Tamai, I.; Morita, K.; Kouda, T.; Miyamoto, K.; Takeda, E.; Tsuji, A. Hepatic sinusoidal membrane transport of anionic drugs mediated by anion transporter Npt1. J. Pharmacol. Exp. Ther. 1998, 286, 1391–1396. [Google Scholar]
- Birnbaum, J.; Kahan, F.M.; Kropp, H.; MacDonald, J.S. Carbapenems, a new class of beta-lactam antibiotics. Discovery and development of imipenem/cilastatin. Am. J. Med. 1985, 78, 3–21. [Google Scholar]
- Tune, B.M.; Fravert, D.; Hsu, C.Y. Thienamycin nephrotoxicity. Mitochondrial injury and oxidative effects of imipenem in the rabbit kidney. Biochem. Pharmacol. 1989, 38, 3779–3783. [Google Scholar] [CrossRef] [PubMed]
- Norrby, S.R.; Björnegård, B.; Ferber, F.; Jones, K.H. Pharmacokinetics of imipenem in healthy volunteers. J. Antimicrob. Chemother. 1983, 12 (Suppl. D), 109–124. [Google Scholar] [PubMed]
- Drusano, G.L.; Standiford, H.C. Pharmacokinetic profile of imipenem/cilastatin in normal volunteers. Am. J. Med. 1985, 78, 47–53. [Google Scholar]
- Takeda, M.; Narikawa, S.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of organic anion transport inhibitors using cells stably expressing human organic anion transporters. Eur. J. Pharmacol. 2001, 419, 113–120. [Google Scholar]
- Goa, K.L.; Noble, S. Panipenem/betamipron. Drugs 2003, 63, 913–925. [Google Scholar]
- Hirouchi, Y.; Naganuma, H.; Kawahara, Y.; Okada, R.; Kamiya, A.; Inui, K.; Hori, R. Preventive effect of betamipron on nephrotoxicity and uptake of carbapenems in rabbit renal cortex. Jpn. J. Pharmacol. 1994, 66, 1–6. [Google Scholar]
- Topham, J.C.; Murgatroyd, L.B.; Jones, D.V.; Goonetilleke, U.R.; Wright, J. Safety evaluation of meropenem in animals: studies on the kidney. J. Antimicrob. Chemother. 1989, 24 (Suppl. A), 287–306. [Google Scholar] [PubMed]
- Izzedine, H.; Launay-Vacher, V.; Deray, G. Antiviral drug-induced nephrotoxicity. Am. J. Kidney Dis. 2005, 45, 804–817. [Google Scholar]
- Fisher, E.J.; Chaloner, K.; Cohn, D.L.; Grant, L.B.; Alston, B.; Brosgart, C.L.; Schmetter, B.; El-Sadr, W.M.; Sampson, J. The safety and efficacy of adefovir dipivoxil in patients with advanced HIV disease: a randomized, placebo-controlled trial. AIDS 2001, 15, 1695–1700. [Google Scholar]
- Kahn, J.; Lagakos, S.; Wulfsohn, M.; Cherng, D.; Miller, M.; Cherrington, J.; Hardy, D.; Beall, G.; Cooper, R.; Murphy, R.; Basgoz, N.; Ng, E.; Deeks, S.; Winslow, D.; Toole, J.J.; Coakley, D. Efficacy and safety of adefovir dipivoxil with antiretroviral therapy: a randomized controlled trial. JAMA 1999, 282, 2305–2312. [Google Scholar]
- Earle, K.E.; Seneviratne, T.; Shaker, J.; Shoback, D. Fanconi's syndrome in HIV+ adults: report of three cases and literature review. J. Bone Miner. Res. 2004, 19, 714–721. [Google Scholar]
- Vittecoq, D.; Dumitrescu, L.; Beaufils, H.; Deray, G. Fanconi syndrome associated with cidofovir therapy. Antimicrob. Agents Chemother. 1997, 41, 1846. [Google Scholar]
- Meier, P.; Dautheville-Guibal, S.; Ronco, P.M.; Rossert, J. Cidofovir-induced end-stage renal failure. Nephrol. Dial. Transplant. 2002, 17, 148–149. [Google Scholar]
- Cundy, K.C.; Barditch-Crovo, P.; Walker, R.E.; Collier, A.C.; Ebeling, D.; Toole, J.; Jaffe, H.S. Clinical pharmacokinetics of adefovir in human immunodeficiency virus type 1-infected patients. Antimicrob. Agents Chemother. 1995, 39, 2401–2405. [Google Scholar]
- Cundy, K.C.; Petty, B.G.; Flaherty, J.; Fisher, P.E.; Polis, M.A.; Wachsman, M.; Lietman, P.S.; Lalezari, J.P.; Hitchcock, M.J.; Jaffe, H.S. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 1995, 39, 1247–1252. [Google Scholar]
- Perazella, M.A. Drug-induced renal failure: update on new medications and unique mechanisms of nephrotoxicity. Am. J. Med. Sci. 2003, 325, 349–362. [Google Scholar]
- Aslamkhan, A.G.; Thompson, D.M.; Perry, J.L.; Bleasby, K.; Wolff, N.A.; Barros, S.; Miller, D.S.; Pritchard, J.B. The flounder organic anion transporter fOat has sequence, function, and substrate specificity similarity to both mammalian Oat1 and Oat3. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1773–R1780. [Google Scholar]
- Uwai, Y.; Ida, H.; Tsuji, Y.; Katsura, T.; Inui, K. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 2007, 24, 811–815. [Google Scholar]
- Gallant, J.E.; Staszewski, S.; Pozniak, A.L.; DeJesus, E.; Suleiman, J.M.; Miller, M.D.; Coakley, D.F.; Lu, B.; Toole, J.J.; Cheng, A.K. Efficacy and safety of tenofovir DF vs stavudine in combination therapy in antiretroviral-naive patients: a 3-year randomized trial. JAMA 2004, 292, 191–201. [Google Scholar]
- Gallant, J.E.; Deresinski, S. Tenofovir disoproxil fumarate. Clin. Infect. Dis. 2003, 37, 944–950. [Google Scholar]
- Birkus, G.; Hitchcock, M.J.; Cihlar, T. Assessment of mitochondrial toxicity in human cells treated with tenofovir: comparison with other nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2002, 46, 716–723. [Google Scholar]
- Coca, S.; Perazella, M.A. Rapid communication: acute renal failure associated with tenofovir: evidence of drug-induced nephrotoxicity. Am. J. Med. Sci. 2002, 324, 342–344. [Google Scholar]
- Gaspar, G.; Monereo, A.; Garcia-Reyne, A.; de Guzman, M. Fanconi syndrome and acute renal failure in a patient treated with tenofovir: a call for caution. AIDS 2004, 18, 351–352. [Google Scholar]
- James, C.W.; Steinhaus, M.C.; Szabo, S.; Dressier, R.M. Tenofovir-related nephrotoxicity: case report and review of the literature. Pharmacotherapy 2004, 24, 415–418. [Google Scholar]
- Malik, A.; Abraham, P.; Malik, N. Acute renal failure and Fanconi syndrome in an AIDS patient on tenofovir treatment--case report and review of literature. J. Infect. 2005, 51, E61–E65. [Google Scholar]
- Izzedine, H.; Hulot, J.S.; Villard, E.; Goyenvalle, C.; Dominguez, S.; Ghosn, J.; Valantin, M.A.; Lechat, P.; Deray, A.G. Association between ABCC2 gene haplotypes and tenofovir-induced proximal tubulopathy. J. Infect. Dis. 2006, 194, 1481–1491. [Google Scholar]
- Rollot, F.; Nazal, E.M.; Chauvelot-Moachon, L.; Kelaidi, C.; Daniel, N.; Saba, M.; Abad, S.; Blanche, P. Tenofovir-related Fanconi syndrome with nephrogenic diabetes insipidus in a patient with acquired immunodeficiency syndrome: the role of lopinavir-ritonavir-didanosine. Clin. Infect. Dis. 2003, 37, e174–e176. [Google Scholar]
- Zimmermann, A.E.; Pizzoferrato, T.; Bedford, J.; Morris, A.; Hoffman, R.; Braden, G. Tenofovir-associated acute and chronic kidney disease: a case of multiple drug interactions. Clin. Infect. Dis. 2006, 42, 283–290. [Google Scholar]
- Hirouchi, M.; Suzuki, H.; Itoda, M.; Ozawa, S.; Sawada, J.; Ieiri, I.; Ohtsubo, K.; Sugiyama, Y. Characterization of the cellular localization, expression level, and function of SNP variants of MRP2/ABCC2. Pharm. Res. 2004, 21, 742–748. [Google Scholar]
- Haenisch, S.; Zimmermann, U.; Dazert, E.; Wruck, C.J.; Dazert, P.; Siegmund, W.; Kroemer, H.K.; Warzok, R.W.; Cascorbi, I. Influence of polymorphisms of ABCB1 and ABCC2 on mRNA and protein expression in normal and cancerous kidney cortex. Pharmacogenomics J. 2007, 7, 56–65. [Google Scholar]
- Cihlar, T.; Ray, A.S.; Laflamme, G.; Vela, J.E.; Tong, L.; Fuller, M.D.; Roy, A.; Rhodes, G.R. Molecular assessment of the potential for renal drug interactions between tenofovir and HIV protease inhibitors. Antivir. Ther. 2007, 12, 267–272. [Google Scholar]
- Deeks, S.G.; Barditch-Crovo, P.; Lietman, P.S.; Hwang, F.; Cundy, K.C.; Rooney, J.F.; Hellmann, N.S.; Safrin, S.; Kahn, J.O. Safety, pharmacokinetics, and antiretroviral activity of intravenous 9-[2-(R)-(Phosphonomethoxy)propyl]adenine, a novel anti-human immunodeficiencyvirus (HIV) therapy, in HIV-infected adults. Antimicrob. Agents Chemother. 1998, 42, 2380–2384. [Google Scholar]
- Kearney, B.P.; Yale, K.; Shah, J.; Zhong, L.; Flaherty, J.F. Pharmacokinetics and dosing recommendations of tenofovir disoproxil fumarate in hepatic or renal impairment. Clin. Pharmacokinet. 2006, 45, 1115–1124. [Google Scholar]
- Kiser, J.J.; Carten, M.L.; Aquilante, C.L.; Anderson, P.L.; Wolfe, P.; King, T.M.; Delahunty, T.; Bushman, L.R.; Fletcher, C.V. The effect of lopinavir/ritonavir on the renal clearance of tenofovir in HIV-infected patients. Clin. Pharmacol. Ther. 2008, 83, 265–272. [Google Scholar]
- House, A.A.; Silva, O.S.; Ronco, C. Anti-inflammatory drugs and the kidney. Int. J. Artif. Organs 2007, 30, 1042–1046. [Google Scholar]
- Muhalwas, K.K.; Shah, G.M.; Winer, R.L. Renal papillary necrosis caused by long-term ingestion of pentazocine and aspirin. JAMA 1981, 246, 867–868. [Google Scholar]
- Rosenberger, C.; Rosen, S.; Heyman, S.N. Renal parenchymal oxygenation and hypoxia adaptation in acute kidney injury. Clin. Exp. Pharmacol. Physiol. 2006, 33, 980–988. [Google Scholar]
- Esteve, J.B.; Launay-Vacher, V.; Brocheriou, I.; Grimaldi, A.; Izzedine, H. COX-2 inhibitors and acute interstitial nephritis: case report and review of the literature. Clin. Nephrol. 2005, 63, 385–389. [Google Scholar]
- Halbritter, J.; Mayer, C.; Rasche, F.M.; Amann, K.; Lindner, T.H. Interstitial nephritis. Internist (Berl) 2009, 50, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Whelton, A. Nephrotoxicity of nonsteroidal anti-inflammatory drugs: physiologic foundations and clinical implications. Am. J. Med. 1999, 106, 13S–24S. [Google Scholar]
- Silva, F.G. Chemical-induced nephropathy: a review of the renal tubulointerstitial lesions in humans. Toxicol. Pathol. 2004, 32 (Suppl. 2), 71–84. [Google Scholar] [CrossRef] [PubMed]
- D'Agati, V. Does aspirin cause acute or chronic renal failure in experimental animals and in humans? Am. J. Kidney Dis. 1996, 28, S24–S29. [Google Scholar] [CrossRef] [PubMed]
- Braden, G.L.; O'Shea, M.H.; Mulhern, J.G. Tubulointerstitial diseases. Am. J. Kidney Dis. 2005, 46, 560–572. [Google Scholar]
- Rocha, G.M.; Michea, L.F.; Peters, E.M.; Kirby, M.; Xu, Y.; Ferguson, D.R.; Burg, M.B. Direct toxicity of nonsteroidal antiinflammatory drugs for renal medullary cells. Proc. Natl. Acad. Sci. USA 2001, 98, 5317–5322. [Google Scholar]
- Bakos, E.; Evers, R.; Sinko, E.; Varadi, A.; Borst, P.; Sarkadi, B. Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. Mol. Pharmacol. 2000, 57, 760–768. [Google Scholar]
- Vanherweghem, J.L.; Tielemans, C.; Abramowicz, D.; Depierreux, M.; Vanhaelen-Fastre, R.; Vanhaelen, M.; Dratwa, M.; Richard, C.; Vandervelde, D.; Verbeelen, D.; Jadoul, M. Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet 1993, 341, 387–391. [Google Scholar]
- Nortier, J.L.; Martinez, M.C.; Schmeiser, H.H.; Arlt, V.M.; Bieler, C.A.; Petein, M.; Depierreux, M.F.; De Pauw, L.; Abramowicz, D.; Vereerstraeten, P.; Vanherweghem, J.L. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). N. Engl. J. Med. 2000, 342, 1686–1692. [Google Scholar]
- Jackson, L.; Kofman, S.; Weiss, A.; Brodovsky, H. Aristolochic acid (NSC-50413): Phase I clinical study. Cancer Chemother. Rep. 1964, 42, 35–37. [Google Scholar]
- Debelle, F.D.; Vanherweghem, J.L.; Nortier, J.L. Aristolochic acid nephropathy: a worldwide problem. Kidney Int. 2008, 74, 158–169. [Google Scholar]
- Depierreux, M.; Van Damme, B.; Vanden Houte, K.; Vanherweghem, J.L. Pathologic aspects of a newly described nephropathy related to the prolonged use of Chinese herbs. Am. J. Kidney Dis. 1994, 24, 172–180. [Google Scholar]
- Nortier, J.L.; Deschodt-Lanckman, M.M.; Simon, S.; Thielemans, N.O.; de Prez, E.G.; Depierreux, M.F.; Tielemans, C.L.; Richard, C.; Lauwerys, R.R.; Bernard, A.M.; Vanherweghem, J.L. Proximal tubular injury in Chinese herbs nephropathy: monitoring by neutral endopeptidase enzymuria. Kidney Int. 1997, 51, 288–293. [Google Scholar]
- Cosyns, J.P.; Dehoux, J.P.; Guiot, Y.; Goebbels, R.M.; Robert, A.; Bernard, A.M.; van Ypersele, D.S. Chronic aristolochic acid toxicity in rabbits: a model of Chinese herbs nephropathy? Kidney Int. 2001, 59, 2164–2173. [Google Scholar]
- Pozdzik, A.A.; Salmon, I.J.; Debelle, F.D.; Decaestecker, C.; Van den, B.C.; Verbeelen, D.; Deschodt-Lanckman, M.M.; Vanherweghem, J.L.; Nortier, J.L. Aristolochic acid induces proximal tubule apoptosis and epithelial to mesenchymal transformation. Kidney Int. 2008, 73, 595–607. [Google Scholar]
- Cosyns, J.P.; Goebbels, R.M.; Liberton, V.; Schmeiser, H.H.; Bieler, C.A.; Bernard, A.M. Chinese herbs nephropathy-associated slimming regimen induces tumours in the forestomach but no interstitial nephropathy in rats. Arch. Toxicol. 1998, 72, 738–743. [Google Scholar]
- Schmeiser, H.H.; Bieler, C.A.; Wiessler, M.; van Ypersele, D.S.; Cosyns, J.P. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res. 1996, 56, 2025–2028. [Google Scholar]
- Lebeau, C.; Arlt, V.M.; Schmeiser, H.H.; Boom, A.; Verroust, P.J.; Devuyst, O.; Beauwens, R. Aristolochic acid impedes endocytosis and induces DNA adducts in proximal tubule cells. Kidney Int. 2001, 60, 1332–1342. [Google Scholar]
- Hori, R.; Okamura, M.; Takayama, A.; Hirozane, K.; Takano, M. Transport of organic anion in the OK kidney epithelial cell line. Am. J. Physiol. 1993, 264, F975–F980. [Google Scholar]
- Hammann, C.; Guelpa, G. Drug-induced calculi. Schweiz. Rundsch. Med. Prax. 1993, 82, 1129–1132. [Google Scholar]
- Jolivet, J.; Cowan, K.H.; Curt, G.A.; Clendeninn, N.J.; Chabner, B.A. The pharmacology and clinical use of methotrexate. N. Engl. J. Med. 1983, 309, 1094–1104. [Google Scholar]
- Borchers, A.T.; Keen, C.L.; Cheema, G.S.; Gershwin, M.E. The use of methotrexate in rheumatoid arthritis. Semin. Arthritis Rheum. 2004, 34, 465–483. [Google Scholar]
- Patel, V.; Macdonald, J.K.; McDonald, J.W.; Chande, N. Methotrexate for maintenance of remission in Crohn's disease. Cochrane Database Syst. Rev. 2009, CD006884. [Google Scholar]
- El-Matary, W.; Vandermeer, B.; Griffiths, A.M. Methotrexate for maintenance of remission in ulcerative colitis. Cochrane Database Syst. Rev. 2009, CD007560. [Google Scholar]
- Widemann, B.C.; Adamson, P.C. Understanding and managing methotrexate nephrotoxicity. Oncologist 2006, 11, 694–703. [Google Scholar]
- Maiche, A.G. Acute renal failure due to concomitant action of methotrexate and indomethacin. Lancet 1986, 1, 1390. [Google Scholar]
- Thyss, A.; Milano, G.; Kubar, J.; Namer, M.; Schneider, M. Clinical and pharmacokinetic evidence of a life-threatening interaction between methotrexate and ketoprofen. Lancet 1986, 1, 256–258. [Google Scholar]
- Ellison, N.M.; Servi, R.J. Acute renal failure and death following sequential intermediate-dose methotrexate and 5-FU: a possible adverse effect due to concomitant indomethacin administration. Cancer Treat. Rep. 1985, 69, 342–343. [Google Scholar]
- Frenia, M.L.; Long, K.S. Methotrexate and nonsteroidal antiinflammatory drug interactions. Ann. Pharmacother. 1992, 26, 234–237. [Google Scholar]
- Hulot, J.S.; Villard, E.; Maguy, A.; Morel, V.; Mir, L.; Tostivint, I.; William-Faltaos, D.; Fernandez, C.; Hatem, S.; Deray, G.; Komajda, M.; Leblond, V.; Lechat, P. A mutation in the drug transporter gene ABCC2 associated with impaired methotrexate elimination. Pharmacogenet. Genomics 2005, 15, 277–285. [Google Scholar]
- Shen, D.D.; Azarnoff, D.L. Clinical pharmacokinetics of methotrexate. Clin. Pharmacokinet. 1978, 3, 1–13. [Google Scholar]
- Hendel, J.; Nyfors, A. Nonlinear renal elimination kinetics of methotrexate due to saturation of renal tubular reabsorption. Eur. J. Clin. Pharmacol. 1984, 26, 121–124. [Google Scholar]
- Uwai, Y.; Taniguchi, R.; Motohashi, H.; Saito, H.; Okuda, M.; Inui, K. Methotrexate-loxoprofen interaction: involvement of human organic anion transporters hOAT1 and hOAT3. Drug Metab. Pharmacokinet. 2004, 19, 369–374. [Google Scholar]
- Crews, K.R.; Liu, T.; Rodriguez-Galindo, C.; Tan, M.; Meyer, W.H.; Panetta, J.C.; Link, M.P.; Daw, N.C. High-dose methotrexate pharmacokinetics and outcome of children and young adults with osteosarcoma. Cancer 2004, 100, 1724–1733. [Google Scholar]
- Konno, T.; Ebihara, T.; Hisaeda, K.; Uchiumi, T.; Nakamura, T.; Shirakusa, T.; Kuwano, M.; Wada, M. Identification of domains participating in the substrate specificity and subcellular localization of the multidrug resistance proteins MRP1 and MRP2. J. Biol. Chem. 2003, 278, 22908–22917. [Google Scholar]
- Chen, Z.S.; Lee, K.; Walther, S.; Raftogianis, R.B.; Kuwano, M.; Zeng, H.; Kruh, G.D. Analysis of methotrexate and folate transport by multidrug resistance protein 4 (ABCC4): MRP4 is a component of the methotrexate efflux system. Cancer Res. 2002, 62, 3144–3150. [Google Scholar]
- van Aubel, R.A.; Smeets, P.H.; Peters, J.G.; Bindels, R.J.; Russel, F.G. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J. Am. Soc. Nephrol. 2002, 13, 595–603. [Google Scholar]
- Mikkaichi, T.; Suzuki, T.; Onogawa, T.; Tanemoto, M.; Mizutamari, H.; Okada, M.; Chaki, T.; Masuda, S.; Tokui, T.; Eto, N.; Abe, M.; Satoh, F.; Unno, M.; Hishinuma, T.; Inui, K.; Ito, S.; Goto, J.; Abe, T. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc. Natl. Acad. Sci. USA 2004, 101, 3569–3574. [Google Scholar]
- VanWert, A.L.; Srimaroeng, C.; Sweet, D.H. Organic anion transporter 3 (oat3/slc22a8) interacts with carboxyfluoroquinolones, and deletion increases systemic exposure to ciprofloxacin. Mol. Pharmacol. 2008, 74, 122–131. [Google Scholar]
- Tanihara, Y.; Masuda, S.; Sato, T.; Katsura, T.; Ogawa, O.; Inui, K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem. Pharmacol. 2007, 74, 359–371. [Google Scholar]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Kobayashi, Y.; Yamamoto, T.; Cha, S.H.; Sekine, T.; Endou, H. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J. Pharmacol. Exp. Ther. 2002, 300, 918–924. [Google Scholar]
- Wang, Y.; Zhao, R.; Russell, R.G.; Goldman, I.D. Localization of the murine reduced folate carrier as assessed by immunohistochemical analysis. Biochim. Biophys. Acta 2001, 1513, 49–54. [Google Scholar]
- Moscow, J.A.; Connolly, T.; Myers, T.G.; Cheng, C.C.; Paull, K.; Cowan, K.H. Reduced folate carrier gene (RFC1) expression and anti-folate resistance in transfected and non-selected cell lines. Int. J. Cancer 1997, 72, 184–190. [Google Scholar]
- Williams, F.M.; Flintoff, W.F. Isolation of a human cDNA that complements a mutant hamster cell defective in methotrexate uptake. J. Biol. Chem. 1995, 270, 2987–2992. [Google Scholar]
- Nozaki, Y.; Kusuhara, H.; Endou, H.; Sugiyama, Y. Quantitative evaluation of the drug-drug interactions between methotrexate and nonsteroidal anti-inflammatory drugs in the renal uptake process based on the contribution of organic anion transporters and reduced folate carrier. J. Pharmacol. Exp. Ther. 2004, 309, 226–234. [Google Scholar]
- Morshed, K.M.; McMartin, K.E. Reabsorptive and secretory 5-methyltetrahydrofolate transport pathways in cultured human proximal tubule cells. Am. J. Physiol. 1997, 272, F380–F388. [Google Scholar]
- Morshed, K.M.; Ross, D.M.; McMartin, K.E. Folate transport proteins mediate the bidirectional transport of 5-methyltetrahydrofolate in cultured human proximal tubule cells. J. Nutr. 1997, 127, 1137–1147. [Google Scholar]
- Ashokkumar, B.; Mohammed, Z.M.; Vaziri, N.D.; Said, H.M. Effect of folate oversupplementation on folate uptake by human intestinal and renal epithelial cells. Am. J. Clin. Nutr. 2007, 86, 159–166. [Google Scholar]
- Subramanian, V.S.; Reidling, J.C.; Said, H.M. Differentiation-dependent regulation of the intestinal folate uptake process: studies with Caco-2 cells and native mouse intestine. Am. J. Physiol. Cell Physiol. 2008, 295, C828–C835. [Google Scholar]
- Zhao, R.; Qiu, A.; Tsai, E.; Jansen, M.; Akabas, M.H.; Goldman, I.D. The proton-coupled folate transporter: impact on pemetrexed transport and on antifolates activities compared with the reduced folate carrier. Mol. Pharmacol. 2008, 74, 854–862. [Google Scholar]
- Badagnani, I.; Castro, R.A.; Taylor, T.R.; Brett, C.M.; Huang, C.C.; Stryke, D.; Kawamoto, M.; Johns, S.J.; Ferrin, T.E.; Carlson, E.J.; Burchard, E.G.; Giacomini, K.M. Interaction of methotrexate with organic-anion transporting polypeptide 1A2 and its genetic variants. J. Pharmacol. Exp. Ther. 2006, 318, 521–529. [Google Scholar]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004, 447, 653–665. [Google Scholar]
- Kouzuki, H.; Suzuki, H.; Stieger, B.; Meier, P.J.; Sugiyama, Y. Characterization of the transport properties of organic anion transporting polypeptide 1 (oatp1) and Na(+)/taurocholate cotransporting polypeptide (Ntcp): comparative studies on the inhibitory effect of their possible substrates in hepatocytes and cDNA- transfected COS-7 cells. J. Pharmacol. Exp. Ther. 2000, 292, 505–511. [Google Scholar]
- Thorsteinsson, S.B.; Bergan, T.; Oddsdottir, S.; Rohwedder, R.; Holm, R. Crystalluria and ciprofloxacin, influence of urinary pH and hydration. Chemotherapy 1986, 32, 408–417. [Google Scholar]
- Hootkins, R.; Fenves, A.Z.; Stephens, M.K. Acute renal failure secondary to oral ciprofloxacin therapy: a presentation of three cases and a review of the literature. Clin. Nephrol. 1989, 32, 75–78. [Google Scholar]
- Connor, J.P.; Curry, J.M.; Selby, T.L.; Perlmutter, A.D. Acute renal failure secondary to ciprofloxacin use. J. Urol. 1994, 151, 975–976. [Google Scholar]
- Sedlacek, M.; Suriawinata, A.A.; Schoolwerth, A.; Remillard, B.D. Ciprofloxacin crystal nephropathy—a 'new' cause of acute renal failure. Nephrol. Dial. Transplant. 2006, 21, 2339–2340. [Google Scholar]
- Stratta, P.; Lazzarich, E.; Canavese, C.; Bozzola, C.; Monga, G. Ciprofloxacin crystal nephropathy. Am. J. Kidney Dis. 2007, 50, 330–335. [Google Scholar]
- Traa, B.S.; Walker, C.L.; Munos, M.; Black, R.E. Antibiotics for the treatment of dysentery in children. Int. J. Epidemiol. 2010, 39 (Suppl. 1), i70–i74. [Google Scholar] [PubMed]
- Höffken, G.; Lode, H.; Prinzing, C.; Borner, K.; Koeppe, P. Pharmacokinetics of ciprofloxacin after oral and parenteral administration. Antimicrob. Agents Chemother. 1985, 27, 375–379. [Google Scholar]
- Jaehde, U.; Sorgel, F.; Reiter, A.; Sigl, G.; Naber, K.G.; Schunack, W. Effect of probenecid on the distribution and elimination of ciprofloxacin in humans. Clin. Pharmacol. Ther. 1995, 58, 532–541. [Google Scholar]
- Terada, T.; Inui, K. Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A). Biochem. Pharmacol. 2008, 75, 1689–1696. [Google Scholar]
- Ohta, K.Y.; Imamura, Y.; Okudaira, N.; Atsumi, R.; Inoue, K.; Yuasa, H. Functional characterization of multidrug and toxin extrusion protein 1 as a facilitative transporter for fluoroquinolones. J. Pharmacol. Exp. Ther. 2009, 328, 628–634. [Google Scholar]
- Sörgel, F.; Kinzig, M. Pharmacokinetics of gyrase inhibitors, Part 1: Basic chemistry and gastrointestinal disposition. Am. J. Med. 1993, 94, 44S–55S. [Google Scholar]
- Mohkam, M.; Karimi, A.; Gharib, A.; Daneshmand, H.; Khatami, A.; Ghojevand, N.; Sharifian, M. Ceftriaxone associated nephrolithiasis: a prospective study in 284 children. Pediatr. Nephrol. 2007, 22, 690–694. [Google Scholar]
- Avci, Z.; Koktener, A.; Uras, N.; Catal, F.; Karadag, A.; Tekin, O.; Degirmencioglu, H.; Baskin, E. Nephrolithiasis associated with ceftriaxone therapy: a prospective study in 51 children. Arch. Dis. Child. 2004, 89, 1069–1072. [Google Scholar]
- Stojanovic, V.; Djuric, V.G. Nephrolithiasis caused by ceftriaxone in a 3-year-old child with ureteropelvic junction obstruction. Case Report. Med. 2009, 2009, 365962:1–365962:3. [Google Scholar]
- Monte, S.V.; Prescott, W.A.; Johnson, K.K.; Kuhman, L.; Paladino, J.A. Safety of ceftriaxone sodium at extremes of age. Expert Opin. Drug Saf. 2008, 7, 515–523. [Google Scholar]
- Patel, I.H.; Chen, S.; Parsonnet, M.; Hackman, M.R.; Brooks, M.A.; Konikoff, J.; Kaplan, S.A. Pharmacokinetics of ceftriaxone in humans. Antimicrob. Agents Chemother. 1981, 20, 634–641. [Google Scholar]
- Stoeckel, K.; Trueb, V.; Dubach, U.C.; McNamara, P.J. Effect of probenecid on the elimination and protein binding of ceftriaxone. Eur. J. Clin. Pharmacol. 1988, 34, 151–156. [Google Scholar]
- Fukumoto, K.; Aida, S.; Oishi, T.; Ueno, K. Pharmacokinetics of ceftriaxione, a third-generation cephalosporin, in pediatric patients. Biol. Pharm. Bull. 2009, 32, 1139–1141. [Google Scholar]
- Blum, M.R.; Liao, S.H.; de Miranda, P. Overview of acyclovir pharmacokinetic disposition in adults and children. Am. J. Med. 1982, 73, 186–192. [Google Scholar]
- Laskin, O.L.; de Miranda, P.; King, D.H.; Page, D.A.; Longstreth, J.A.; Rocco, L.; Lietman, P.S. Effects of probenecid on the pharmacokinetics and elimination of acyclovir in humans. Antimicrob. Agents Chemother. 1982, 21, 804–807. [Google Scholar]
- De Bony, F.; Tod, M.; Bidault, R.; On, N.T.; Posner, J.; Rolan, P. Multiple interactions of cimetidine and probenecid with valaciclovir and its metabolite acyclovir. Antimicrob. Agents Chemother. 2002, 46, 458–463. [Google Scholar]
- Hilgendorf, C.; Ahlin, G.; Seithel, A.; Artursson, P.; Ungell, A.L.; Karlsson, J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab. Dispos. 2007, 35, 1333–1340. [Google Scholar]
- Gorboulev, V.; Ulzheimer, J.C.; Akhoundova, A.; Ulzheimer-Teuber, I.; Karbach, U.; Quester, S.; Baumann, C.; Lang, F.; Busch, A.E.; Koepsell, H. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997, 16, 871–881. [Google Scholar]
- Adachi, M.; Sampath, J.; Lan, L.B.; Sun, D.; Hargrove, P.; Flatley, R.; Tatum, A.; Edwards, M.Z.; Wezeman, M.; Matherly, L.; Drake, R.; Schuetz, J. Expression of MRP4 confers resistance to ganciclovir and compromises bystander cell killing. J. Biol. Chem. 2002, 277, 38998–39004. [Google Scholar]
- de Miranda, P.; Blum, M.R. Pharmacokinetics of acyclovir after intravenous and oral administration. J. Antimicrob. Chemother. 1983, 12 (Suppl. B), 29–37. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hagos, Y.; Wolff, N.A. Assessment of the Role of Renal Organic Anion Transporters in Drug-Induced Nephrotoxicity. Toxins 2010, 2, 2055-2082. https://doi.org/10.3390/toxins2082055
Hagos Y, Wolff NA. Assessment of the Role of Renal Organic Anion Transporters in Drug-Induced Nephrotoxicity. Toxins. 2010; 2(8):2055-2082. https://doi.org/10.3390/toxins2082055
Chicago/Turabian StyleHagos, Yohannes, and Natascha A. Wolff. 2010. "Assessment of the Role of Renal Organic Anion Transporters in Drug-Induced Nephrotoxicity" Toxins 2, no. 8: 2055-2082. https://doi.org/10.3390/toxins2082055